
Abstract
Limitations in the applicability, accuracy, and precision of individual structure characterization methods can sometimes be overcome via an integrative modeling approach that relies on information from all available sources, including all available experimental data and prior models. The open-source Integrative Modeling Platform (IMP) is one piece of software that implements all computational aspects of integrative modeling. To maximize the impact of integrative structures, the coordinates should be made publicly available, as is already the case for structures based on X-ray crystallography, NMR spectroscopy, and electron microscopy. Moreover, the associated experimental data and modeling protocols should also be archived, such that the original results can easily be reproduced. Finally, it is essential that the integrative structures are validated as part of their publication and deposition. A number of research groups have already developed software to implement integrative modeling and have generated a number of structures, prompting the formation of an Integrative/Hybrid Methods Task Force. Following the recommendations of this task force, the existing PDBx/mmCIF data representation used for atomic PDB structures has been extended to address the requirements for archiving integrative structural models. This IHM-dictionary adds a flexible model representation, including coarse graining, models in multiple states and/or related by time or other order, and multiple input experimental information sources. A prototype archiving system called PDB-Dev (https://pdb-dev.wwpdb.org) has also been created to archive integrative structural models, together with a Python library to facilitate handling of integrative models in PDBx/mmCIF format.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Overview of integrative structure modeling
Interactions among molecules lead to the emergence of biological phenomena—most evidently in the forms of macromolecular machines and dynamic liaisons that transmit information and control behaviors. Thus, the structures of proteins and their complexes are generally helpful in understanding their function, modulating their activities, and mapping their evolution. Experimental determination of the structures of biomolecular systems is often rather difficult, as no single experimental method is universally applicable. For example, crystals suitable for X-ray crystallography cannot always be produced, especially for large assemblies of multiple components (Blundell and Johnson 1976; Holcomb et al. 2017). Although cryo-electron microscopy (cryo-EM) can be used to study large assemblies, the resolution can be limited (Chiu et al. 2005; Lucic et al. 2008; Stahlberg and Walz 2008). Finally, molecular biology, biochemistry, and proteomics techniques, such as yeast two-hybrid (Parrish et al. 2006), affinity purification (Fernandez-Martinez et al. 2012), and mass spectrometry (Gingras et al. 2007), can yield information about the interactions between proteins, but not the positions of these proteins within the assembly or the structures of the proteins themselves.
Limitations in the applicability, accuracy, and precision of individual structure characterization methods can sometimes be overcome via an integrative modeling approach that relies on information from all available sources, including all available experimental data and prior models (Sali et al. 2003; Ward et al. 2013; Joseph et al. 2017; Kim et al. 2018; Rout and Sali 2019) Integrative modeling is cast as a computational optimization problem in which information can be used in the following five ways, guided by maximizing the accuracy and precision of the model while remaining computationally feasible: (i) representing components of a model with some variables (e.g., atomic coordinates, coarse-grained representations), (ii) scoring alternative models for their consistency with input information, (iii) searching for good-scoring models, (iv) filtering models based on input information, and (v) validation of models. Much of the input information about the modeled system is encoded into data-based restraints comprising a scoring function ((ii) above) used to evaluate candidate models produced by structural sampling ((iii) above). In this regard, integrative modeling is similar to protein structure determination by nuclear magnetic resonance (NMR) spectroscopic methods in which spatial restraints implied by the NMR data, such as nuclear overhauser effects (NOE) and J-coupling constants, must be satisfied. By simultaneously considering all available information, the integrative approach maximizes the accuracy, precision, completeness, and efficiency of structure determination.
Numerous static structures of large complexes have already been solved using integrative methods; for example, the 26S proteasome (Lasker et al. 2012), the type III secretion system needle (Loquet et al. 2012), chromatin comprising the alpha-globin gene neighborhood (Bau et al. 2011), the yeast core spindle pole body (Viswanath et al. 2017a), and the yeast nuclear pore complex (NPC) (Kim et al. 2018). Moreover, the integrative approach can be extended from modeling a single static structure to computing models of multiple structural states in a heterogeneous sample (e.g., the two states in the functional cycle of PhoQ kinase (Molnar et al. 2014)), spatiotemporal models of dynamic processes (e.g., macromolecular transport through the NPC (Raveh et al. 2016; Timney et al. 2016)), and models of molecular networks (e.g., metabolic pathway for gulonate synthesis (Calhoun et al. 2018)).
Modeling with IMP
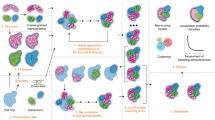
There are multiple software packages that can be useful for integrative modeling. The open-source Integrative Modeling Platform (IMP) software (https://integrativemodeling.org) (Alber et al. 2007a, b; Russel et al. 2009, 2012; Lasker et al. 2010a; Webb et al. 2018) is our attempt to implement all computational aspects of integrative modeling. The modeling process proceeds through four stages (Fig. 1) (Alber et al. 2007a, 2008a; Russel et al. 2012).

The four-step modeling workflow as implemented in the Integrative Modeling Platform. The workflow is illustrated by its application to structure determination of the Nup84 heptamer (Shi et al. 2014). In this application, crystallographic structures and comparative models are used to represent the seven components of the Nup84 complex. The scoring function incorporates data extracted from CX-MS experiments and 2DEM class average images. The sampling explores both the conformations of the components and their configuration, searching for those assembly structures that satisfy the spatial restraints as accurately as possible. In this case, the result is an ensemble of many good-scoring models that satisfy the input data within acceptable thresholds. The sampling is then assessed for convergence, models are clustered, and evaluated by the degree to which they satisfy the data used to construct them as well as omitted data. The protocol can iterate through the four stages, until the models are judged to be satisfactory, most often based on their precision and the degree to which they satisfy the data. The resulting models are deposited in PDB-Dev (Burley et al. 2017; Vallat et al. 2018) with accession number PDBDEV_ 00000001
In the first stage, all information that describes the system of interest is collected. This information can include data from various experiments, structural propensities such as atomic statistical potentials (Sippl 1990; Shen and Sali 2006), physical principles such as those encoded in molecular mechanics force fields (Brooks et al. 2009), and other models, such as atomic structures of the subunits in a modeled complex.
In the second stage, a suitable representation of the system is chosen depending on the quantity and resolution of the available information. Different parts of a model may be represented at different resolutions, and a given part of the model may be represented in several different ways simultaneously. Next, information is translated into a set of spatial restraints on the components of the system. For example, in early characterizations of the molecular architecture of the NPC (Alber et al. 2007a, b), atomic structures of the protein subunits were not available, but the approximate size and shape of each protein was known, so each protein was represented as a ‘string’ of connected spheres whose volumes were consistent with the protein size and shape. A simple distance between two proteins can be restrained by a harmonic function of the distance, while the fit of a model into a three-dimensional Electron Microscopy (3DEM) density map can be scored by means of the cross-correlation between the model and experimental densities. Next, the spatial restraints are combined into a single scoring function that ranks alternative models based on their agreement with input information.
In the third stage, alternative models are sampled, using a method such as conjugate gradients, molecular dynamics, Brownian dynamics (Chen and Kim 2004), Monte Carlo (Metropolis and Ulam 1949), and divide-and-conquer message passing (Lasker et al. 2009). This sampling generally generates not a single structure, but an ensemble of models that are as consistent with the input information as possible. There may be many different models that score well if the data are incomplete, or none if the uncertainty of the data is underestimated or the representation does not include appropriate degrees of freedom (e.g., too coarse a representation is used, a flexible subunit is modeled as rigid, or a single-state model is used instead of a multiple-state model). Models produced by sampling can be optionally filtered by some information that cannot be feasibly evaluated many times during sampling (e.g., a match between a model and a two-dimensional Electron Microscopy (2DEM) class average (Velazquez-Muriel et al. 2012)).
In the fourth stage, input information and output structures need to be analyzed to estimate structure precision and accuracy, detect inconsistent and missing information, and to suggest most informative future experiments. Assessment begins with structural clustering of the modeled structures produced by sampling, followed by assessment of the thoroughness of structural sampling, estimating structure precision based on variability in the ensemble of good-scoring structures, quantification of the structure fit to the input information, structure assessment by cross-validation, and structure assessment by data not used to compute it (Viswanath et al. 2017b).
Integrative modeling can iterate through these four stages until a satisfactory model is built. Many iterations of the cycle may be required, given the need to gather more data as well as to resolve errors and inconsistent data.
Integrative modeling problems vary in size and scope. Thus, IMP offers a great deal of flexibility and several abstraction levels as part of a multi-tiered platform. At the lowest level, IMP is designed as a toolkit or set of “building blocks,” providing components and tools to allow method developers to convert data from new experimental methods into spatial restraints, to implement sampling and analysis techniques, and to implement an integrative modeling procedure from scratch, using the C ++ and Python programming languages. IMP is freely available as open source software under the terms of the GNU Lesser General Public License. To allow a community of developers to easily add sources of information, sampling schemes and analysis methods, IMP is structured as a collection of self-contained modules that can be developed and distributed independently.
In IMP, models are encoded as collections of particles, each representing a piece of the system. Depending on the data available, particles can be used to create atomic, coarse-grained, and/or hierarchical representations. It is straightforward to represent a protein at any resolution, from fully flexible atomic models (one particle per atom), to rigid bodies, to coarse-grained models consisting of only one or a few particles for the whole protein. Different parts of the model can be represented differently, as dictated by the available information. Each particle has associated attributes, such as coordinates, radius, residue information, and mass. Likewise, an IMP model can consist of one or more states of the same system (e.g., PhoQ kinase in two functional states (Molnar et al. 2014)) and/or multiple similar systems related via an alignment (Echeverria and Sali 2018).
Candidate IMP models are evaluated by a scoring function composed of terms called spatial restraints, each of which measures how well a model agrees with the information from which the restraint was derived. A restraint encodes what is known about structures in general (e.g., a molecular mechanics force field) or what is known about this particular structure (e.g., a distance restraint from NMR measurement). Thus, a candidate model that scores well is generally consistent with all used information. The precision and accuracy of the resulting model ensemble increases with the amount and quality of information that is encoded in the representation, restraints, sampling, and filtering after sampling. IMP’s growing set of restraints supports small angle X-ray (SAXS) profiles (Schneidman-Duhovny et al. 2011), various proteomics data such as data from affinity co-purifications and yeast two-hybrid experiments (Alber et al. 2008b), EM single particle images, 2DEM class averages (Schneidman-Duhovny et al. 2012; Velazquez-Muriel et al. 2012), and 3DEM density maps (Lasker et al. 2010a, b), most of the NMR spectroscopy-derived restraints (Simon et al. 2010), the CHARMM force-field (Brooks et al. 2009), restraints implied by an alignment with related structures (Sali and Blundell 1993), chemical crosslinking (Erzberger et al. 2014), hydrogen–deuterium exchange (Saltzberg et al. 2017), chromosome conformation capture (Bau et al. 2011), Förster resonance energy transfer (FRET) (Bonomi et al. 2014), a variety of statistical potentials (Shen and Sali 2006), and others. A common and powerful application of IMP involves the combination of information on local inter-particle distances and angles, such as that derived from NMR or crosslinking experiments, with overall shape information, such as that provided from 3DEM density maps (Zeng-Elmore et al. 2014; Luo et al. 2015; Robinson et al. 2015; Kim et al. 2018).
For most applications, the full flexibility of defining a system from the bottom up as sets of particles is unnecessary. IMP provides a higher-level interface called Python Modeling Interface (PMI) that allows for a top-down representation of the system, using biological names for protein subunits (Saltzberg et al. 2019). It provides simple mechanisms to set up higher order structure, such as multiple copies of subunits or symmetry-related subsets of the system, at multiple resolutions. It also allows easy setup of the myriad advanced restraints available in IMP. Finally, it provides ready-built protocols and other utilities, for example to generate publication-ready plots. Using PMI, the entire modeling protocol can be described with a set of Python scripts, which are typically deposited, together with the input data and output models, in a publicly available repository, such as GitHub and the Worldwide Protein Data Bank (wwPDB) prototype archive for integrative structures called PDB-Dev (Burley et al. 2017; Vallat et al. 2018); for examples, see references (Algret et al. 2014; Erzberger et al. 2014; Shi et al. 2014; Luo et al. 2015; Robinson et al. 2015; Shi et al. 2015; Chen et al. 2016; Fernandez-Martinez et al. 2016; Wang et al. 2017b). Finally, at the highest abstraction levels, for users with limited programming experience, IMP provides less flexible but more user-friendly applications to handle specific tasks, such as fitting of proteins into a density map of their assembly (Lasker et al. 2009), scoring protein–ligand interactions (Fan et al. 2011), combining multiple SAXS profiles (Spill et al. 2014), comparing a structure with the corresponding SAXS profile (Schneidman-Duhovny et al. 2010, 2013, 2016), or enriching pairwise docking using SAXS data (Schneidman-Duhovny et al. 2016); these functionalities can be accessed through web interfaces, from Chimera (Pettersen et al. 2004), or from the command line.
IMP has been used to produce structural models of more than 30 varied biomolecular systems; for example, a eukaryotic ribosome (Taylor et al. 2009), aryanodine receptor channel (Serysheva et al. 2008), the yeast Mediator complex (Robinson et al. 2015), the Hsp90 chaperonin (Krukenberg et al. 2008), a yeast exosome in multiple states (Shi et al. 2015), the actin-scruin complex (Cong et al. 2008), deoxyribose nucleic acid (DNA) transcription factor II H (TFIIH) (Luo et al. 2015), chromatin (Bau et al. 2011; Tjong et al. 2016), and the NPC and its subcomplexes (Alber et al. 2007b; Fernandez-Martinez et al. 2012, 2016; Kim et al. 2014, 2018; Shi et al. 2014; Upla et al. 2017).
Requirements for archiving integrative models
To maximize the impact of integrative structures, the coordinates should be made publicly available, at least upon publication, as is already the case for structures based on X-ray crystallography, NMR spectroscopy, and 3DEM maps. Moreover, the associated experimental data and modeling protocols should also be archived, such that both the authors and others can easily reproduce the original results. Finally, it is essential that the integrative structures are validated as part of their publication and deposition, as is already the case for other structures currently archived in the Protein Data Bank (PDB) (Gore et al. 2017; Young et al. 2017).
In recognition of the challenges involved in archiving integrative models, the wwPDB convened an Integrative/Hybrid Methods Task Force workshop in 2014. The IHM Task Force made several recommendations to facilitate the archiving of integrative structural models (Sali et al. 2015). A fundamental requirement is the development of a flexible model representation that allows us to represent ensembles of multi-scale, multi-state, and ordered collections of structural models. The representation should also provide support for spatial restraints derived from diverse types of experimental data obtained from different samples, used as input in the modeling. Another requirement is creating the software infrastructure required for deposition, curation, validation, archiving, and dissemination of integrative structures. The development of a flexible data representation and a prototype system for archiving integrative structural models are discussed in sects. “Standards for archiving integrative models” and “The IHM-dictionary”.
Another recommendation from the Task Force was to build a Federation of structural model and experimental data repositories that interoperate with one another. This requires development of well-aligned data standards and data exchange protocols that enable efficient and automated interoperation. Lastly, the Task Force recommended the creation of methods for evaluating and validating integrative structures so that they can be appropriately used for downstream applications. A reasonable starting point for structure validation is the model assessment process outlined in sect. “Modeling with IMP”. However, much more research effort on the part of the entire community is needed to define the necessary validation criteria and implement them in robust software, eventually leading to a validation pipeline that can be part of the archiving process. Work is currently in progress to build an interoperating network of repositories as well as to develop the validation pipeline for integrative models.
Multi-method structures in the Protein Data Bank
The PDB is the sole international repository for experimentally-determined 3D atomic structures of biological macromolecules (Berman et al. 2000, 2003). When the resource was first established in 1971, X-ray crystallography was the principal method for determining the structures of these molecules and therefore the PDB archived structures determined from diffraction experiments, initially using X-ray and later from neutron radiation. Over time, the structural biology field grew and newer methods of structure determination using NMR spectroscopy and 3DEM were developed. Simultaneously, the PDB expanded itself to serve the needs of the structural biology community and started archiving structures determined using NMR spectroscopy (Borah et al. 1985) and 3DEM (Henderson et al. 1990). In 2008, the PDB began to require the deposition of structure factors for X-ray structures and the deposition of NMR chemical shifts for NMR structures (wwPDB consortium 2007). BioMagResBank (BMRB (Ulrich et al. 2008)) and Electron Microscopy Data Bank (EMDB (Tagari et al. 2002; Lawson et al. 2016; Patwardhan and Lawson 2016)) have been created independently to archive NMR data and 3DEM maps. The availability of the underlying experimental data made it possible to create better validation standards for the structural models archived in the PDB. The wwPDB consortium (Berman et al. 2007) that manages the PDB archive has recently developed the OneDep system (Young et al. 2017) to provide a unified portal for the deposition of structural models determined using X-ray crystallography, NMR spectroscopy, and 3DEM along with associated experimental data that aids structure validation.
In recent times, structural biologists have started to combine data from two or more experimental methods to build structural models of macromolecules. The PDB archives structures determined using multiple methods, where the experiments are carried out on samples of similar composition. Usually, methods capable of resolving atomistic features, such as X-ray crystallography, neutron crystallography, NMR spectroscopy, and 3DEM, can be combined with each other or used in combination with methods that provide coarse-grained information, such as small angle solution scattering (SAS) methods, solid-state NMR spectroscopy, and electron paramagnetic resonance (EPR) spectroscopy. The multi-method experimental structures are distinct from the integrative models where complex computational algorithms combine data obtained from an unrestricted set of experimental observations on a potentially diverse set of experimental samples, although the distinction is more of a degree than kind. Figure 2 shows the historical growth of multi-methods structures in the PDB, which highlights the increase in the deposition of multi-method structures over the last 10 years. Table 1 shows the breakdown of method combinations in multi-methods structures currently released by the PDB. Not surprisingly, multi-method structures in the PDB frequently use X-ray crystallography in combination with neutron diffraction and solution NMR in combination with SAS. To support the facile deposition of structures that use solution NMR in combination with SAS, the wwPDB OneDep team recently extended the deposition infrastructure to handle SAS data. This work has been carried out in collaboration with the SASBDB repository, which archives SAS data (Valentini et al. 2015).

Number of multi-method structures archived in the PDB over the years (data as of December 6, 2018)
As more structures were determined by combining data from multiple methods, integrative modelers began exploring the application of additional biophysical techniques such as chemical crosslinking mass spectrometry (CX-MS), SAS, EPR spectroscopy, FRET, hydrogen/deuterium exchange mass spectrometry (HDX-MS), and others, to derive spatial restraints that can be combined to determine structures of complex macromolecular assemblies (Sali et al. 2003; Alber et al. 2007a, b) (Ward et al. 2013; Sali et al. 2015; Rout and Sali 2019). These integrative modeling methods became especially useful to model structures of macromolecular assemblies that are elusive to the traditional methods of structure determination. To adapt to the growing needs of the structural biology community, the PDB, in collaboration with the integrative modeling community, began developing the infrastructure required to archive, validate, visualize, and disseminate integrative structural models.
Standards for archiving integrative models
A primary requirement for archiving data is the creation of a standard representation of the data to be archived. These data standards provide the foundation for building an archive. Under the auspices of the IUCr, the mmCIF data representation (Fitzgerald et al. 2005) was developed for structures of macromolecules determined using X-ray crystallography. That data dictionary is based on a robust framework that supports the representation of macromolecular structure data and associated metadata. The framework also provides mechanisms to include metadata used for assessing and maintaining data consistency, such as definitions of data types, boundary conditions, controlled vocabularies, and parent–child relationships with other data items.
Extensions of the mmCIF data representation have been created to represent different kinds of experimental data and structural restraints derived from them. These extensions are now embodied in the PDBx/mmCIF dictionary that is the standard for data archiving by the PDB (Westbrook 2013). For example, the NMR community has extended the PDBx/mmCIF dictionary to represent NMR restraints in the NMR Exchange Format (NEF) dictionary (Gutmanas et al. 2015) and the SAS community has created the sasCIF extension dictionary (Malfois and Svergun 2000; Kachala et al. 2016), which is used by the SASBDB repository (Valentini et al. 2015) to archive SAS data. We have extended the existing PDBx/mmCIF data representation to address the requirements for archiving integrative structural models. This extended data representation, called the IHM-dictionary (Vallat et al. 2018), is developed as a collaborative project that is distributed freely through a public GitHub repository (https://github.com/ihmwg/IHM-dictionary). Building an extension dictionary based on the PDBx/mmCIF representation allows us to use a single robust framework to create new definitions while retaining the existing definitions in the main dictionary where applicable. This design helps avoiding duplication while allowing us to focus on the new definitions that address the current requirements.
The IHM-dictionary
The IHM-dictionary is an extension of the PDBx/mmCIF dictionary and therefore only defines those terms required for representing integrative structural models that are not already included in the PDBx/mmCIF dictionary. For instance, the nomenclature and chemistry of small molecules, polymeric macromolecules, and molecular complexes consisting of small molecules and macromolecules are already defined in the PDBx/mmCIF dictionary. Similarly, the definitions of the molecular structure in terms of atomic coordinates are also clearly represented in the PDBx/mmCIF dictionary. Although these definitions provide the foundation for representing the chemistry and structure of a molecular system, they are not sufficient for representing the complexities of integrative models.
Therefore, the IHM-dictionary extends the definitions in the PDBx/mmCIF dictionary in five significant aspects that address the requirements for archiving integrative models (Vallat et al. 2018).
-
1.
It allows for a flexible model representation with atomic and coarse-grained objects consisting of single and multi-residue spherical beads and three-dimensional Gaussian objects.
-
2.
It supports constitutionally diverse structural assemblies and conformationally diverse ensembles, thereby providing representations for multi-state structural models and models related by time or other order.
-
3.
It captures the spatial restraints derived from different kinds of biophysical techniques, such as CX-MS, SAS methods, EPR spectroscopy, DNA footprinting, mutagenesis, and others. Experimental restraints already captured in the PDBx/mmCIF dictionary and other related extensions are retained and reused where applicable. Several kinds of experimental data provide spatial restraints in the form of distances between atoms or residues (e.g., distances from NMR NOE, FRET, and CX-MS experiments). To address the broad range of experimentally derived distance restraints, the IHM-dictionary includes a general representation of distance restraints between different kinds of features (e.g., atoms, single and multiple residues, contiguous residue ranges) and the corresponding uncertainties associated with these distance measurements. The specifications for different types of spatial restraints are encoded in different data categories within the dictionary. An mmCIF file corresponding to an integrative model derived using restraints from multiple experimental sources will contain several data tables that capture the relevant restraint information. Representation of the spatial restraints in the dictionary enables the visualization of the restraints along with the structural models as well as the validation of integrative models based on the experimental restraints.
-
4.
It provides a generic representation for referencing related data from external resources via stable identifiers, such as accession codes or persistent digital object identifiers (DOIs). This is useful for referencing related data that either lives in an external repository (via stable accession codes) or does not yet have a primary repository (via standard DOIs).
-
5.
It promotes reproducibility by incorporating simplified definitions for the modeling workflow and providing mechanisms to link modeling scripts and software program files.
The IHM-dictionary thus provides a comprehensive set of standardized definitions for representing multi-scale, multi-state, and ordered ensembles of complex macromolecular assemblies. The dictionary has been developed using diverse sets of examples and requirements gathered from the integrative modeling community. Collaborative tools provided by the GitHub platform have been used effectively to gather feedback from the scientific community regarding the definitions in the IHM-dictionary and incorporate their recommendations.
Representation of NMR restraints
The contents of the PDBx/mmCIF dictionary (Fitzgerald et al. 2005; Westbrook 2013) grew from a core set of mmCIF definitions describing macromolecular structure and the X-ray diffraction experiment to its current scope through an incremental process of building compatible content extensions (e.g., NMR and 3DEM) in collaboration with community specialists. The development of the IHM-dictionary has followed a similar path by integrating existing definitions in the PDBx/mmCIF dictionary and compatible community extension dictionaries. For example, the IHM-dictionary takes advantage of an existing data dictionary developed to facilitate the programmatic exchange of NMR restraint data, the NEF dictionary (Gutmanas et al. 2015). The IHM-dictionary does not include new definitions for NMR restraints. Rather, definitions from the NEF dictionary are reused to describe NMR restraints used in integrative models.
Creating a consensus representation of NMR restraint data with broad adoption by NMR application developers has proved to be challenging. In part owing to the complexity and diversity of NMR restraint data, neither the NMR-STAR (Markley et al. 2003) representation used by the NMR experimental archive, BioMagResBank (BMRB (Ulrich et al. 2008)), nor the representation adopted by Collaborative Computational Project for NMR (CCPN (Vranken et al. 2005)) gained wide adoption among developers of NMR structure determination and refinement software.
In 2013, a group of NMR experts assembled by the wwPDB, the wwPDB NMR Validation Task Force (VTF), published a set of recommendations for the validation of NMR structure and experimental data archived by the PDB (Montelione et al. 2013). This report included recommendations for restraint-based model-versus-data validation comparing each member of the ensemble of NMR models to the available NMR restraints. Lacking a community consensus representation and format, the wwPDB has historically collected and archived NMR restraint data in native programmatic format. While there have been efforts to retrospectively standardize these native restraint data files using NMR-STAR (Doreleijers et al. 2009), these approaches were not fully automatable and proved difficult to sustain. A Working Group of the wwPDB NMR VTF, including developers of the principal NMR structure determination packages, was subsequently created to revisit the challenges of representing and exchanging NMR restraints and supporting experimental data. In 2015, this Working Group published the first set of recommendations for the NEF dictionary (Gutmanas et al. 2015). In addition to the NMR distance, dihedral, and residual dipolar coupling (RDC) restraint data, the NEF dictionary also includes definitions describing chemical shift and observed spectral peaks. While these data definitions have long been represented in the BMRB NMR-STAR reference dictionary, they are reorganized in the NEF dictionary to simplify their production and exchange by NMR software.
The representation of NMR-specific distance restraints in the NEF dictionary has also informed the development of the representation of generic derived distance restraints for experiment types such CX-MS and FRET in the IHM-dictionary. Work is in progress to build software tools that support the NEF dictionary for the IHM data pipeline.
The PDB-Dev prototype archiving system
Based on the data standards provided by the IHM-dictionary, we have built a prototype archiving system called PDB-Dev (https://pdb-dev.wwpdb.org) to archive integrative structural models (Burley et al. 2017; Vallat et al. 2018). The integrative structures archived in PDB-Dev conform to the definitions in the IHM-dictionary (Vallat et al. 2018). In order to deposit structures to PDB-Dev, users are required create an account on the PDB-Dev website and upload an mmCIF file that is compliant with the IHM-dictionary. Optionally, supporting files such as images can be included with the deposition. After a structure is deposited, compliance to the IHM-dictionary is checked using software tools built for the PDBx/mmCIF dictionary. If the deposited file is not compliant, communication is initiated with the authors to obtain any missing or incomplete information regarding the deposition. Once a compliant mmCIF file is obtained, the structure is either released immediately or kept on hold until publication. At present, we do not carry out any automated or manual curation of the data or validation of the structural models. The development of a comprehensive deposition, data harvesting, curation and model validation pipeline is the focus of ongoing research.
PDB-Dev currently archives twenty-two integrative structures that have been released along with five additional structures that have been processed and placed on hold for publication. A snapshot of the structures archived in PDB-Dev is shown in Fig. 3. These structures include several macromolecular assemblies, such as the nuclear pore complex (Kim et al. 2018), the mediator complex (Robinson et al. 2015), the exosome complex (Shi et al. 2015), the mitochondrial cysteine desulfurase complex (van Zundert et al. 2015), and others. The integrative structures in PDB-Dev have been obtained by satisfying spatial restraints from different experimental techniques, such as CX-MS, SAS, 2DEM, 3DEM, NMR, EPR, FRET, DNA footprinting, mutagenesis, hydroxyl radical footprinting and predicted contacts from coevolution data (Fig. 4a). Evidently, CX-MS is emerging as a dominant experimental technique to define distance restraints on pairs of cross-linked residues used in integrative modeling, often in combination with 3DEM density maps. Furthermore, the CX-MS field is rapidly evolving to identify novel crosslinking agents and develop better methods for deriving the spatial restraints. Figure 4b shows that the structures archived in PDB-Dev have been modeled using a variety of integrative modeling software tools, including IMP (Russel et al. 2012), Rosetta (Leaver-Fay et al. 2011), Haddock (Dominguez et al. 2003), TADbit (Trussart et al. 2015; Serra et al. 2017), FPS (Kalinin et al. 2012), XPLOR-NIH (Schwieters et al. 2018), PatchDock (Schneidman-Duhovny et al. 2005), and iSPOT (Hsieh et al. 2017). The diversity of software applications that produced the PDB-Dev structures shows that the data standards captured in the IHM-dictionary are generic enough to work with different integrative modeling methods. The model of mitochondrial cysteine desulfurase complex (Fig. 3) built by Haddock (Dominguez et al. 2003) using spatial restraints derived from NMR chemical shift perturbations, SAS, and CX-MS is currently the only example in PDB-Dev that uses NMR data. However, as the integrative modeling methods evolve and the PDB-Dev archive grows, we expect more structures that use restraints derived from NMR experiments to be deposited in PDB-Dev, especially since NMR restraints are inherently amenable to being used in integrative modeling.

A snapshot of integrative structural models deposited in PDB-Dev. a Nup84 sub-complex (PDBDEV_ 00000001 (Shi et al. 2014)), b Nup133 sub-complex (PDBDEV_ 00000016 (Kim et al. 2014)), c Nup82 sub-complex (PDBDEV_ 00000020 (Fernandez-Martinez et al. 2016)), d Pom152 sub-complex (PDBDEV_ 00000017 (Upla et al. 2017)), e, f, g Nuclear pore complex 1-spoke, 3-spokes & 8-spokes (PDBDEV_ 00000010, PDBDEV_ 00000011, PDBDEV_ 00000012 (Kim et al. 2018)), h Mediator complex (PDBDEV_ 00000003 (Robinson et al. 2015)), i Exosome complex (PDBDEV_ 00000002 (Shi et al. 2015)), j 16 s RNA—Methyl transferase A complex (PDBDEV_ 00000014 (van Zundert et al. 2015)), k Human complement system C3(H2O) (PDBDEV_ 00000021 (Chen et al. 2016)), l Fruit fly chromosome 2L segment (PDBDEV_ 00000008 (Trussart et al. 2015)), m Ecm29 protein with 26S proteasome complex (PDBDEV_00000026 (Wang et al. 2017a)), n Pol II(G) complex (PDBDEV_00000025 (Jishage et al. 2018)), o Mitochondrial cysteine desulfurase complex (PDBDEV_ 00000015 (Cai et al. 2018)), p Diubiquitin (PDBDEV_ 00000004 (Liu et al. 2018)), q, r, s Human serum albumin domains A, B & C (PDBDEV_ 00000005, PDBDEV_ 00000006, PDBDEV_ 00000007 (Belsom et al. 2016)), t Human Rev7 dimer (PDBDEV_ 00000009 (Rizzo et al. 2018)), u E6AP-E6-p53 enzyme–substrate complex (PDBDEV_00000023 (Sailer et al. 2018))

Statistics of current structures in PDB-Dev (including structures released and structures on-hold for publication as of December 6, 2018). a Plot of number of entries in PDB-Dev as a function of the type of input experimental restraints. b Plot of number of entries in PDB-Dev as a function of the integrative modeling software application used
The integrative models archived in PDB-Dev can be visualized using the ChimeraX software (Goddard et al. 2018). ChimeraX supports the visualization of multi-scale structural models as well as different types of experimental restraints used in the modeling such as crosslinking distances, 3DEM maps and 2DEM class averages. The images in Fig. 3 have been generated using ChimeraX.
The IHM-dictionary and the PDB-Dev system are under continuous development to address the emerging needs of the integrative modeling community along with a growing range of experimental data types and software applications used to model integrative structures. This effort is carried out in collaboration with the modelers, who provide us with up-to-date examples of integrative models and the associated spatial restraints. We have used these examples as building blocks to develop the IHM-dictionary and the PDB-Dev system. We are also working with the integrative modeling community to build support for the IHM-dictionary within their modeling software, so that these software can easily read and write data files compliant with the IHM-dictionary, thereby streamlining the deposition process of integrative models into PDB-Dev as well as using multiple software programs in one application. The project highlights a concerted community endeavor to create the data standards, develop supporting software tools, and build a prototype system for deposition and archiving integrative structural models.
Python-ihm library
We have developed the python-ihm software library (https://github.com/ihmwg/python-ihm) to support reading, writing, and managing data files that comply with the IHM-dictionary (Vallat et al. 2018). The python-ihm library implements software support for the IHM-dictionary as a set of Python classes. This implementation allows an integrative model to be represented as a hierarchy of Python objects, and supports reading and writing these hierarchies as IHM-dictionary-compliant mmCIF data files, as well as binary representations such as BinaryCIF (Sehnal 2016). It is available under a permissive open source license, and is designed to be used either standalone or as part of an integrative modeling package. By providing a software implementation of the dictionary, developers of integrative modeling software are relieved of the burden of developing their own support for IHM-dictionary; this service should lower the barrier to entry to PDB-Dev (Burley et al. 2017; Vallat et al. 2018). For example, both IMP (Russel et al. 2012) and Haddock (Dominguez et al. 2003) already use python-ihm to output their models in a format compliant with the IHM-dictionary for deposition in PDB-Dev. Furthermore, the ChimeraX visualization software (Goddard et al. 2018) uses the python-ihm library to support visualization of integrative models archived in PDB-Dev.
Challenges and future perspectives
In the last 4 years, there has been substantial progress in creating the framework for archiving integrative structure models. The creation of an extensible dictionary has made this archival possible as has the development of the PDB-Dev test platform that allows for prototyping an archiving system. There are considerable challenges ahead. The first is the creation of standards for all the experimental methods that contribute restraints to the modeling. Achieving this goal will require that each experimental community reach consensus on their own standards. The second is to find a mechanism to exchange these data among all the relevant communities and with the PDB archive. The last and most difficult challenge is to come up with methods to validate each model so that it will be possible for users of these models to understand their limits. Meeting these challenges will require further scientific research, technology development and implementation, and most of all a spirit of collaboration and cooperation among the very heterogeneous communities.
References
Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, Rout MP, Sali A (2007a) Determining the architectures of macromolecular assemblies. Nature 450:683–694
Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, Sali A, Rout MP (2007b) The molecular architecture of the nuclear pore complex. Nature 450:695–701
Alber F, Chait BT, Rout MP, Sali A (2008a) Integrative structure determination of protein assemblies by satisfaction of spatial restraints. Protein-protein interactions and networks. Panchenko A, Przytycka T Springer, London, p 99–114
Alber F, Forster F, Korkin D, Topf M, Sali A (2008b) Integrating diverse data for structure determination of macromolecular assemblies. Annu Rev Biochem 77:443–477
Algret R, Fernandez-Martinez J, Shi Y, Kim SJ, Pellarin R, Cimermancic P, Cochet E, Sali A, Chait BT, Rout MP, Dokudovskaya S (2014) Molecular architecture and function of the SEA complex, a modulator of the TORC1 pathway. Mol Cell Proteomics 13:2855–2870
Bau D, Sanyal A, Lajoie BR, Capriotti E, Byron M, Lawrence JB, Dekker J, Marti-Renom MA (2011) The three-dimensional folding of the alpha-globin gene domain reveals formation of chromatin globules. Nat Struct Mol Biol 18:107–114
Belsom A, Schneider M, Fischer L, Brock O, Rappsilber J (2016) Serum albumin domain structures in human blood serum by mass spectrometry and computational biology. Mol Cell Proteomics 15:1105–1116
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28:235–242
Berman HM, Henrick K, Nakamura H (2003) Announcing the worldwide protein data bank. Nat Struct Biol 10:980
Berman H, Henrick K, Nakamura H, Markley JL (2007) The Worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res 35:D301–D303
Blundell TL, Johnson LN (1976) Protein crystallography. Academic Press, Massachusetts
Bonomi M, Pellarin R, Kim SJ, Russel D, Sundin BA, Riffle M, Jaschob D, Ramsden R, Davis TN, Muller EG, Sali A (2014) Determining protein complex structures based on a Bayesian model of in vivo Forster resonance energy transfer (FRET) data. Mol Cell Proteomics 13:2812–2823
Borah B, Chen CW, Egan W, Miller M, Wlodawer A, Cohen JS (1985) Nuclear magnetic resonance and neutron diffraction studies of the complex of ribonuclease A with uridine vanadate, a transition-state analogue. Biochemistry 24:2058–2067
Brooks BR, Brooks CL 3rd, Mackerell AD Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30:1545–1614
Burley SK, Kurisu G, Markley JL, Nakamura H, Velankar S, Berman HM, Sali A, Schwede T, Trewhella J (2017) PDB-Dev: a prototype system for depositing integrative/hybrid structural models. Structure 25:1317–1318
Cai K, Frederick RO, Dashti H, Markley JL (2018) Architectural features of human mitochondrial cysteine desulfurase complexes from crosslinking mass spectrometry and small-angle X-ray scattering. Structure 26(1127–1136):e1124
Calhoun S, Korczynska M, Wichelecki DJ, San Francisco B, Zhao S, Rodionov DA, Vetting MW, Al-Obaidi NF, Lin H, O’Meara MJ, Scott DA, Morris JH, Russel D, Almo SC, Osterman AL, Gerlt JA, Jacobson MP, Shoichet BK, Sali A (2018) Prediction of enzymatic pathways by integrative pathway mapping. Elife 7:e31097
Chen JC, Kim AS (2004) Brownian dynamics, molecular dynamics, and monte carlo modeling of colloidal systems. Adv Colloid Interface Sci 112:159–173
Chen ZA, Pellarin R, Fischer L, Sali A, Nilges M, Barlow PN, Rappsilber J (2016) Structure of complement C3(H2O) revealed By quantitative cross-linking/mass spectrometry and modeling. Mol Cell Proteomics 15:2730–2743
Chiu W, Baker ML, Jiang W, Dougherty M, Schmid MF (2005) Electron cryomicroscopy of biological machines at subnanometer resolution. Structure 13:363–372
Cong Y, Topf M, Sali A, Matsudaira P, Dougherty M, Chiu W, Schmid MF (2008) Crystallographic conformers of actin in a biologically active bundle of filaments. J Mol Biol 375:331–336
Dominguez C, Boelens R, Bonvin AM (2003) HADDOCK: a protein–protein docking approach based on biochemical or biophysical information. J Am Chem Soc 125:1731–1737
Doreleijers JF, Vranken WF, Schulte C, Lin J, Wedell JR, Penkett CJ, Vuister GW, Vriend G, Markley JL, Ulrich EL (2009) The NMR restraints grid at BMRB for 5266 protein and nucleic acid PDB entries. J Biomol NMR 45:389–396
Echeverria I, Sali A (2018) Integrative structure determination of protein complexes by inferred structural equivalence. Biophys J 114(3):61a
Erzberger JP, Stengel F, Pellarin R, Zhang S, Schaefer T, Aylett CHS, Cimermancic P, Boehringer D, Sali A, Aebersold R, Ban N (2014) Molecular architecture of the 40SeIF1eIF3 translation initiation complex. Cell 158:1123–1135
Fan H, Schneidman-Duhovny D, Irwin JJ, Dong G, Shoichet BK, Sali A (2011) Statistical potential for modeling and ranking of protein–ligand interactions. J Chem Inf Model 51:3078–3092
Fernandez-Martinez J, Phillips J, Sekedat MD, Diaz-Avalos R, Velazquez-Muriel J, Franke JD, Williams R, Stokes DL, Chait BT, Sali A, Rout MP (2012) Structure–function mapping of a heptameric module in the nuclear pore complex. J Cell Biol 196:419–434
Fernandez-Martinez J, Kim SJ, Shi Y, Upla P, Pellarin R, Gagnon M, Chemmama IE, Wang J, Nudelman I, Zhang W, Williams R, Rice WJ, Stokes DL, Zenklusen D, Chait BT, Sali A, Rout MP (2016) Structure and function of the nuclear pore complex cytoplasmic mRNA export platform. Cell 167:1215–1228.e1225
Fitzgerald PMD, Westbrook JD, Bourne PE, McMahon B, Watenpaugh KD, Berman HM (2005) 4.5 Macromolecular dictionary (mmCIF). International Tables for Crystallography G. Definition and exchange of crystallographic data. Hall SR, McMahon B Springer Science and Business Media, Berlin, p 295–443
Gingras AC, Gstaiger M, Raught B, Aebersold R (2007) Analysis of protein complexes using mass spectrometry. Nat Rev Mol Cell Biol 8:645–654
Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE (2018) UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci 27:14–25
Gore S, Sanz Garcia E, Hendrickx PMS, Gutmanas A, Westbrook JD, Yang H, Feng Z, Baskaran K, Berrisford JM, Hudson BP, Ikegawa Y, Kobayashi N, Lawson CL, Mading S, Mak L, Mukhopadhyay A, Oldfield TJ, Patwardhan A, Peisach E, Sahni G, Sekharan MR, Sen S, Shao C, Smart OS, Ulrich EL, Yamashita R, Quesada M, Young JY, Nakamura H, Markley JL, Berman HM, Burley SK, Velankar S, Kleywegt GJ (2017) Validation of structures in the protein data bank. Structure 25:1916–1927
Gutmanas A, Adams PD, Bardiaux B, Berman HM, Case DA, Fogh RH, Guntert P, Hendrickx PM, Herrmann T, Kleywegt GJ, Kobayashi N, Lange OF, Markley JL, Montelione GT, Nilges M, Ragan TJ, Schwieters CD, Tejero R, Ulrich EL, Velankar S, Vranken WF, Wedell JR, Westbrook J, Wishart DS, Vuister GW (2015) NMR Exchange Format: a unified and open standard for representation of NMR restraint data. Nat Struct Mol Biol 22:433–434
Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH (1990) Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J Mol Biol 213:899–929
Holcomb J, Spellmon N, Zhang Y, Doughan M, Li C, Yang Z (2017) Protein crystallization: eluding the bottleneck of X-ray crystallography. AIMS Biophys 4:557–575
Hsieh A, Lu L, Chance MR, Yang S (2017) A practical guide to iSPOT modeling: an integrative structural biology platform. Adv Exp Med Biol 1009:229–238
Jishage M, Yu X, Shi Y, Ganesan SJ, Chen WY, Sali A, Chait BT, Asturias FJ, Roeder RG (2018) Architecture of Pol II(G) and molecular mechanism of transcription regulation by Gdown1. Nat Struct Mol Biol 25:859–867
Joseph AP, Polles G, Alber F, Topf M (2017) Integrative modelling of cellular assemblies. Curr Opin Struct Biol 46:102–109
Kachala M, Westbrook J, Svergun D (2016) Extension of the sasCIF format and its applications for data processing and deposition. J Appl Crystallogr 49:302–310
Kalinin S, Peulen T, Sindbert S, Rothwell PJ, Berger S, Restle T, Goody RS, Gohlke H, Seidel CA (2012) A toolkit and benchmark study for FRET-restrained high-precision structural modeling. Nat Methods 9:1218–1225
Kim SJ, Fernandez-Martinez J, Sampathkumar P, Martel A, Matsui T, Tsuruta H, Weiss TM, Shi Y, Markina-Inarrairaegui A, Bonanno JB, Sauder JM, Burley SK, Chait BT, Almo SC, Rout MP, Sali A (2014) Integrative structure–Structure-function mapping of a heptameric function mapping of the nucleoporin nup133 suggests a conserved mechanism for membrane anchoring of the nuclear pore complex. Mol Cell Proteomics 13:2911–2926
Kim SJ, Fernandez-Martinez J, Nudelman I, Shi Y, Zhang W, Raveh B, Herricks T, Slaughter BD, Hogan JA, Upla P, Chemmama IE, Pellarin R, Echeverria I, Shivaraju M, Chaudhury AS, Wang J, Williams R, Unruh JR, Greenberg CH, Jacobs EY, Yu Z, de la Cruz MJ, Mironska R, Stokes DL, Aitchison JD, Jarrold MF, Gerton JL, Ludtke SJ, Akey CW, Chait BT, Sali A, Rout MP (2018) Integrative structure and functional anatomy of a nuclear pore complex. Nature 555:475–482
Krukenberg KA, Forster F, Rice LM, Sali A, Agard DA (2008) Multiple conformations of E. coli Hsp90 in solution: insights into the conformational dynamics of Hsp90. Structure 16:755–765
Lasker K, Topf M, Sali A, Wolfson HJ (2009) Inferential optimization for simultaneous fitting of multiple components into a CryoEM map of their assembly. J Mol Biol 388:180–194
Lasker K, Phillips JL, Russel D, Velazquez-Muriel J, Schneidman-Duhovny D, Tjioe E, Webb B, Schlessinger A, Sali A (2010a) Integrative structure modeling of macromolecular assemblies from proteomics data. Mol Cell Proteomics 9:1689–1702
Lasker K, Sali A, Wolfson HJ (2010b) Determining macromolecular assembly structures by molecular docking and fitting into an electron density map. Proteins 78:3205–3211
Lasker K, Forster F, Bohn S, Walzthoeni T, Villa E, Unverdorben P, Beck F, Aebersold R, Sali A, Baumeister W (2012) Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci USA 109:1380–1387
Lawson CL, Patwardhan A, Baker ML, Hryc C, Garcia ES, Hudson BP, Lagerstedt I, Ludtke SJ, Pintilie G, Sala R, Westbrook JD, Berman HM, Kleywegt GJ, Chiu W (2016) EMDataBank unified data resource for 3DEM. Nucleic Acids Res 44:D396–403
Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban YE, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popovic Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P (2011) ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol 487:545–574
Liu Z, Gong Z, Cao Y, Ding YH, Dong MQ, Lu YB, Zhang WP, Tang C (2018) Characterizing protein dynamics with integrative use of bulk and single-molecule techniques. Biochemistry 57:305–313
Loquet A, Sgourakis NG, Gupta R, Giller K, Riedel D, Goosmann C, Griesinger C, Kolbe M, Baker D, Becker S, Lange A (2012) Atomic model of the type III secretion system needle. Nature 486:276–279
Lucic V, Leis A, Baumeister W (2008) Cryo-electron tomography of cells: connecting structure and function. Histochem Cell Biol 130:185–196
Luo J, Cimermancic P, Viswanath S, Ebmeier CC, Kim B, Dehecq M, Raman V, Greenberg CH, Pellarin R, Sali A, Taatjes DJ, Hahn S, Ranish J (2015) Architecture of the human and yeast general transcription and DNA repair factor TFIIH. Mol Cell 59:794–806
Malfois M, Svergun DI (2000) sasCIF: an extension of core crystallographic information file for SAS. J Appl Crystallogr 33:812–816
Markley JL, Ulrich EL, Westler WM, Volkman BF (2003) Macromolecular structure determination by NMR spectroscopy. Structural Bioinformatics. Bourne PE, Weissig H (ed) John Wiley & Sons, Inc., New Jersey, p 89–113
Metropolis N, Ulam S (1949) The monte carlo method. J Am Statis Assoc 44:335–341
Molnar KS, Bonomi M, Pellarin R, Clinthorne GD, Gonzalez G, Goldberg SD, Goulian M, Sali A, DeGrado WF (2014) Cys-scanning disulfide crosslinking and bayesian modeling probe the transmembrane signaling mechanism of the histidine kinase, PhoQ. Structure 22:1239–1251
Montelione GT, Nilges M, Bax A, Guntert P, Herrmann T, Richardson JS, Schwieters CD, Vranken WF, Vuister GW, Wishart DS, Berman HM, Kleywegt GJ, Markley JL (2013) Recommendations of the wwPDB NMR validation task force. Structure 21:1563–1570
Parrish JR, Gulyas KD, Finley RL Jr (2006) Yeast two-hybrid contributions to interactome mapping. Curr Opin Biotechnol 17:387–393
Patwardhan A, Lawson CL (2016) Databases and archiving for CryoEM. Methods Enzymol 579:393–412
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Raveh B, Karp JM, Sparks S, Dutta K, Rout MP, Sali A, Cowburn D (2016) Slide-and-exchange mechanism for rapid and selective transport through the nuclear pore complex. Proc Natl Acad Sci USA 113:E2489–2497
Rizzo AA, Vassel FM, Chatterjee N, D’Souza S, Li Y, Hao B, Hemann MT, Walker GC, Korzhnev DM (2018) Rev7 dimerization is important for assembly and function of the Rev1/Polzeta translesion synthesis complex. Proc Natl Acad Sci USA 115:E8191–E8200
Robinson PJ, Trnka MJ, Pellarin R, Greenberg CH, Bushnell DA, Davis R, Burlingame AL, Sali A, Kornberg RD (2015) Molecular architecture of the yeast Mediator complex. Elife 4:e08719
Rout MP, Sali A (2019) Principles for integrative structural biology studies. Cell 177:1384–1403
Russel D, Lasker K, Phillips J, Schneidman-Duhovny D, Velazquez-Muriel JA, Sali A (2009) The structural dynamics of macromolecular processes. Curr Opin Cell Biol 21:97–108
Russel D, Lasker K, Webb B, Velazquez-Muriel J, Tjioe E, Schneidman-Duhovny D, Peterson B, Sali A (2012) Putting the pieces together: integrative modeling platform software for structure determination of macromolecular assemblies. PLoS Biol 10:e1001244
Sailer C, Offensperger F, Julier A, Kammer KM, Walker-Gray R, Gold MG, Scheffner M, Stengel F (2018) Structural dynamics of the E6AP/UBE3A-E6-p53 enzyme-substrate complex. Nat Commun 9:4441
Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234:779–815
Sali A, Glaeser R, Earnest T, Baumeister W (2003) From words to literature in structural proteomics. Nature 422:216–225
Sali A, Berman HM, Schwede T, Trewhella J, Kleywegt G, Burley SK, Markley J, Nakamura H, Adams P, Bonvin AM, Chiu W, Peraro MD, Di Maio F, Ferrin TE, Grunewald K, Gutmanas A, Henderson R, Hummer G, Iwasaki K, Johnson G, Lawson CL, Meiler J, Marti-Renom MA, Montelione GT, Nilges M, Nussinov R, Patwardhan A, Rappsilber J, Read RJ, Saibil H, Schroder GF, Schwieters CD, Seidel CA, Svergun D, Topf M, Ulrich EL, Velankar S, Westbrook JD (2015) Outcome of the first wwPDB hybrid/integrative methods task force workshop. Structure 23:1156–1167
Saltzberg DJ, Broughton HB, Pellarin R, Chalmers MJ, Espada A, Dodge JA, Pascal BD, Griffin PR, Humblet C, Sali A (2017) A residue-resolved Bayesian approach to quantitative interpretation of hydrogen–deuterium exchange from mass spectrometry: application to characterizing protein–ligand interactions. J Phys Chem B 121:3493–3501
Saltzberg D, Greenberg CH, Viswanath S, Chemmama IE, Webb B, Pellarin R, Echeverria I, Sali A (2019) Modeling biological complexes using integrative modeling platform. Methods Mol Biol
Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ (2005) PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res 33:W363–367
Schneidman-Duhovny D, Hammel M, Sali A (2010) FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res 38:W540–544
Schneidman-Duhovny D, Hammel M, Sali A (2011) Macromolecular docking restrained by a small angle X-ray scattering profile. J Struct Biol 173:461–471
Schneidman-Duhovny D, Rossi A, Avila-Sakar A, Kim SJ, Velazquez-Muriel J, Strop P, Liang H, Krukenberg KA, Liao M, Kim HM, Sobhanifar S, Dotsch V, Rajpal A, Pons J, Agard DA, Cheng Y, Sali A (2012) A method for integrative structure determination of protein–protein complexes. Bioinformatics 28:3282–3289
Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A (2013) Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys J 105:962–974
Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A (2016) FoXS, FoXSDock and MultiFoXS: single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res 44:W424–429
Schwieters CD, Bermejo GA, Clore GM (2018) Xplor-NIH for molecular structure determination from NMR and other data sources. Protein Sci 27:26–40
Sehnal, D. (2016). “BinaryCIF.” 2019, from https://github.com/dsehnal/BinaryCIF
Serra F, Bau D, Goodstadt M, Castillo D, Filion GJ, Marti-Renom MA (2017) Automatic analysis and 3D-modelling of Hi-C data using TADbit reveals structural features of the fly chromatin colors. PLoS Comput Biol 13:e1005665
Serysheva II, Ludtke SJ, Baker ML, Cong Y, Topf M, Eramian D, Sali A, Hamilton SL, Chiu W (2008) Subnanometer-resolution electron cryomicroscopy-based domain models for the cytoplasmic region of skeletal muscle RyR channel. Proc Natl Acad Sci USA 105:9610–9615
Shen MY, Sali A (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci 15:2507–2524
Shi Y, Fernandez-Martinez J, Tjioe E, Pellarin R, Kim SJ, Williams R, Schneidman-Duhovny D, Sali A, Rout MP, Chait BT (2014) Structural characterization by cross-linking reveals the detailed architecture of a coatomer–related heptameric module from the nuclear pore complex. Mol Cell Proteomics 13:2927–2943
Shi Y, Pellarin R, Fridy PC, Fernandez-Martinez J, Thompson MK, Li Y, Wang QJ, Sali A, Rout MP, Chait BT (2015) A strategy for dissecting the architectures of native macromolecular assemblies. Nat Methods 12:1135–1138
Simon B, Madl T, Mackereth CD, Nilges M, Sattler M (2010) An efficient protocol for NMR-spectroscopy-based structure determination of protein complexes in solution. Angew Chem Int Ed Engl 49:1967–1970
Sippl MJ (1990) Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J Mol Biol 213:859–883
Spill YG, Kim SJ, Schneidman-Duhovny D, Russel D, Webb B, Sali A, Nilges M (2014) SAXS Merge: an automated statistical method to merge SAXS profiles using Gaussian processes. J Synchrotron Radiat 21:203–208
Stahlberg H, Walz T (2008) Molecular electron microscopy: state of the art and current challenges. ACS Chem Biol 3:268–281
Tagari M, Newman R, Chagoyen M, Carazo JM, Henrick K (2002) New electron microscopy database and deposition system. Trends Biochem Sci 27:589
Taylor DJ, Devkota B, Huang AD, Topf M, Narayanan E, Sali A, Harvey SC, Frank J (2009) Comprehensive molecular structure of the eukaryotic ribosome. Structure 17:1591–1604
Timney BL, Raveh B, Mironska R, Trivedi JM, Kim SJ, Russel D, Wente SR, Sali A, Rout MP (2016) Simple rules for passive diffusion through the nuclear pore complex. J Cell Biol 215:57–76
Tjong H, Li W, Kalhor R, Dai C, Hao S, Gong K, Zhou Y, Li H, Zhou XJ, Le Gros MA, Larabell CA, Chen L, Alber F (2016) Population-based 3D genome structure analysis reveals driving forces in spatial genome organization. Proc Natl Acad Sci USA 113:E1663–E1672
Trussart M, Serra F, Bau D, Junier I, Serrano L, Marti-Renom MA (2015) Assessing the limits of restraint-based 3D modeling of genomes and genomic domains. Nucleic Acids Res 43:3465–3477
Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J, Livny M, Mading S, Maziuk D, Miller Z, Nakatani E, Schulte CF, Tolmie DE, Kent Wenger R, Yao H, Markley JL (2008) BioMagResBank. Nucleic Acids Res 36:D402–D408
Upla P, Kim SJ, Sampathkumar P, Dutta K, Cahill SM, Chemmama IE, Williams R, Bonanno JB, Rice WJ, Stokes DL, Cowburn D, Almo SC, Sali A, Rout MP, Fernandez-Martinez J (2017) Molecular architecture of the major membrane ring component of the nuclear pore complex. Structure 25:434–445
Valentini E, Kikhney AG, Previtali G, Jeffries CM, Svergun DI (2015) SASBDB, a repository for biological small-angle scattering data. Nucleic Acids Res 43:D357–D363
Vallat B, Webb B, Westbrook JD, Sali A, Berman HM (2018) Development of a prototype system for archiving integrative/hybrid structure models of biological macromolecules. Structure 26(894–904):e892
van Zundert GCP, Melquiond ASJ, Bonvin A (2015) Integrative modeling of biomolecular complexes: HADDOCKing with cryo-electron microscopy data. Structure 23:949–960
Velazquez-Muriel J, Lasker K, Russel D, Phillips J, Webb BM, Schneidman-Duhovny D, Sali A (2012) Assembly of macromolecular complexes by satisfaction of spatial restraints from electron microscopy images. Proc Natl Acad Sci USA 109:18821–18826
Viswanath S, Bonomi M, Kim SJ, Klenchin VA, Taylor KC, Yabut KC, Umbreit NT, Van Epps HA, Meehl J, Jones MH, Russel D, Velazquez-Muriel JA, Winey M, Rayment I, Davis TN, Sali A, Muller EG (2017a) The molecular architecture of the yeast spindle pole body core determined by Bayesian integrative modeling. Mol Biol Cell 28:3298–3314
Viswanath S, Chemmama IE, Cimermancic P, Sali A (2017b) Assessing exhaustiveness of stochastic sampling for integrative modeling of macromolecular structures. Biophys J 113:2344–2353
Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59:687–696
Wang X, Chemmama IE, Yu C, Huszagh A, Xu Y, Viner R, Block SA, Cimermancic P, Rychnovsky SD, Ye Y, Sali A, Huang L (2017a) The proteasome-interacting Ecm29 protein disassembles the 26S proteasome in response to oxidative stress. J Biol Chem 292:16310–16320
Wang X, Cimermancic P, Yu C, Schweitzer A, Chopra N, Engel JL, Greenberg C, Huszagh AS, Beck F, Sakata E, Yang Y, Novitsky EJ, Leitner A, Nanni P, Kahraman A, Guo X, Dixon JE, Rychnovsky SD, Aebersold R, Baumeister W, Sali A, Huang L (2017b) Molecular details underlying dynamic structures and regulation of the human 26S proteasome. Mol Cell Proteomics 16:840–854
Ward AB, Sali A, Wilson IA (2013) Biochemistry. Integrative structural biology. Science 339:913–915
Webb B, Viswanath S, Bonomi M, Pellarin R, Greenberg CH, Saltzberg D, Sali A (2018) Integrative structure modeling with the integrative modeling platform. Protein Sci 27:245–258
Westbrook, J. (2013). “PDBx/mmCIF Dictionary Resources.” Retrieved August 25, 2015, from http://mmcif.wwpdb.org
wwPDB consortium (2007). Announcement: Experimental Data Will Be Required for Depositions Starting February 1, 2008. wwPDB Newsletter, wwPDB consortium,
Young JY, Westbrook JD, Feng Z, Sala R, Peisach E, Oldfield TJ, Sen S, Gutmanas A, Armstrong DR, Berrisford JM, Chen L, Chen M, Di Costanzo L, Dimitropoulos D, Gao G, Ghosh S, Gore S, Guranovic V, Hendrickx PMS, Hudson BP, Igarashi R, Ikegawa Y, Kobayashi N, Lawson CL, Liang Y, Mading S, Mak L, Mir MS, Mukhopadhyay A, Patwardhan A, Persikova I, Rinaldi L, Sanz-Garcia E, Sekharan MR, Shao C, Swaminathan GJ, Tan L, Ulrich EL, van Ginkel G, Yamashita R, Yang H, Zhuravleva MA, Quesada M, Kleywegt GJ, Berman HM, Markley JL, Nakamura H, Velankar S, Burley SK (2017) OneDep: unified wwPDB system for deposition, biocuration, and validation of macromolecular structures in the PDB archive. Structure 25:536–545
Zeng-Elmore X, Gao XZ, Pellarin R, Schneidman-Duhovny D, Zhang XJ, Kozacka KA, Tang Y, Sali A, Chalkley RJ, Cote RH, Chu F (2014) Molecular architecture of photoreceptor phosphodiesterase elucidated by chemical cross-linking and integrative modeling. J Mol Biol 426:3713–3728
Acknowledgements
We acknowledge research support from NIH grants U54DK107981, R01GM083960, P41GM109824, P01AG002132 and NSF award DBI-1756250 (AS); and the NSF awards DBI-1519158 and DBI-1756248 (HMB). We thank the wwPDB leadership and the members of the wwPDB IHM Task Force for their support. We thank the developers of the ChimeraX software, Thomas Ferrin and Thomas Goddard for enabling the visualization of integrative models archived in PDB-Dev.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Vallat, B., Webb, B., Westbrook, J. et al. Archiving and disseminating integrative structure models. J Biomol NMR 73, 385–398 (2019). https://doi.org/10.1007/s10858-019-00264-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-019-00264-2