
Abstract
Simple and convenient method of protein dynamics evaluation from the insufficient experimental 15N relaxation data is presented basing on the ratios, products, and differences of longitudinal and transverse 15N relaxation rates obtained at a single magnetic field. Firstly, the proposed approach allows evaluating overall tumbling correlation time (nanosecond time scale). Next, local parameters of the model-free approach characterizing local mobility of backbone amide N–H vectors on two different time scales, S2 and R ex , can be elucidated. The generalized order parameter, S2, describes motions on the time scale faster than the overall tumbling correlation time (pico- to nanoseconds), while the chemical exchange term, R ex , identifies processes slower than the overall tumbling correlation time (micro- to milliseconds). Advantages and disadvantages of different methods of data handling are thoroughly discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Analysis of nuclear magnetic relaxation provides insight into the molecular motions of proteins (Palmer 2004; Mayo and Daragan 2005; Ejchart 2007; Charlier et al. 2016; Lisi and Loria 2016). One of the most successful and widely used approaches to the analysis of relaxation data is the model-free approach, MFA (Lipari and Szabo 1982). Two other model-independent descriptions of the internal molecular motions have been developed independently: the two-step model (Halle and Wennerström 1981) and the slowly relaxing local structure model (Tugarinov et al. 2001). In the frame of the MFA, local mobility is described by two parameters, the generalized order parameter, S2, which corresponds to the spatial freedom of motion, and the internal correlation time, τint, corresponding to the rate of this motion in the pico- to nanosecond time scale, which is smaller than the single correlation time describing an isotropic overall molecular tumbling, τ R . The additional term, R ex , accounts for the conformational exchange contribution to R2 resulting from processes in the micro- to millisecond time scale, often referred to as chemical exchange effects (Stone et al. 1992; Korzhnev et al. 2001).
The most frequently measured protein relaxation parameters are the longitudinal, R1, and transverse, R2, relaxation rates of backbone amide 15N nuclei complemented with 15N-{1H} NOEs (Palmer 2004; Reddy and Rainey 2010). Nuclear magnetic relaxation measurements are long lasting, their data reduction is demanding, and interpretation of the results complex (Zhukov and Ejchart 1999; Pawley et al. 2006; Jaremko et al. 2015). Especially NOE measurements are very time consuming owing to their inherently low sensitivity (Fushman 2003) and susceptibility to systematic errors resulting from not fully relaxed spectra and/or saturation transfer due to the exchange with water protons (Ferrage et al. 2008; Gong and Ishima 2007; Grzesiek and Bax 1993; Renner et al. 2002). So it often happens that limited relaxation data (e.g., R1 and R2 at a single magnetic field), precluding the determination of all MFA parameters, are solely available. Then, commonly used procedure is to examine the ratio between transverse and longitudinal rates, R2/R1, for the estimation of global isotropic correlation time of a protein (Kay et al. 1989). As a prerequisite this procedure requires an efficient rejection from calculation the residues exhibiting skewed R2/R1 values owing to intense fast local motions and/or slow conformational exchange. Residues with an NOE (if available) < 0.6 are usually excluded, since in this case the internal motions are likely to skew significantly the R2/R1 value. Usually such residues exhibit low experimental R2/R1 values. On the other hand, residues undergoing conformational exchange are characterized by high R2/R1 values. Routine method of appropriate residue selection can be done by excluding those residues with a R2/R1 value outside of ± 1 standard deviation of the mean (Clore et al. 1990).
With the τ R already estimated, three methods of further analysis of limited relaxation data were independently developed. One of them is based on the observation that one of the MFA parameters, the generalized order parameter, S2, can be extracted with a reasonable accuracy from a linear combination of relaxation rates, 2R2 − R1 (Habazettl and Wagner 1995). Another method investigates the product of relaxation rates R1R2 also giving access to the S2 parameter (Kneller et al. 2002). The last method of the S2 parameter extraction from R1 and R2, measured with the CEST technique to save experimental time and named lean MFA (LMFA), relies on the least squares minimization (Gu et al. 2016). Since all these methods base on the determination of the overall correlation time τ R from the R2/R1 ratio an appropriate data selection seems to be crucial. Therefore, it was proposed to use for this purpose the product of relaxation rates R1R2 and claimed that in contrary to the R2/R1 ratio, the former allows to distinguish between the effects of motional anisotropy and chemical exchange (Kneller et al. 2002). For convenience, R2/R1, 2R2 − R1, and R1R2 will be further denoted as Q, D, and P, respectively.
This paper describes a comparative analysis of three mentioned above combinations of relaxation rates, which allows to select the optimal method of the MFA parameter elucidation from deficient relaxation data. The utility of this approach is presented by applying it to the relaxation data of four proteins of different size: immunoglobulin-binding domain of streptococcal protein G, GB1 (Idiyatullin et al. 2003), human ubiquitin (Lee and Wand 1999), human S100A1 calcium binding protein in apo state (Nowakowski et al. 2011), and β-lactamase PSE-4 (Morin and Gagné 2009). First limited use of this approach was applied to the 15N relaxation rates of BacSp222 peptide (Nowakowski et al. 2018).
Method
Equations describing relaxation rates of 15N nuclei relaxing by dipolar and chemical shift anisotropy mechanisms in terms of spectral density functions are given as (Abragam 1989; Korzhnev et al. 2001):
where \(d=\frac{{{\mu _0}}}{{8{\pi ^2}}}\frac{{{\gamma _N}{\gamma _H}h}}{{\left\langle {r_{{NH}}^{3}} \right\rangle }},\quad c={\omega _N}\Delta \sigma\) and other symbols have their usual meaning. It has to be mentioned that in all calculations the vibrationally averaged N–H distance, rNH = 1.04 Å (Ottiger and Bax 1998) and the chemical shift anisotropy of the 15N chemical shift tensor Δδ = − 170 ppm (Yao et al. 2010) were used.
The conformational exchange contribution to the transverse relaxation rate, R ex , is proportional to the square of the 15N Larmor frequency, ω N . This term can be written as \({R_{ex}}=\Phi \omega _{N}^{2}\) (Peng and Wagner 1995). The proportionality factor Φ represents the effectiveness of conformational exchange processes and is independent on magnetic field strength facilitating direct comparison of chemical exchange terms determined at different magnetic field strengths for different proteins.
Model-free approach spectral density function takes the form (Lipari and Szabo 1982):
where \({\tau ^{ - 1}}=\tau _{R}^{{ - 1}}+\tau _{{\operatorname{int} }}^{{ - 1}}\). Performing the complete MFA analysis of relaxation data for N-residue protein one has to determine 3N local parameters, S2, τint, and R ex for each residue. Additionally one global parameter τ R or six parameters characterizing either isotropic or fully anisotropic overall tumbling, respectively, have to be determined. Extension of the spectral density function for isotropic motion (Eq. 3) to the anisotropic one, based on the formalism developed by Woessner (1962), was implemented to the protein relaxation studies (Tjandra et al. 1995; Baber et al. 2001). Allowing for the positive degree of freedom of a computational task it means that besides R1, R2, and 15N-{1H} NOE at a single magnetic field, at least one additional set of relaxation parameters has to be measured. It happens, however, that the number of available relaxation data is insufficient, due to sample instability or lack of experimental time and additional data processing has to be applied (Jaremko et al. 2014). Often only R1 and R2 relaxation rates at a single magnetic field are at one’s disposal.
A joined analysis of Q = R2/R1 and D = 2R2 − R1 or P = R1R2 values allows obtaining semi-quantitative insight into the protein dynamics owing to the different relations of these quantities to the MFA parameters and, therefore, untangling these parameters from experimental data. The Q parameter is quasi-insensitive to both local MFA parameters, S2 and τint, in a reasonably broad range of their values (Fig. 1A, B). Therefore, it is well suited for the evaluation of the overall tumbling correlation times of proteins comprising residues with diverse local mobility. On the other hand, the P values are quasi-insensitive to τint but decrease considerably with the increased amplitude of local motions, as manifested at smaller values of the S2 order parameter (Fig. 1B). The D parameter is even less sensitive to τint changes than Q and P, but it displays a modest sensitivity to S2 changes. All three quantities, Q, D, and P, are sensitive to the chemical exchange term and increase with the R ex enlargement (Fig. 1C). Simultaneous effect of S2 and τint, changes on Q, P and D is shown in Figs. S1–S3 (Supporting Information). One has to be aware of the opposite effects of fast (ps–ns) and slow (µs–ms) motions on the P values. Both these effects can compensate one another leaving the P value unchanged and, thus, hiding chemical exchange effect. The D values are also sensitive to such compensation. They are, however, less sensitive to fast motions and more sensitive to slow ones than P values and, therefore, should retain at least partially the ability of detection of R ex terms.

Calculated relationships between normalized Q, D, and P quantities and local parameters of MFA. The presented quantities are normalized in relation to their counterparts in rigid molecules. A Q(τint), D(τint), and P(τint) functions with S2 = 0.85 and R ex = 0. B Q(S2), D(S2), and P(S2) functions with τint = 50 ps and R ex = 0. C Q(R ex ), D(R ex ), and P(R ex ) with τint = 50 ps and S2 = 0.85. Additional input data: τR = 5 ns and B0 = 16.4 T were used in all calculations. Take note that Q and P are superposed in part C
Use of Q, D, and P values in the analysis of a backbone protein dynamics requires several simplifying assumptions bearing a number of consequences. It has been noticed (Peng and Wagner 1992) that the spectral density functions at three highest frequencies J(ω H + ω N ), J(ω H ), and J(ω H − ω N ) are only a small fraction of two other component J(0) and J(ω N ) and can be neglected in Eqs. (1) and (2) describing D values (Habazettl and Wagner 1995) or P values (Kneller et al. 2002). As a result following expressions can be written:
In the approach utilizing the Q ratio for the estimation of global correlation time, the assumption τint = 0.0 is made resulting in a simplified spectral density function (Kay et al. 1989).
In order to obtain so estimated global correlation time, τ R (Q), one has to compute it from Eq. (8) given by Kay et al. (1989). Additionally, neglecting second term in Eq. (4c) and assuming \({({\omega _N}{\tau _R})^2}>>1\) one obtains:
Use of the Q values in the evaluation of an overall correlation time results in the τR underestimation provided the overall tumbling is isotropic (Korzhnev et al. 1997). Influence of the input τR and magnetic field strength values on the value of the apparent τR is demonstrated in Fig. 2. In the utmost situations (parts of plots below the dashed line in the Fig. 2 corresponding to intense internal motion: S2 = 0.7, τint = 100 ps, slow overall tumbling: τR = 32 ns and very high magnetic field: 23.5 T) the τR evaluation derived from the Q values breaks down; relative errors exceed 25%. Use of Q values retains the sense only if correlation time of internal motion, τint, is short and its amplitude small (Fig. S4).

Normalized values of apparent τR evaluated from the Q values, given as a fraction of the synthetic τR used in simulations. The τR,app/τR ratio is shown as a function of B0 for several τR values. Calculations were performed applying sizeable internal motion: S2 = 0.7 and τint = 100 ps. Performance of the Q-based method is poor for B0 and τR corresponding to plots below the dashed line marking 15% deviation of τR,app
In the case of anisotropic tumbling, the determined value of the orientation averaged overall correlation time τR = 0.5/(D1 + D2 + D3), can be either larger or smaller than the τR value estimated from Q values, depending on the orientation of the N–H vector. Appropriate comparison is presented in Table 1 and Fig. 3.

Molecular tumbling is anisotropic (prolate ellipsoid \(\left\langle {{\tau _{\text{R}}}} \right\rangle\) = 8 ns, ΔD = 1.5 and η = 0). A sizeable internal motion is assumed: S2 = 0.8 and τint = 100 ps. The τR estimated from the derived Q value can deviate significantly from the expected value of 8 ns marked by a horizontal black line. The deviations depend on the N–H vector orientation relative to the unique axis of diffusion tensor given by an angle α. Deviations depend on the magnetic field strength (red and blue circles). Field dependence nearly disappears for rigid N–H vector (S2 = 1.0; red and blue triangles). The tendency of τ R (α) dependence for oblate ellipsoid (\(\left\langle {{\tau _{\text{R}}}} \right\rangle\) = 8 ns, ΔD = 0.67 and η = 0) is opposite in comparison with a prolate ellipsoid (red and blue squares)
Estimation of the average generalized order parameters \(S_{{av}}^{2}\) from the experimentally observed P values was proposed by Kneller et al. (2002) using formula:
where \(\left\langle P \right\rangle\) is experimentally observed 10% trimmed mean value and Pmax is determined from the relaxation parameters calculated for a rigid molecule (S2 = 1.0, R ex = 0.0) which reorients with τR(Q). This formula results directly from Eq. (6c). Use of medians is superior to the trimmed mean values since the distributions of Q, D, and P data are most commonly non gaussian (Table S1) and robust statistics has to be used in their description (Maronna et al. 2006). Medians allow not only avoiding influence of outliers but also eliminating residues from the unstructured segments of protein characterized by inherently small Q values and resulting in extremely skewed Q distributions. Robust statistics also facilitates identifying residues undergoing chemical exchange as outliers in Q, D, and P sets. In the following text Q, D, and P medians are solely used and denoted as \(\tilde {Q}\), \(\tilde {D}\), and \(\tilde {P}\). Therefore, the Kneller et al. formula is rewritten as
This approach is also extended to the estimation of site specific generalized order parameters, \(S_{i}^{2}\) utilizing either D or P values:
One has to be aware of possible systematic deviations of so estimated \(S_{i}^{2}\) values. As it was shown earlier, the Q-derived τR values are underestimated in isotropically tumbling molecules. Accordingly, the Pmax is underestimated as well, resulting in the overestimation of \(S_{i}^{2}\) values (Fig. 4). This effect is especially pronounced for slower internal motions (long τint) with large amplitudes (small S2) at high magnetic fields. A similar effect was reported for the relaxation data in ATPase α-domain (Gu et al. 2016).

Normalized values of apparent S2 evaluated from D or P values (Eqs. 8a and 8b), given as a fraction of the input S2 used in simulations. The \(S_{{app}}^{2}/{S^2}\) ratio is shown as a function of the input S2 for three B0 (9.4, 14.1, and 18.8 T) and two τint values (10 and 100 ps). Calculations were performed applying overall correlation time τ R = 16 ns. Performance of this method becomes poor above the horizontal dashed line representing 10% deviation of the \(S_{{app}}^{2}\) values
It is stated that the Q values do not distinguish between the effects of motional anisotropy and chemical exchange (Kneller et al. 2002), while the analysis of P data significantly attenuates the effects of motional anisotropy (c.f. Table 1) permitting rapid identification of residues undergoing chemical exchange, R ex . It will be shown in the next section, however, that the attenuation of motional anisotropy is not sufficient to identify unequivocally the R ex influenced residues. The elevated P values not always allow identifying residues affected by chemical exchange. In fact, only simultaneous outlying Q, D, and P values point out unequivocally to the chemical exchange.
Results and discussion
Combined analysis of Q, D, and P values is applied to four proteins for which the large relaxation data sets are available in the literature. Essential information on these proteins is collected in the Table 2.
Overall correlation time
The Q, D, and P site specific values for the analyzed proteins at all available magnetic fields are shown in Figs. 5 and S5–S14.The medians of experimentally observed Q = R1/R2 values were used for evaluating overall correlation times. Vizualization of this procedure is shown in Figs. S15–S18. Some details are explained in the captions to these figures.

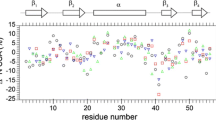
Sequence specific Q, D, and P values calculated from R1 and R2 relaxation rates determined for S100A1 protein at 16.4 T. Solid lines represent medians: \(\tilde {Q}\) = 10.31, \(\tilde {P}\) = 14.68, and \(\tilde {D}\) = 23.49. Dashed lines mark the limit of outliers calculated from the formula Q3 + 1.5·IQR, where Q3 is third quartile and IQR is the interquartile range. Residue Glu22 undergoing a chemical exchange is marked with a red circle. Blue circles mark residues with a questionable presence of chemical exchange mechanism
For the proteins analyzed here, the determined τR(Q) values are larger than the corresponding values obtained from the MFA analysis as shown in the Fig. 6. Corresponding numerical values are reported in Table S2. The averaged deviations are equal to 18, 5, 2, and 5% for GB1, ubiquitin, S100A1, and PSE4, respectively. The opposite results would be expected from the simulation shown in Fig. 2. However, it is demonstrated in the “Method” section that the anisotropic tumbling can result in the overestimation of τR values and one should note that all four proteins reorient anisotropically (Table 3). It is worth noting that the largest discrepancy between Q-based and MFA derived τR values appears for GB1 characterized by the strongest tumbling anisotropy. Anisotropic tumbling and, therefore, the possible overestimation of τR values can be anticipated from the ratios of principal values of inertia tensor provided the protein structure is available (Mandel et al. 1995).

Comparison of the overall correlation times τR determined by the model-free approach with those obtained from the appropriate \(\tilde {Q}\) values. Determination of uncertainties represented by error bars is described in the caption to Table S2
Anisotropic tumbling as a reason of the divergence between τR values obtained from the MFA analysis and Q-based method can be demonstrated analyzing two residues of GB1, Thr17 and Asp40. Their N–H vectors are nearly perpendicular one another. The Q values have been obtained from the R1 and R2 relaxation rates and next used in the τ R (Q) determination. Results are presented in Table 4. The orientation of N–H vectors strongly influences the value of pseudo-isotropic overall correlation time determined using Q value. It has to be pointed out that the median, \(\tilde {Q}\), can be skewed due to the non uniform distribution of N–H vector orientations. Regardless of anisotropic tumbling the presented examples of τR estimation by means of the Q-based method give suitable results with the average deviation 7% and the largest one 21% for GB1 at 11.7 T as compared with the MFA results.
Chemical exchange
Residues exhibiting slow conformational mobility in the micro- to millisecond time scale have to be selected and next skipped in the further analysis of fast motions. Elevated Q, P, or D values owing to the chemical exchange mechanism result in non physical values of the generalized order parameter, S2 > 1.
None of GB1 residues shows outlying Q, P, or D values being a hallmark of the chemical exchange (c.f. Figs. S5–S7). This observation is consistent with the MFA analysis performed for R1, R2, and NOE data at three magnetic fields (Table S3). Both, standard deviations of Φ factors multiplied by an appropriate Student’s t-value and F-test applied to the partial target functions characterizing the fit quality for a given residue with and without assumption of chemical exchange (three or two local parameters) were applied to verify the significance of the MFA-derived Φ factors.
Among the Q, P, and D values calculated for ubiquitin one residue, Asn25, displays the largest values pointing out to the effective chemical exchange mechanism in the relaxation of this residue (c.f. Figs. S8 and S9). This result is consistent with the results of the MFA analysis. Asn25 is the unique residue exhibiting meaningful Φ value (Table S4). Three other residues show large but erratic Q, P, D values. None of them, however, possesses a meaningful Φ value in the MFA analysis.
Glu22 is the unique residue in the S100A1 protein displaying simultaneous large Q, P, and D values, thus clearly exhibiting the chemical exchange mechanism (Figs. 5, S10, and S11). This fact is also consistent with the MFA analysis (Table S5). However, three other residues, Glu3, Gly23 (only at 16.4 T), and Lys25, similarly as Glu22, display distinct P values. So, they can be suspected to undergo a chemical exchange. On the other hand, their Q values and majority of D values are very close to the corresponding medians. Φ factors of Gly23 and Lys25 residues obtained in the MFA analysis reveal the chemical exchange mechanism for these residues but definitely exclude it for Glu3 residue. Summing up, Q values fail to recognize chemical exchange for Gly23 and Lys25 but P values can lead to false recognition of non existing slow motions. D values perform the best at 16.4 T but fail at 9.4 T. None of Q, P, and D parameters is fully immune to the erroneous identification of chemical exchange.
In PSE4 protein (Figs. S12–S14) Q, P, D values for two residues, Thr128 and Ser235, at 11.7 T exceed corresponding outliers’ limits indicating chemical exchange. They also display the largest Φ values in the MFA analysis (Table S6). Nevertheless, not all of Q, P, D values at two higher magnetic fields confirm the efficient chemical exchange. Several other residues show non systematic, outlying Q, P, D values. Three of them, Thr57, Leu221, and Gly236, exhibit meaningful Φ values indicating chemical exchange while for the remaining residues, protruding Q, P, or D values seem to be misleading.
All the above discussed results point out to the possibility of ambiguous or false recognition of chemical exchange involved residues. None of Q, P, and D parameters is fully immune to the erroneous identification of chemical exchange but their values inspected simultaneously usually identify residues influenced by chemical exchange with a high probability.
Generalized order parameters
There are three methods allowing to estimate site specific generalized order parameters S2 which describe local fast mobility of a protein backbone. Since such information is important in study of protein function and interactions with other molecules, it is necessary to compare results of these methods. Determination of the isotropic, overall tumbling correlation time, τ R , from the properly selected Q value makes a starting point of all these methods. Two methods utilizing Eqs. (8a) or (8b) are based on D or P values, respectively. The third method, LMFA, requires fit of back calculated relaxation rates R1 and R2 with fixed τ R (Q) and adjustable S2 parameters to the experimental data.
Comparison of the results of these methods with those obtained for the MFA analysis of all available relaxation data as a reference, was performed. Similar conclusions can be reached from the results obtained for each of four investigated proteins. General tendencies can be discussed and presented on the examples of several selected GB1 residues. They are shown in Fig. 7.

Comparison of S2 values and their confidence limits for selected residues of GB1 protein. S2 obtained from D values, P values, and LMFA approach are represented by red, green, and blue circles, respectively. The leftmost values are reference MFA derived results. Confidence limits for the MFA and LMFA calculations were obtained as standard deviations from 200 Monte Carlo simulations. Symmetrized confidence limits for S2(D) and S2(P) values were evaluated applying standard method of error propagation
An important conclusion drawn from data shown in Fig. 7 is that confidence ranges of S2(D) are roughly twice larger than corresponding ranges for S2(P) and S2(LMFA). This is a general feature obtained for all residues in all analyzed proteins (total 399 residues). Confidence ranges for S2(P) and S2(LMFA) are comparable, but always larger than those characterizing MFA results for obvious reason of a larger number of data and parameters used in the latter method. Another important factor concerns dispersion of S2 values resulting from different methods for the same residue. In the majority of cases S2(D), S2(P), and S2(LMFA) are close each other at a given magnetic field but differ from the S2(MFA) and among the data determined at different magnetic fields (Fig. 7, Thr11 and Glu19). In a number of cases all the data are close each other (Fig. 7, Ala23). Such situation takes place when all individual Q values are close to the corresponding medians \(\tilde {Q}\). In the opposite case, when individual Q values differ markedly from medians, all S2 values are broadly dispersed (Fig. 7, Asp40).
Whole sets of S2 values estimated as the sums of squared differences between S2(MFA) and S2(X) (X = D, P, LMFA) allow to estimate accuracy of each method. Once more P-based and LMFA approaches are comparable and slightly more accurate than the D-based one.
Concluding, P-based and LMFA methods of S2 determination perform similarly but the former one is less demanding from the computational standpoint.
Conclusions
The R1 and R2 relaxation rates of 15N nuclei measured at a single magnetic field strength in proteins are not sufficient to perform a formal MFA analysis, but can be utilized for the semi-quantitative evaluation of the overall tumbling correlation time from the median of the set of transverse to longitudinal relaxation rates Q = R2/R1. Generalized order parameters S2 characterizing amplitude of internal local motions, faster than the overall tumbling, can be site selectively evaluated using either linear combination D = 2R2 − R1 or product P = R2·R1 or LMFA method. Efficiency of these methods was carefully compared and the P and LMFA methods are comparably accurate while the former is less demanding from the computational standpoint. As the final result one obtains estimation of the overall correlation time, τR, and parameters characterizing internal motions, S2, on the time scales faster than τR. Additionally, residues undergoing conformational motions in the micro- to millisecond time scale can be selected basing on the Q, D, and P outliers.
References
Abragam A (1989) Principles of nuclear magnetism. Clarendon Press, Oxford
Baber JL, Szabo A, Tjandra N (2001) Analysis of slow interdomain motion of macromolecules using NMR relaxation data. J Am Chem Soc 123:3953–3959
Charlier C, Cousin SF, Ferrage F (2016) Protein dynamics from nuclear magnetic relaxation. Chem Soc Rev 45:2410 – 2422
Clore GM, Driscoll PC, Wingfield PT, Gronenborn AM. (1990) Analysis of the backbone dynamics of interleukin-16 using two-dimensional inverse detected heteronuclear 15N-1H NMR spectroscopy. Biochemistry 29:7387–7401
Ejchart A (2007) Insight into protein dynamics from nuclear magnetic relaxation studies. Polimery 52:745–751
Ferrage F, Piserchio A, Cowburn D, Ghose R (2008) On the measurement of 15N–{1H} nuclear Overhauser effects. J Magn Reson 192:302 – 313
Fushman D (2003) In BioNMR in drug research. In: Zerbe O (ed) Methods and principles in medicinal chemistry, vol 16. Wiley, Weinheim, pp 283–308
Gong Q, Ishima R (2007) 15N–{1H} NOE experiment at high magnetic field strengths. J Biomol NMR 37:147–157
Grzesiek S, Bax A (1993) The importance of not saturating H2O in protein NMR. Application to sensitivity enhancement and NOE measurements. J Am Chem Soc 115:12593 – 12594
Gu Y, Hansen AL, Peng Y, Brüschweiler R (2016) Rapid determination of fast protein dynamics from NMR chemical exchange saturation transfer data. Angew Chem Int Ed 55:3117–3119
Habazettl J, Wagner G (1995) A simplified method for analyzing nitrogen-15 nuclear magnetic resonance data of proteins. J Magn Reson B 109:100–104
Halle B, Wennerström H (1981) Interpretation of magnetic-resonance data from water nuclei in heterogeneous systems. J Chem Phys 75:1928–1943
Idiyatullin D, Nesmelova I, Daragan VA, Mayo KH (2003) Heat capacities and a snapshot of the energy landscape in protein GB1 from the pre-denaturation temperature dependence of backbone NH nanosecond fluctuations. J Mol Biol 325:149–162
Jaremko M, Jaremko L, Nowakowski M, Wojciechowski M, Szczepanowski RH, Panecka R, Zhukov I, Bochtler M, Ejchart A (2014) NMR structural studies of the first catalytic half-domain of ubiquitin activating enzyme. J Struct Biol 185:69–78
Jaremko L, Jaremko M, Nowakowski M, Ejchart A (2015) The quest for simplicity: remarks on the free-approach models. J Phys Chem B 119:11978 – 11987
Kay LE, Torchia DA, Bax A (1989) Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry 28:8972–8979
Kneller MK, Lu M, Bracken C (2002) An effective method for the discrimination of motional anisotropy and chemical exchange. J Am Chem Soc 124:1852–1853
Korzhnev DM, Orekhov VY, Arseniev AS (1997) Model-free approach beyond the borders of its applicability. J Magn Reson 127:184–191
Korzhnev DM, Billeter M, Arseniev AS, Orekhov VY (2001) NMR studies of Brownian tumbling and internal motions in proteins. Prog Nucl Magn Reson Spectrosc 38:197–266
Lee AL, Wand AJ (1999) Assessing potential bias in the determination of rotational correlation times of proteins by NMR relaxation. J Biomol NMR 13:101–112
Lipari G, Szabo A (1982) Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc 104:4546–4559
Lisi GP, Loria JP (2016) Using NMR spectroscopy to elucidate the role of molecular motions in enzyme function. Prog NMR Spectrosc 92–93:1–17
Mandel AM, Akke M, Palmer AG III (1995) Backbone dynamics of Escherichia coli ribonuclease HI: correlation with structure and function in an active enzyme. J Mol Biol 246:144–163
Maronna RA, Martin RD, Yohai VJ (2006) Robust statistics: theory and methods. Wiley, Hoboken
Mayo KH, Daragan VA (2005) Protein dynamics NMR relaxation. World Scientific Publishing, London
Nowakowski M, Jaremko Ł, Jaremko M, Zhukov I, Belczyk A, Bierzyński A, Ejchart A (2011) Solution NMR structure and dynamics of human apo-S100A1 protein. J Struct Biol 174:391–399
Nowakowski M, Jaremko Ł, Władyka B, Dubin G, Ejchart A, Mak P (2018) Spatial attributes of the four-helix bundle group of bacteriocins—the high-resolution structure of BacSp222 in solution. Int J Biol Macromol 107:2715–2724
Ottiger M, Bax A (1998) Determination of relative NHN, N-C′, Cα-C′, and Cα-Hα effective bond lengths in a protein by NMR in a dilute liquid crystalline phase. J Am Chem Soc 120:12334–12341
Palmer AG III (2004) NMR characterization of the dynamics of biomolecules. Chem Rev 104:3623–3640
Pawley NH, Clark MD. Michalczyk R (2006) Rectifying system-specific errors in NMR relaxation measurements. J Magn Reson 178:77–87
Peng JW, Wagner G (1992) Mapping of the spectral densities of N-H bond motions in Eglin c using heteronuclear relaxation experiments. Biochemistry 31:8571–8583
Peng JW, Wagner G (1995) Frequency spectrum of NH bonds in eglin c from spectral density mapping at multiple fields.Biochemistry 34:16733–16752
Reddy T, Rainey JK (2010) Interpretation of biomolecular NMR spin relaxation parameters. Biochem Cell Biol 88:131–142
Renner C, Schleicher M, Moroder L, Holak TA (2002) Practical aspects of the 2D 15N-{1H}-NOE experiment. J Biomol NMR 23:23–33
Stone MJ, Fairbrother WJ, Palmer AG III, Reizer J, Saier MH Jr, Wright PE (1992) Backbone dynamics of the Bacillus subtilis glucose permease IIA domain determined from 15N NMR relaxation measurements. Biochemistry 31:4394–4406
Tjandra N, Feller SE, Pastor RW, Bax A (1995) Rotational diffusion anisotropy of human ubiquitin from 15N NMR relaxation. J Am Chem Soc 117:12562–12566
Tugarinov V, Liang Z, Shapiro YE, Freed JH, Meirovitch E (2001) A structural mode-coupling approach to 15N NMR relaxation in proteins. J Am Chem Soc 123:3055–3063
Woessner DE (1962) Nuclear spin relaxation in ellipsoids undergoing rotational Brownian motion. J Chem Phys 37:647–654
Yao L, Grishaev A, Cornilescu G, Bax A (2010) Site-specific backbone amide 15N chemical shift anisotropy tensors in a small protein from liquid crystal and cross-correlated relaxation measurements. J Am Chem Soc 132:4295 – 4309
Zhukov I, Ejchart A (1999) Factors improving the accuracy of determination of 15N relaxation parameters in proteins. Acta Biochim Pol 46:665–671
Acknowledgements
The research by LJ and MJ reported in this publication was supported by funding from King Abdullah University of Science and Technology (KAUST).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jaremko, Ł., Jaremko, M., Ejchart, A. et al. Fast evaluation of protein dynamics from deficient 15N relaxation data. J Biomol NMR 70, 219–228 (2018). https://doi.org/10.1007/s10858-018-0176-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-018-0176-3