
Abstract
The anti-proliferative activities of Novel series of 2-(4-fluorophenyl) imidazol-5-ones against MCF-7 breast cancer cell line were explored via in-slico studies which includes Quantitative structure–activity relationship QSAR, molecular docking studies, designing new compounds, and analyzing the pharmacokinetics properties of the designed compounds. From the QSAR analysis, model number one emerged the best as seen from the arithmetic assessments of (R2) = 0.6981, (R2adj) = 0.6433, (Q2) = 0.5460 and (R2pred) of 0.5357. Model number one was used in designing new derivative compounds, with higher effectiveness against estrogen positive breast cancer (MCF-7 cell line). The Molecular docking studies between the derivatives and Polo-like kinases (Plk1) receptor proved that the derivatives of 2-(4-fluorophenyl) imidazol-5-ones bind tightly to the receptor, thou ligand 24 and 27 had the highest binding affinities of −8.8 and − 9.1 kcal/mol, which was found to be higher than Doxorubicin with a docking score of −8.0 kcal/mol. These new derivatives of 2-(4-fluorophenyl) imidazol-5-ones shall be excellent inhibitors against (plk1). The pharmacokinetics analysis performed on the new structures revealed that all the structures passed the test and also the Lipinski rule of five, and they could further proceed to pre-clinical tests. They both revealed a revolution in medicine for developing novel anti-breast cancer drugs against MCF-7 cell line.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cancer is described by uncontrolled cell proliferation, thereby affecting the surrounding tissue, and over again spreading throughout the body. It is a complicated disease. (Bajaj et al. 2018). Despite the vast high-tech and social enhancement, cancer remains the most alarming disease and a leading cause of pain and death in humans (Bhaumik et al. 2019).
Among women, after lung cancer, cancer of the breast is the second source of mortality. About 40,610 women passed away from cancer of the breast and about 252,710 more diagnoses were projected in 2017 (Bajaj et al. 2018). Cancer of the breast accounts for about 24% of all cancer types in females (Xiao et al. 2018). A likely population of people with breast cancer has similar features such as older age, lack of prolonged breast feeding, adding weight, overdue age at first birth, lack of exercise, etc. (Liu et al. 2018). Detailed investigation of pathways and mechanisms on how cancer spreads and discovering many anti-cancer agents have made a breakthrough in the treatment of cancer (Bhaumik et al. 2019).
Luminal type of breast cancer is Estrogen receptor (ER)−/progesterone receptor (PR) - the positive type which is caused by the overexpression of estrogen receptor α (ERα). It is made up of about 70% of the mammary tumor patients tagged as ER-positive (ER+). The endless stimulation of ERα by estrogens induce the multiplication of cancer cells, MCF-7 cell line (Jordan et al. 2007). The master mitotic regulator, Polo-like kinase 1 (Plk1), is an important gene cell division and a known cancer drug target. It is found overexpressed in a large collection of different cancer types and this tumoral overexpression often correlates with poor patient prognosis (De Cárcer 2019).
In the study of (Sanhaji et al. 2012), it showed that Polo-like kinases (Plk1) causes destructive proliferation in tumor cells and strongly stimulates the development of cell circle. Plk1 overexpression allows cells to supersede barriers, causing genomic uncertainty and stimulating the alteration of mammalian cells. Plk1 was proven as amongst the utmost striking receptor for breast cancer treatment. Plk1 mediates estrogen receptor (ER) which regulates gene overexpression in human breast cancer cells. Recently, (Abo-Elanwar et al. 2019) reported novel Thirty-nine derivatives of Imidazolones connected to chalone moiety which showed a great anti-breast cancer activity against MCF-7 cell line. Imidazole and its derivatives have much significance in both natural products and synthetic molecules. The exclusive molecules have great electron-rich features which allow them to bind easily with diverse enzymes and receptors, thus showing wide anti-proliferative activities (Zhang et al. 2020). Several activities such as anticancer, antimicrobial (Premakumari et al. 2014), cardio-activity, and angiotensin II receptor antagonistic activity have been described explicitly in compounds containing imidazolone moiety (Abo-Elanwar et al. 2019).
Chemotherapy remains one of the best and fast clinical options though it is often limited due to undesirable toxic effect including weight loss, fatigue, nausea, loss of appetite, and so on, making it urgent to develop more effective drug candidates with less toxicity to eradicate this disease (Iqbal et al. 2019). The computer-aided drug design approach saves time and ensures better effectiveness of the drug candidate. This research would be aimed at exploring the novel derivatives of imidazolone by building a mathematical model (QSAR) that predicts the anti-proliferative activities from its compound and using plk1 receptor with the derivatives to understand their interactions via molecular docking studies towards anti-breast cancer drug discovery, concentrating on breast cancer treatment with less toxicity and more effectiveness.
Methodology
Computer applications
The software includes; Chemdraw version 12.0.2, Spartan’14 (version 1.1.2), Material studio (V8) software, Pyrex software, PADEL V2.20, DTC data lab software version, and Auto dock visualizer version 4.2.
QSAR studies
Dataset
39 derivatives of Imidazolones connected to chalone moiety with anti-proliferative activities (IC50) on MCF-7 cancer cell lines were obtained from (Abo-Elanwar et al. 2019) writings. The anti-proliferative activities were measured in inhibitory concentration (IC50) and then converted to the logarithm scale (pIC50). The tabulated form of the IC50 is measured in concentration of micromolar (μM) and the pIC50 is shown in Table 1.
Molecular optimization
The geometric optimization is executed such that the countable electronic and molecular parameters could depict the original physicochemical properties of the observed molecule. (Putri et al. 2019). The derivatives were sketched using Chemdraw (V12.0.2) software in 2D format. They were then converted to 3D format and further optimized using Spartan 14 (V1.1.4) software, with the parameters, Density Functional Theory (DFT) at B3LYP, 6-31G/ basis set (Ibrahim et al. 2018; Abdullahi et al. 2019).
Molecular descriptors
Pharmaceutical Data Exploration Laboratory Software V (2.20) was used in calculating molecular descriptors for the thirty-nine (39) optimized compounds of Imidazolones derivatives which were then converted to SDF format after optimization. (Yap 2011).
Pretreatment and division of data set
The outcomes obtained from PADEL-software were pretreated to remove persistent values and undesirable descriptors using Data Pre-treatment software GUI 1.2. (Abdulrahman et al. 2020). Kennard-Stone algorithm (Kennard and Stone 1969) method divided the derivatives into 27 calibration and 12 validation sets to get a mathematical equation.
QSAR equation
Material studio software (V8) was used in constructing a model with Genetic Function Approximation (GFA) procedure. The dependent variable is the anti-proliferative activities (pIC50) and the independent variable is model parameters.
Validating the equation (internal)
The validity of the built model should be tested on an external set of data that has not yet been used during the process of developing the model (Tropsha et al. 2003). The built equations were evaluated using the Friedman formula; (Friedman 1991).
Where: SEE is the Standard Estimated Error. If SEE is small, it suggests an enhanced equation. SEE is expressed below;
d equals user-defined smoothing parameter, C equals to the sum of the model terms, M equals the sum of train set compounds and p is the equation parameters (Ibrahim et al. 2018). The correlation coefficient (R2) accounts for the fragment of the total difference of the equation. The nearer the R2 value is to 1, the more enhanced the model is built. R2 is given as:
Where Yexp and Ypred are the means of biological and calculated activities of the calibration set (Tropsha et al. 2003).
R2 rises as the number of descriptors increase. Hence R2 does not assure the effectiveness of the equations. R2adj is used to reconfirm the strength and effectiveness of the equation.
Where p and n are the descriptors from the equation and calibration set. Validation coefficient test (Q2cv) was used in assessing the robustness of the model and prediction power of the derivatives, it’s given as:
Ymintraining Yexp, and Ypred equals to the mean activities (pIC50) of calibration set, bio-activities (IC50) and calculated of the calibration set (Brandon and Orr 2015).
External model validation
Mean effect
The mean effect shows the descriptors or model parameters that influence the generated equation.
The symbols on the model parameters show the various impact of each parameter in the overall derived equation, either an increase or decrease of the model parameter. Thus it’s expressed as;
Where m equals the model parameters, Bj equals to descriptors coefficient j, n equals to the prediction set molecules and Dj is the matrix value of the model parameter in the prediction set (Minovski et al. 2013).
Variance inflation factor (VIF)
The VIF takes into account the amount of co-linearity amongst the descriptors in an equation. It is calculated as
R2 is the correlation coefficient. (Myers 1990).
The greater the value, the bigger the link amongst the model parameters. The VIF values of less than 10 show the equation is stable while the values above 10 indicate the equation is not effective and cannot be used.
Applicability domain
The applicability domain approach is aimed at estimating independently, the reliability of every generated equation (Eriksson et al. 2003). Model validation should be within the training domain and the compounds need to be assessed as fitting within the domain to ascertain the model. The applicability domain is evaluated by the leverage values for each compound. Leverage defines the applicability domain of the built equation (Veerasamy et al. 2011). It is given as;
Where XT is the matrix transpose of X used in constructing the equation, Xi is the matrix of calibration set of I and X is the n x k matrix of train set descriptors. (d*) is the warning leverage, d* searches for outliers. It is shown as;
k equals to the total model parameters and n equals to the calibration compounds. William’s plot (A plot of standardized values vs. leverages) of the calibration and validation compounds. Molecules found in the warning leverages within the graph are the calculated molecules.
Quality assurance model generated
Table 2 shows the least required values in assessing the mathematical equation (Ibrahim et al. 2018). The table parameters were used in conforming to the effectiveness and prediction power of the derived equations.
Molecular docking studies
Five compounds with high pIC50 underwent molecular docking studies with the receptor Polo-like kinase 1(PKL1) in complex with B16727. The receptor used was obtained from Protein Data Bank (Code: 3FC2) and was set using Discovery studio software, the ligand (compounds) were also converted to PBD format as shown in Fig. 4. Autodock Vina of Pyrx software was employed in calculating the binding affinity of the ligand and receptor (Abdulfatai et al. 2018).
Pharmacokinetics (drug-likeness)
SwissADME; an online tool, used in investigating the ADME property physicochemical, pharmacokinetic, and medicinal chemistry responsiveness of smalls compounds (Daina et al. 2017) was employed in assessing the Pharmacokinetic parameters of the new structures.
Again, the designed compounds were checked for their adaptability with Lipinski’s rule of five (Hou et al. 2019), well-used criteria to comprehend if a compound can be taken orally or not, such as molecular weight (MW) ≤ 500, octanol/water partition coefficient (AlogP) ≤ 5, number of hydrogen bond donors (HBDs) ≤ 5 and number of hydrogen bond acceptors (HBAs) ≤ 10.6. According to the rule of five, a compound cannot be taken orally if it does not meet up to two or rules out of the rules of five (Guan et al. 2019).
Results and discussion
QSAR of 2-(4-fluorophenyl) imidazol-5-ones derivatives
QSAR examination was used to verify the relationship of 2-(4-fluorophenyl) imidazol-5-ones derivatives with its anti-proliferative activities. Using the Genetic Function Approximation (GFA) method, four QSAR equations were built to predict the anti-proliferative activities of imidazole derivatives. From both internal and external validation parameters, model number 1 passed with correlation coefficient squared (R2) of 0.6981, correlation coefficient adjusted squared (R2adj) of 0.6433, cross-validation coefficient (Q2) of 0.5460 and external validation (R2pred) of 0.5390. Tables 3 and 4 shows how the external validation of model 1 was calculated using the validation set (test set) and model descriptors.
-
Model 1
-
Model 2
-
Model 3
-
Model 4
The biological, calculated and the residual values of 2-(4-fluorophenyl) imidazol-5-ones compounds are seen in Table 5.
The descriptors obtained from the mathematical model 1 were defined and classified as seen in Table 6.
Further statistical analysis was carried out on the model parameters to find the correlation between the individual descriptors and also the impact of each descriptor in the model. The results are shown in Table 7.
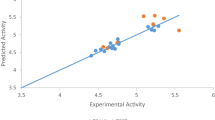
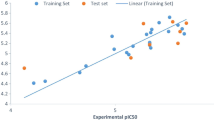
A graph of the calculated activities against the biological activities was drawn to show the relationship between derivatives as seen in Fig. 1.

Plot of calculated activity versus inhibition concentration (biological activities)
Figure 2 shows a plot of standardized residual values versus the experimental activities of the derivatives.

Plot of standardized residual against bioactivities (Experimental activity).
A graph of standardized residual against the leverages was plotted to show the derivatives that fell within the applicability domain as seen in Figs. 3 and 4.

The William’s plot

3D representation of prepared ligand and receptor
Molecular docking analysis
A summary of the relationship between some 2-(4-fluorophenyl) imidazole-5-ones derivatives and the receptor was shown in Table 8. The pictorial analysis of docked compounds was accessed by assessing the hydrogen bond interactions, hydrogen bond length, and hydrophobic interactions. Both 2D representations of the binding pose of compounds 24 and 27 to the active pocket of the protein target are shown in Figs. 5 and 6 respectively.

2D and 3D representation of complex 27

3D and 2D representation of complex 24
Ligand Base drug design
Ligand based approach was used in designing 18 new imidazole derivative compounds with higher calculated activities than that of the template compounds as shown in Table 9.
Pharmacokinetics of designed 2-(4-fluorophenyl) imidazole-5-ones compounds
The newly designed compounds were further explored to ascertain their drug-friendliness. The pharmacokinetic analysis of the new compounds are shown in Table 10, all the compounds pass the Lipinski rule of five test. The bioavailability radar of molecules 11, 13, and 17 are shown in Fig. 7.

The bioavailability radar for molecules 11, 13, and 17
Discussion
QSAR of 2-(4-fluorophenyl) imidazol-5-ones derivatives
The results obtained from the QSAR analysis showed that both internal and external validation of the model were in agreement with the minimum proposed values used in assessing the equation as seen in Table 2 above. The external validation of model 1 was achieved using the model parameters from the validation compounds as seen in Tables 3 and 4. The effectiveness of the equation was measured by the reliability of the validation compounds and calculated pIC50 of the calibration compounds. The biological, calculated and the residual values of 2-(4-fluorophenyl) imidazol-5-ones compounds are seen in Table 5. The low residual values are obtained from the difference between the biological and calculated activities, showing the high prediction power of eq. 1. Both internal and external validation conforms eq. 1 to be greatly effective, strong, and extremely predictive. Table 6 shows the definition of the model 1 parameters. The mean effect obtained from the model parameters, shows (GATS5e, MATS4e, and SpMax4_Bhs) carries a positive coefficient showing that an increase in those factors would increase the bioactivities of the derivatives while (RDF150u) carrying a negative coefficient indicates a decrease in the descriptor would also increase the experimental activities of 2-(4-fluorophenyl) imidazol-5-ones derivative compounds. The statistical analysis shows that there is no much collinearity amongst the model parameters ensuring that the equation is highly robust as seen in Table 7.
The graph of calculated activities (pIC50) against biological activities (IC50) as shown in Fig. 1 indicates the pIC50 has been in good agreement with the biological activities as seen in Table 3. Figure 2 shows the values of both calibration and validation compounds spread on both sides of the graph, showing no systematic errors between the standardized residual versus bio-activities (Experimental activity) (Jalali-Heravi and Kyani 2004). Fig. 3 shows William’s graph (standardized residuals against leverages), indicating that all the molecules fell in the warning leverage area, calculated to be (h = 0.56).
Molecular docking analysis
The docking analysis on compounds of 2-(4-fluorophenyl) imidazole-5-ones derivatives with the protein target, Polo-like kinase 1(PKL1) in complex with B16727 was performed. 5 compounds with high pIC50 were chosen for these studies, amongst the 5, compound 24 and 27 had the highest docking score of −8.8 and − 9.1 kcal/mol as seen in Table 8.
Compound 27 showed backbone conventional hydrogen bonding interaction between -NH group with LYS82 (2.7685A0) and carbonyl group with LEU59 (2.0889A0). Three amino acids showed electrostatic interaction at LYS82 (4.57869A0) which is a pi-orbital cation interaction then GLU101 (4.83015A0), ASP194 (3.54145A0) are pi-orbital anion interaction. Furthermore, the compound formed a hydrophobic bond with three amino acids of LEU59 at distance 3.8345A0, 3.93488A0, and 4.89515A0, then GLY62 (3.80969A0), ARG136 (4.58314A0), and ALA65 (4.75156A0).
Again in compound 24, ARG136 and ASP 139 gave covalent hydrogen interaction at a distance of 2.60978A0 and 1.99045A0. Two electrostatic bonds occur with the compound at LYS82 (4.16479A0) and ASP194 (1.99045A0). It formed a hydrophobic bond with PHE64 (5.25545A0), MET98 (4.1282A0) also, the pi-orbital containing delocalized electrons in the benzene ring interact with the alkyl groups of PHE64 (4.85386A0), CYS67 (5.47383A0), ALA65 (5.17103A0), MET98 (5.26908A0), ALA80 (5.26908A0) three amino acids of PRO384 (5.1107A0, 4.7845A0 and 4.7531A0) to form a hydrophobic bond. Both the hydrogen bond and the hydrophobic interactions in the complexes showed that ligand 24 and 27 of 2-(4-fluorophenyl) imidazole-5-ones derivatives are most active against Polo-like kinase 1(PKL1) in complex with B16727 respectively.
Ligand based design
Eighteen (18) new 2-(4-fluorophenyl) imidazole-5-ones derivative compounds were designed, their predicted activities were higher than that of the chosen template (compounds 4 with pIC50−5.0620 and compound 2 with pIC50–4.9846) as shown in Table 9. From the mean effect of the descriptors, SpMax4_Bhs had a greater positive impact followed by GATS5e and MATS4e while RDF150u had the least negative impact on the model. According to MATS4e (Moran autocorrelation - lag 4 / weighted by Sanderson electronegativities) and GATS5e (Geary autocorrelation - lag 5 / weighted by Sanderson electronegativities) descriptors, adding more electronegative elements (GATS5e and MATS4e) would increase the potency of the designed compounds. The modification occurred by adding more electronegative elements to the template (compounds 2 and 4).
Pharmacokinetics of designed 2-(4-fluorophenyl) imidazole-5-ones compounds
The designed compounds were assessed for their drug-friendliness. The molecules passed the drug- friendliness assessment (ADME and physicochemical properties) as shown in Table 10, none of the designed compounds violated two rules out of the Lipinski rule of five; a famous benchmark utilized in invalidating the drug-likeness of a molecule (as stated in “Molecular Docking Studies” section). The bioavailability radar of molecules 11, 13, and 17 are shown in Fig. 7. The bioavailability radar gives a quick and easy summary of the pharmacokinetic properties of a compound. The pink area signifies the ideal ranges for each property (lipophilicity: XLOGP3 from −0.7 to +5.0, size: molecular weight from 150 to 500 g/mol, polarity: TPSA from 20 to 130 Å2, solubility: log S less than 6, saturation: the fraction of carbons in the sp3 hybridization not higher than 0.25, and flexibility: less than 9 rotatable bonds) (Daina et al. 2017).
Conclusion
2-(4-fluorophenyl) imidazole-5-ones derivatives showed a more reliable anti-cancer drug candidate against MCF-7 cell line using QSAR analysis, molecular docking assessment, and pharmacokinetics analysis. The model 1 parameters obtained from QSAR showed that increasing MATS4e and RDF150u, and decreasing GATS5e and SPMAX4_Bhs would proliferation the biological activities of the inhibitors 2-(4-fluorophenyl) imidazol-5-ones derivatives as an effective drug for curing breast cancer. The strength and predictive capability of the generated equation was explored for both internal and external validation assessment which conforms with the least approved values, indicating that model number one parameters could be used in developing new 2-(4-fluorophenyl) imidazol-5-ones drug compounds with higher effectiveness. The model parameters (MATS4e and GATS5e) had more significant and based on their mean effect, adjustment were made on the fragments of the lead compounds (2 and 4) to design 18 new imidazole derivative compounds with a higher calculated activity against MCF-7 cell line.
The molecular docking result showed that compound 24 and 27 had the highest docking score of −8.8 and − 9.1 kcal/mol. From the research it is proved that some series of 2-(4-fluorophenyl) imidazol-5-ones derivatives compounds bind tightly to the binding pose of the target, stabilizing the receptor Polo-like kinase 1(PKL1) in complex with B16727 which is proven from the complexes as seen above. The compounds would serve as the most capable inhibitors against (PKL1) and this shows a revolution in medicine to design and develop new estrogen-positive (MCF-7 cell line) breast cancer drugs.
Additionally, the pharmacokinetics analysis (drug-likeliness test) executed on the designed molecules revealed that all the compounds can move on to the next step of pre-clinical trial because they passed drug-friendliness analysis (ADME and other physicochemical properties) and they also adhered to the Rule of Five: a benchmark used in assessing the drug-likeness of compounds. This shows a great discovery for medicine in finding permanent solutions to breast cancer (MCF-7 cell line).
Data availability
The data set was obtained from (Abo-Elanwar et al. 2019).
References
Abdulfatai U, Uzairu A, Uba S (2018) Molecular docking and quantitative structure-activity relationship study of anticonvulsant activity of aminobenzothiazole derivatives. Beni-Suef Univ JBasic App Sci 7(2):204–214
Abdullahi M, Uzairu A, Shallangwa GA, Mamza P, Arthur DE, Ibrahim MT (2019) In-silico modeling studies on some C14-urea-tetrandrine derivatives as potent anti-cancer agents against prostate (PC3) cell line. J King Saud Univ-Sci
Abdulrahman HL, Uzairu A, Uba S (2020) In silico studies of some 2-anilinopyrimidine derivatives as anti-triple-negative breast cancer agents. Beni-Suef University Journal of Basic and Applied Sciences 9(1):1–12
Abo-Elanwar YA, Mostafa AS, El-Sayed MA, Nasr MN (2019) Synthesis and biological evaluation of new 2-(4-fluorophenyl) imidazol-5-ones as anticancer agents. J Appl Pharm Sci 9(05):001–011
Bajaj S, Roy PP, Singh J (2018) Synthesis, thymidine phosphorylase inhibitory, and computational study of novel 1, 3, 4-oxadiazole-2-thione derivatives as potential anticancer agents. Comput Biol Chem 76:151–160
Bhaumik I, Pal K, Debnath U, Karmakar P, Jana K, Misra AK (2019) Natural products inspired allicin analogs as novel anti-cancer agents. Bioorg Chem 86:259–272
Brandon V, Orr A (2015) Comprehensive R archive network (CRAN): http://CRAN.Rproject.org
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness, and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717
De Cárcer G (2019) The mitotic cancer target polo-like kinase 1: oncogene or tumor suppressor? Genes 10(3):208
Eriksson L, Jaworska J, Worth AP, Cronin MT, McDowell RM, Gramatica P (2003) Methods for reliability and uncertainty assessment and applicability evaluations of classification-and regression-based QSARs. Environ Health Perspect 111(10):1361–1375
Friedman JH (1991) Multivariate adaptive regression splines. Ann Stat 1–67
Guan L, Yang H, Cai Y, Sun L, Di P, Li W, ..., Tang Y (2019). ADMET-score–a comprehensive scoring function for the evaluation of chemical drug-likeness. MedChemComm 10(1): 148–157
Hou Y, Zhu L, Li Z, Shen Q, Xu Q, Li W, …, Gong P (2019). Design, synthesis and biological evaluation of novel 7-amino-[1,2,4] triazolo [4,3-f] pteridinone and 7-aminotetrazolo [1,5-f] pteridinone derivative as potent antitumor agents. Eur J Med Chem 163: 690–709
Ibrahim MT, Uzairu A, Shallangwa GA, Ibrahim A (2018) In-silico studies of some oxadiazoles derivatives as anti-diabetic compounds. J King Saud Univ-Sci
Iqbal J, Ejaz SA, Khan I, Ausekle E, Miliutina M, Langer P (2019) Exploration of quinolone and quinoline derivatives as potential anticancer agents. DARU J Pharm Sci 1–14
Jordan VC, Brodie AM (2007) Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids 72(1):7–25
Kennard RW, Stone LA (1969) Computer-aided design of experiments. Technometrics 11:137–148
Liu L, Tang Z, Wu C, Li X, Huang A, Lu X, You Q, Xiang H (2018) Synthesis and biological evaluation of 4, 6-diaryl-2-pyrimidinamine derivatives as anti-breast cancer agents. Bioorg Med Chem Lett 28(6):1138–1142
Minovski N, Zˇuperl Š, Drgan V, Novicˇ M (2013) Assessment of applicability domain for multivariate counter-propagation artificial neural network predictive models by minimum Euclidean distance space analysis: a casestudy. Anal Chim Acta 759:28–42
Myers RH (1990) Classical and modern regression application. Duxbury Press
Premakumari C, Muralikrishna A, Padmaja A, Padmavathi V, Park SJ, Kim TJ, Reddy GD (2014) Synthesis, antimicrobial and anticancer activities of amido sulfonamido methane linked bis heterocycles. Arab J Chem 7(4):385–395
Putri DE, Pranowo HD, Haryadi WINARTO (2019) Study on anti-tumor activity of novel 3-substituted 4 Anilino-Coumarin derivatives using quantitative structure-activity relationship (QSAR). In Materials Science Forum (Vol. 948, pp. 101-108). Trans Tech Publications
Sanhaji M, Kreis NN, Zimmer B, Berg T, Louwen F, Yuan J (2012) p53 is not directly relevant to the response of polo-like kinase 1 inhibitors. Cell Cycle 11(3):543–553
Tropsha A, Gramatica P, Gombar VK (2003) The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. Mol Inf 22:69–77
Veerasamy R, Rajak H, Jain A, Sivadasan S, Varghese CP, Agrawal RK (2011) Validation of QSAR models-strategies and importance. Int J Drug Des Discov 3:511–519
Xiao X, Xu M, Yang C, Yao Y, Liang LN, Chung PE, ..., Ben-David Y (2018) Novel racemosin B derivatives as new therapeutic agents for aggressive breast cancer. Bioorg Med Chem 26(23–24): 6096–6104
Yap CW (2011) PaDEL-descriptor: an open-source software to calculate molecular descriptors and fingerprints. J Comput Chem 32:1466–1474
Zhang C, Li Q, Meng L, Ren Y (2020) Design of novel dopamine D2 and serotonin 5-HT2A receptors dual antagonists toward schizophrenia: an integrated study with QSAR, molecular docking, virtual screening, and molecular dynamics simulations. J Biomol Struct Dyn 38(3):860–885
Acknowledgments
The authors acknowledge the technical effort of the physical chemistry Department, Ahmadu Bello University Zaria for their immense advice and various contribution toward the successful completion of the research.
Author information
Authors and Affiliations
Contributions
HAL worked on the data set computationally by the methodology to get a mathematical model with high predictive activity and was a major contributor in drafting the manuscript, AU carried out statistical analysis on the built model to ensure its effectiveness and also participated in the write-up and SU re-edited the work to ensure it conforms with the manuscript’s guide. All the Arthurs read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Not applicable.
Code availability
The software’s used includes; Chemdraw software version 12.0.2, Spartan’14 (version 1.1.2), Material studio (V8) software, Pyrex software version, PADEL-Descriptor Software V2.20, and DTC data lab software, Microsoft word Office Excel 2013 version and Auto dock visualizer version 4.2,
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lawal, H.A., Uzairu, A. & Uba, S. QSAR, molecular docking, design, and pharmacokinetic analysis of 2-(4-fluorophenyl) imidazol-5-ones as anti-breast cancer drug compounds against MCF-7 cell line. J Bioenerg Biomembr 52, 475–494 (2020). https://doi.org/10.1007/s10863-020-09858-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10863-020-09858-0