
Abstract
Overlap of NMR signals is the major cause of difficulties associated with NMR structure elucidation of molecules contained in complex mixtures. A 2D homonuclear correlation spectroscopy in particular suffers from low dispersion of 1H chemical shifts; larger dispersion of 13C chemical shifts is often used to reduce this overlap, while still providing the proton–proton correlation information e.g. in the form of a 2D 1H, 13C HSQC-TOCSY experiment. For this methodology to work, 13C chemical shift must be resolved. In case of 13C chemical shifts overlap, 1H chemical shifts can be used to achieve the desired resolution. The proposed (3, 2)D 1H, 13C BIRDr,X-HSQC-TOCSY experiment achieves this while preserving singlet character of cross peaks in the F1 dimension. The required high-resolution in the 13C dimension is thus retained, while the cross peak overlap occurring in a regular HSQC-TOCSY experiment is eliminated. The method is illustrated on the analysis of a complex carbohydrate mixture obtained by depolymerisation of a fucosylated chondroitin sulfate isolated from the body wall of the sea cucumber Holothuria forskali.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Spread of 13C chemical shifts provides much needed separation of resonances in the indirectly detected dimension of 2D heterocorrelated NMR experiments. This separation is crucial for establishing networks of coupled spins by hyphenated techniques that combine homo- and heteronuclear polarisation transfers, such as 2D 1H-13C HSQC-TOCSY. Indeed, 2D 1H-13C HSQC-TOCSY spectra have been used to analyse complex oligosaccharides (Rodriguez-Carvajal et al. 2003; Sato et al. 2008; Panagos et al. 2012), polysaccharides (Uhrin et al. 1994), glycoproteins (Debeer et al. 1994), peptides (Sonti et al. 2012), fatty acids (Willker and Leibfritz 1998), metabolites (Bingol and Bruschweiler 2011; Bingol et al. 2014, 2016), or food material (Kew et al. 2017; Wei et al. 2011). However, as the sample complexity increases, 13C chemical shifts stop being unique at some point, making the interpretation of 2D HSQC-TOCSY spectra problematic. This is often the case for biomolecules with repeating structural units, especially if these are “randomly” modified. For example, methylation, sulfation or acetylation of polysaccharides at irregular position induces chemical shift changes that can affect nuclei at considerable distances from the substitution site, sometimes beyond the monosaccharide unit carrying the modification. When such polysaccharides are partially depolymerised, their natural heterogeneity is further increased due to the size variation of produced oligomers, causing molecular size dependent variations of chemical shift. Taking together, small variations in 1H and/or 13C chemical shifts cause clusters of cross peaks to appear in heterocorrelated spectra of biomolecules and their mixtures. This is of particular concern for complex oligosaccharides, which are characterised by low 13C chemical shift dispersion (Fig. S1) caused by their monotonous structural makeup with the prevalence of CH–OH groups. This complicates analysis of carbohydrate mixtures considerably, and above all, hides subtle, but important structural variations.
Providing the overlap exists only in the 13C dimension, the resulting ambiguity can in principle be resolved by a 3D HSQC-TOCSY experiment (Krishnamurthy 1995). However, given the crowded nature of spectra of complex samples, increased dimensionality of NMR experiments will likely further reduce the achievable resolution and fail to deliver the desired outcome. Avoiding signal overlap may require much longer 3D experiments that are beyond practical limits. A more promising solution is to stay in two dimensions, while relying on the combined 13C and 1H chemical shifts to separate signal. This is one of the principles of the G-matrix and Fourier transformation (GFT) spectroscopy (Kim and Szyperski 2003). Here, more than one frequency is sampled simultaneously during the indirectly detected dimension of an (X,2)D experiment. Providing the 1H chemical shifts are unique, the overlap caused by 13C chemical shift degeneracy will be removed.
The proposed (3, 2)D HSQC-TOCSY experiment thus samples combined 1H and 13C chemical shifts of the directly bonded proton and carbon pairs, while the same F1 frequency labelling is preserved for the TOCSY cross peaks. This concept was recently explored in the context of a dual receiver NMR experiment, which combined simultaneous acquisition of several 2D spectra (Pudakalakatti et al. 2014), including (3, 2)D HSQC-TOCSY. The authors acquired two (3, 2)D HSQC-TOCSY spectra coding for Ω1H ± Ω13C offset frequencies in the indirectly-detected dimension and analysed them using a covariance spectroscopy.
In this work we present an implementation of a (3, 2)D 1H, 13C HSQC-TOCSY experiment that removes modulation of cross peaks in the F1 dimension due to proton–proton scalar couplings that is inherent to a regular (3, 2)D 1H,13C HSQC-TOCSY experiment. Such treatment is essential, as it does not introduce further signal overlap to already crowded spectra. A simple manipulation of the original spectra produces high-resolution spectra with F1 singlets at only one of the Ω13C ± κΩ1H offset frequencies.
The method is illustrated on an analysis of a mixture of oligosaccharides produced by β-eliminative depolymerisation (Gao et al. 2015) of fucosylated chondroitin sulfate (fCS) isolated from the body wall of the sea cucumber Holothuria forskali (Panagos et al. 2014). This mixture is referred to throughout the paper as “the fCS mixture”.Fucosyalted chondroitin sulfates appear in a range of marine organisms (Chen et al. 2011; Ustyuzhanina et al. 2017, 2016) and display a variety of biological roles and potential bio-medical applications (Panagos et al. 2014; Nagase et al. 1995; Mourao et al. 1996; Borsig et al. 2007). The studied fCS is composed of the following repeating trisaccharide unit, [→3)GalNAcβ4,6S(1→4) [FucαX(1→3)]GlcAβ(1→]n, where X stands for different sulfation patterns (S) of fucose. For this species X = 3,4S (46%), 2,4S (39%) and 4S (15%), where the numbers before S refer to individual carbons of fucose (Fig. 1). Only the two major sulfation patterns were present in sufficient concentration to yield detectable signals. As seen in the 1D 1H and 2D 1H, 13C HSQC spectra of the fCS mixture (Fig. 1S), heterogeneous sulfation and a mixture of different sizes results in high degree of signal overlap in both the 1H and 13C dimension. Such overlap complicates structural studies of fCS, an issue that can be addressed by the use of (3, 2)D 1H,13C HSQC-TOCSY experiment.

Fucosylated chondroitin sulfate (I) and derived oligosaccharides (II–IV)
Results and discussion
The pulse sequences
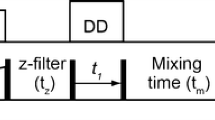
A Bruker implementations of the sensitivity-enhanced 2D 1H-13C HSQC (Kay et al. 1992; Schleucher et al. 1994) and 2D 1H-13C HSQC-TOCSY (Kay et al. 1992; Schleucher et al. 1994; Palmer et al. 1991) were used as a basis for the development of the new experiments referred to here as (3, 2)D BIRDr,X-HSQC and (3, 2)D BIRDr,X-HSQC-TOCSY. Their pulse sequences (Fig. 2) start with Ω1H labelling during the initial variable t1 period. Here, the evolution of proton–proton and proton-carbon couplings is suppressed by a central BIRDr,X pulse (Garbow et al. 1982; Uhrin et al. 1993). This pulse, in addition to 13C spins, also inverts protons attached to 12C, thus allowing Ω1H labelling of 13C-attached protons, while refocusing their evolution due to couplings with 12C-atached protons and 13C nuclei. The BIRDr,X pulse is surrounded by pulsed field gradients of opposite polarity, which helps with the suppression of 12C-attached protons. Depending on the phase of the first 90° 1H pulse of the pulse sequences, the 13C chemical shifts labelled during the regular t1 interval after the transfer of polarisation to carbons, are modulated either by cos(2πΩ1Hκt1) or sin(2πΩHκt1) frequencies. Ω1H is the difference between the 1H chemical shift and the 1H r.f. carrier frequency and κ is a scaling factor between the two simultaneously incremented indirectly detected periods. As a result, cross peaks in (3, 2)D BIRDr,X-HSQC-(TOCSY) spectra appear as in phase or antiphase doublets centred around 13C chemical shifts of the 1H,13C cross peaks and separated by 2κΩ1H in F1. This generates κΩ1H dependent displacement of signals. The cosine and sine modulated spectra are acquired in an interleaved manner and processed to produce two simplified spectra by the addition or subtraction of the original spectra. The simplified spectra thus contain only one part of the F1 doublets each as positive signals with increased intensity. They are referred to here as the Ω13C + κΩ1H and the Ω13C − κΩ1H spectrum. The overall theoretical sensitivity of the (3, 2)D BIRDr,X-HSQC-(TOCSY) experiment is ½ of that of a regular 2D HSQC(-TOCSY) experiment. In practise, additional signal-to-noise reduction is observed, which can be attributed to the 1JCH mismatch and relaxation effects during the BIRDr,X pulse; signal-to-noise of 30–45% relative to 2D HSQC spectra was measured for the corresponding cross peaks in the (3, 2)D BIRDr,X-HSQC spectra of the fCS mixture.

Pulse sequence of (3, 2)D BIRDr,X-HSQC (A + B) and (3, 2)D BIRDr,X-HSQC-TOCSY (A + C). For parameters see Materials and methods
(3, 2)D BIRDr,X HSQC
Acquisition of the (3, 2)D BIRDr,X HSQC spectra is not essential, but may help to identify the position of the direct proton-carbon correlation cross peaks in the (3, 2)D BIRDr,X-HSQC-TOCSY spectra and thus aid in their analysis. An overlay of a 2D 1H, 13C HSQC spectrum and the Ω13C ± κΩ1H (3, 2)D BIRDr,X-HSQC spectra of the fCS mixture focusing on the anomeric region is shown in Fig. 3. A complete spectrum is shown in Fig. S2. These spectra illustrate the essential attributes of the presented methodology. Cross peaks in the indirectly detected dimension of the (3, 2)D BIRDr,X-HSQC are displaced by ± κΩ1H frequencies relative to the cross peaks in the regular HSQC spectrum. In this instance, the 1H carrier frequency was set on the HOD signal, resulting in the (3, 2)D BIRDr,X-HSQC cross peaks to the left (positive ΩH) and the right (negative ΩH) of the water signal to swap the direction of the displacement in the final spectra. Expansions enclosed in rectangles illustrate how the cross peaks overlapping in the HSQC spectrum become resolved in the (3, 2)D BIRDr,X-HSQC spectra. Depending on the local topology of cross peaks, increased resolution is achieved in the Ω13C + κΩ1H or in the Ω13C − κΩ1H spectrum or in both, as discussed latter.

The anomeric region of 2D 1H, 13C HSQC (violet) and two (3, 2)D BIRDr,X-HSQC spectra (red and blue, pulse sequence of Fig. 2) of the fCS mixture; κ = 1 was used. 1D 1H spectrum and the projection of the 2D 1H, 13C HSQC spectrum are shown at the top and the side, respectively. For further discussion see the text
(3, 2)D BIRDr,X-HSQC-TOCSY
The cross peak displacement seen in the (3, 2)D BIRDr,X-HSQC spectra is propagated to the TOCSY peaks in the (3, 2)D BIRDr,X-HSQC-TOCSY spectra. An example of (3, 2)D BIRDr,X-HSQC-TOCSY spectra of fCS mixture is shown in Fig. 4, where an overlay of four partial heterocorrelated spectra is presented. Complete spectra are shown in Figs. S3 and S4. The spectra in Fig. 4 are dominated by the cross peaks in the 4.0–4.4 ppm 1H region that belong to the direct and TOCSY correlations of H5/H6 protons of GlcNAc. These are to be ignored for the purpose of this discussion, as we inspect four groups of HSQC cross peaks (shown in pink) resonating at ~ 66.5 ppm in F1 and around 4.90, 4.36, 4.16 and 3.97 ppm in F2. Their vertical displacement in the (3, 2)D BIRDr,X-HSQC-TOCSY spectra is indicated by black arrows, while their corresponding TOCSY cross peaks, connected by horizontal arrows, are circled. In this example, the TOCSY cross peaks are stronger than the direct correlation peaks, as the magnetisation was efficiently transferred from the 13C- to 12C-attached protons during a 40 ms TOCSY mixing time. The HSQC cross peaks at the δ1H of 4.90 and 4.36 ppm do not show any TOCSY correlations in the displayed region of the spectra and will be discussed later. On the other hand, signals resonating at 4.16 and 3.97 ppm show several HSQC-TOCSY correlations (violet circles), e.g. cross peaks overlapping in the proton dimension of a regular HSQC-TOCSY spectrum between 4.44 and 4.66 ppm. This overlap is removed in the (3, 2)D BIRDr,X-HSQC-TOCSY spectra (cross peaks circled by full and dashed red and blue lines). Analysis of the spectra showed that the HSQC cross peaks belong to protons H3 and H2 of fucose sulfated at positions 2,4 (2,4S, 4.16 ppm) and 3,4 (3,4S, 3.97 ppm), respectively. These protons show TOCSY transfers to protons H2 (4.56–4.44 ppm in 2,4S fucose, dashed circle) and H3 (4.66–4.49 ppm in 3,4S fucose, full line circle), respectively. Their separation in the (3, 2)D BIRDr,X-HSQC-TOCSY spectra is a consequence of unique 1H chemical shifts of H3 (4.16 ppm) and H2 (3.97 ppm) protons.

Overlay of four partial heterocorrelated spectra of the fCS mixture. Regular 2D 1H, 13C HSQC (pink) and 2D 1H, 13C HSQC-TOCSY (violet) spectra and two (3, 2)D BIRDr,X-HSQC-TOCSY spectra (read and blue, pulse sequence of Fig. 2). The vertical and horizontal black arrows show the displacement and the positions of the direct and the TOCSY (circled) cross peaks, respectively. For further commentary see the text
Transfer of magnetisation from the H3 and H2 protons is seen to continue in opposite directions to protons H4 and H1 of their individual monosaccharide rings, an observation that allowed their assignment. These (H4, H1)/C2 and (H4, H1)/C3 cross peaks are also circled in Fig. 4 and appear in pairs between 4.7 and 5.7 ppm. The observed doubling of signals with distinct 1H chemical shifts within each circle is a consequence of depolymerisation. Cleavage of the GalNAc(1→4)GlcA glycosidic bond leads to an appearance of fucose that is now linked to a terminal modified GlcA carrying a C4–C5 double bond (ΔGlcA, compounds II and IV in Fig. 1). At the same time, fucose linked to inner GlcA units (compounds I–III in Fig. 1) is still present to some extent, leading to doubling of signals. A comparison with the chemical shifts of fucose in the fCS polysaccharide showed that the H1 and H4 chemical shifts of the terminal fucose linked to ΔGlcA are always up to 0.2 ppm smaller. These fucose cross peaks are labelled with a Δ symbol in Fig. 4.
(3, 2)D BIRDr,X-HSQC-TOCSY and the conformation of fCS
The other two groups of the above mentioned cross peaks at ~ 66.5 ppm in F1 and 4.90 and 4.36 ppm in F2, were assigned to H5/C5 of fucose based on the H5 to H6 TOCSY transfers that produced H6/C5 cross peaks in the otherwise empty region of the HSQC-TOCSY spectra (1.30–1.45/~ 66.5 ppm). Figure 5 illustrates how the ambiguity of a regular HSQC-TOCSY experiment (Fig. 5b, violet cross peaks), where it is not clear if the transfer originates from one or both of these protons, is removed in the (3, 2)D BIRDr,X-HSQC-TOCSY spectra.

Overlay of 2D 1H, 13C HSQC-TOCSY spectrum (violet) and two (3, 2)D BIRDr,X-HSQC-TOCSY spectra (red and blue) focusing on a H5/C5 and b H6/C5 cross peaks. The black arrows indicate displacement along the vertical axis and horizontal lines connect the H5 and H6 cross peaks. Blue cross peaks labelled by an asterisk in b indicate additional CH3 signals. For further discussion see the text
The (3, 2)D BIRDr,X-HSQC-TOCSY spectra also enabled the assignment of H6 protons of both fucose types. As the J(H4,H5) of fucose is close to zero, no transfer of magnetisation from H5 to H4 protons took place, making the H6/C5 correlations strong. As seen in the spectra in Fig. 5, protons H6 appear at around 1.41 and 1.33 ppm, for the native and terminal fucose, respectively, following the chemical shift trends seen for the other fucose protons in these different environments. Correlations in the opposite direction, from H6/C6 to H5, are also resolved in a (3, 2)D BIRDr,X-HSQC-TOCSY spectrum presented in Fig. S4. Here, the extent of the heterogeneity of the fCS mixture is clearly visible, especially upon inspection of the Ω13C − κΩ1H spectrum.
In the native polysaccharide, the fucose ring is stacked above the neighbouring GlcNAc residue enabling formation of an unusual hydrogen bond from its H5 proton to the ring oxygen of GalNAc, as described previously (Panagos et al. 2014; Aeschbacher et al. 2017). β-elimination breaks the GalNAc(1→4)GlcA glycosidic bond, transforming fucose into a terminal unit of the newly formed oligosaccharides II and IV. This results in significant lowering of the H5 chemical shift of fucose (− 0.51 ppm) and is indicative of successful depolymerisation. As seen above, the H1, H4 and H6 protons of fucose are also affected in a similar manner, although to a lesser extent. Analysis of the (3, 2)D BIRDr,X-HSQC-TOCSY spectra thus allowed to identify chemical shifts changes of fucose protons caused by different conformation of this residue in fCS oligosaccharides.
Which (3, 2)D BIRDr,X-HSQC-TOCSY spectrum to inspect?
As already hinted, it is useful to analyse both Ω13C ± κΩ1H (3, 2)D BIRDr,X-HSQC-TOCSY spectra. This point is elaborated on next in more detail, initially using a cartoon representation (Fig. 6a–c) of possible cross peak displacements in (3, 2)D BIRDr,X-HSQC spectra. It can be seen that when the proton and carbon chemical shift in the overlapped areas are correlated (i.e. both 1H and 13C chemical shifts increase), a better cross peak separation is achieved in the Ω13C − κΩ1H spectrum (Fig. 6b). The opposite is true for the anti-correlated signals (i.e. when 1H chemical shifts increase, but 13C chemical shifts decrease, Fig. 6c), where a better separation is achieved in the Ω13C + κΩ1H spectrum. If the 13C chemical shifts are identical, while the 1H chemical shifts vary, separation of signals is identical in both spectra (Fig. 6a). Nevertheless, as an accidental overlap with other cross peaks may occur in one of the spectra, it is worthwhile to inspect both. These points are illustrated on an overlay of several experimental spectra shown in Fig. 6d. Focusing on the overlapping 2,4S-H2 and 3,4S-H3 fucose HSQC cross peaks at ~ 4.52 ppm (pink circle) and the 2,4S-H3 HSQC-TOCSY cross peaks at ~ 4.16 ppm, it can be seen that that their separation is better in the Ω13C − κΩ1H (red circles) than in the Ω13C + κΩ1H (blue circle) spectrum. This is a consequence of the topology, indicated by a dotted arrow of 2,4S–H2 HSQC cross peaks, which follows the pattern shown in Fig. 6b.

a–c A cartoon of the overlay of four cross peaks in the 2D HSQC (pink), and two (3, 2)D BIRDr,X-HSQC spectra (red and blue) as a function of their 1H and 13C chemical shifts. d Overlay of four partial heterocorrelated spectra of the fCS mixture, 2D 1H, 13C HSQC (pink), 2D 1H, 13C HSQC-TOCSY (violet) and two (3, 2)D BIRDr,X-HSQC-TOCSY spectra (red and blue). The vertical and horizontal black arrows indicate displacement of the direct and TOCSY cross peaks, respectively. The dotted arrows indicate a different degree of cross peak separations in the HSQC and consequently also HSQC-TOCSY in the Ω13C − κΩ1H (red circles) and Ω13C + κΩ1H (blue circle) spectra
Finally, increased separation or elimination of an accidental overlap of cross peaks can be achieved by acquiring spectra with κ ≠ 1. For κ > 1, this can lead to increased relaxation and peak broadening. Depending on the nature of the overlap, the use of κ < 1 may therefore be preferred. Another variable that influences the separation of signals in F1 is the position of the 1H r.f. carrier. This can be explored without changing the duration of the indirectly detected periods.
Conclusions
The presented methodology extends the application of a powerful 2D 1H, 13C HSQC-TOCSY experiment to samples with signal overlap in the 13C dimension. A proposed solution, in the form of (3, 2)D BIRDr,X-HSQC-TOCSY experiment, preserves high digital resolution in the indirectly detected dimension while accepting some signal losses. The method was illustrated on the assignment of fucose resonances in a mixture of oligosaccharides containing FucX(1→3)GlcA and FucX(1→3)ΔGlcA disaccharide fragments. The achieved resolution also made possible a detailed assignment of individual resonances belonging to 2,4 or 3,4 sulfated fucose in different oligosaccharides (I or III and II or IV) and accounted for reducing rings effects resulted from depolymerisation (data not shown). Although illustrated here on the TOCSY transfer, the same principles can also be applied to a NOESY experiment. In conclusion, the (3, 2)D BIRDr,X-HSQC-TOCSY/NOESY are a powerful addition to the limited arsenal of NMR techniques specifically designed to enable structure elucidation of molecule contained in mixtures.
Materials and methods
Materials
Fucosylated chondroitin sulfate polysaccharide was isolated from the body wall of the sea cucumber, Holothuria forskalias, as described previously(Panagos et al. 2014). The sample was depolymerised using a modified protocol (see Supplementary Information) for β-eliminative depolymerisation of carbohydrates in anhydrous solutions (Gao et al. 2015). Structure contained in the studied mixture are shown in Fig. 1.
NMR spectroscopy
The following parameters are associated with pulse sequences shown in Fig. 2. Narrow and wide filled rectangles represent 90° and 180° pulses, respectively. Open rectangles with inclined arrows were 500 µs 13C CHIRP pulses, while filled rectangles with and inclined arrows were 2 ms composite 13C CHIRP pulses. The following delays were used: Δ1 = 0.5/1JCH – p14/2; τg = 1.2 ms (1 ms gradient and 200 µs gradient recovery delay), Δ2 = 0.25/1JCH − τg − p14/2; Δ3 = 0.25/1JCH + τg − p14/2 + k*t1(0); τ = τg + p180 *2* t1(0); Δ4 = 0.25/1JCH + p14/4; Δ5 = 0.25/1JCH − p14/2; Δ6 = τg − 0.78*p90 − DE, where p14 = 500 µs, p90 and p180 are 90° and 180° 1H pulses, κ is the scaling factor for Ω1H frequencies, t1(0) is the initial t1 increment, typically 3 µs and DE is a pre scan delay (10 µs). Unless specified otherwise the pulses were applied from the x axis; ϕ1 = x or y for cosine and sine modulated signals, respectively; ϕ2 = 2x, 2(− x); ϕ3 = x; ϕ4 = x, − x; ϕ5 = 2y, 2(− y); Ψ = x, 2(− x), x. Phases ϕ3, ϕ4 and Ψ were increased in a TPPI manner by 180° simultaneously with t1 incrementation; the real and imaginary points were acquired by changing the polarity of the G1 gradient and increasing by 180° the phase ϕ5. Gradient G3 was 600 µs long, while all other gradients were applied for 1 ms. The following relative strength was used: G1 = 80%, G2 = 20.1%, G3 = 11%, G4 = − 5% and G5 = 7%, where 100% represents 53 Gauss/cm.
The sample (20 mg) was dissolved in D2O (550 µL) and measured at 300 K. The (3, 2)D 1H, 13C HSQC and (3, 2)D 1H, 13C HSQC-TOCSY spectra of the fCS mixture were acquired on a 4-channel Avance III 800 MHz Bruker spectrometer equipped with a 5 mm TCI CryoProbe™ with automated matching and tuning. The following parameters were used: 2048 and 2048 complex points in t2 and t1, respectively, spectral widths of 8 and 130 ppm in F2 and F1, yielding t2 and t1 acquisition times of 160 and 39.17 ms, respectively. Four scans were acquired for each t1 increment using a relaxation time of 1.4 s. The overall acquisition time was 7 h and 47 min (for two interleaved spectra, using 4096 t1 points). A 40 ms mixing time for the TOCSY transfer used DIPSI-2 pulse sequence (Rucker and Shaka 1989). A forward linear prediction to 4096 points was applied in F1. A zero filling to 4096 was applied in F2. A cosine square window function was used for apodization prior to Fourier transformation in both dimensions. Identical parameters were used to acquire regular 2D 1H, 13C HSQC and 2D 1H, 13C HSQC-TOCSY spectra (Bruker pulse programs hsqcedetgpsisp2.3 and hsqcdietgpsisp.2) in half of the time required for the GFT experiments. Spectra were processed using a Bruker AU program provided in the Electronic Supplementary Material.
References
Aeschbacher T et al (2017) A secondary structural element in a wide range of fucosylated glycoepitopes. Chem-A Eur J 23:11598–11610
Bingol K, Bruschweiler R (2011) Deconvolution of chemical mixtures with high complexity by NMR consensus trace clustering. Anal Chem 83:7412–7417
Bingol K, Bruschweiler-Li L, Li DW, Bruschweiler R (2014) Customized metabolomics database for the analysis of NMR H-1-H-1 TOCSY and C-13-H-1 HSQC-TOCSY spectra of complex mixtures. Anal Chem 86:5494–5501
Bingol K, Li DW, Zhang B, Bruschweiler R (2016) Comprehensive metabolite identification strategy using multiple two-dimensional NMR spectra of a complex mixture mplemented in the COLMARm web server. Anal Chem 88:12411–12418
Borsig L et al (2007) Selectin blocking activity of a fucosylated chondroitin sulfate glycosaminoglycan from sea cucumber: effect on tumor metastasis and neutrophil recruitment. J Biol Chem 282:14984–14991
Chen SG et al (2011) Comparison of structures and anticoagulant activities of fucosylated chondroitin sulfates from different sea cucumbers. Carbohyd Polym 83:688–696
Debeer T et al (1994) Rapid and simple approach for the NMR resonance assignment of the carbohydrate chains of an intact glycoprotein: application of gradient-enhanced natural abundance 1H-13C HSQC and HSQC-TOCSY to the α-subunit of human chorionic gonadotropin. Febs Lett 348:1–6
Gao N et al (2015) Beta-eliminative depolymerization of the fucosylated chondroitin sulfate and anticoagulant activities of resulting fragments. Carbohyd Polym 127:427–437
Garbow JR, Weitekamp DP, Pines A (1982) Bilinear rotation decoupling of homonuclear scalar interactions. Chem Phys Lett 93:504–509
Kay LE, Keifer P, Saarinen T (1992) Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J Am Chem Soc 114:10663–10665
Kew W, Bell NGA, Goodall I, Uhrin D (2017) Advanced solvent signal suppression for the acquisition of 1D and 2D NMR spectra of Scotch Whisky. Magn Reson Chem 55:785–796
Kim S, Szyperski T (2003) GFT NMR, a new approach to rapidly obtain precise high-dimensional NMR spectral information. J Am Chem Soc 125:1385–1393
Krishnamurthy VV (1995) Sensitivity-enhanced 3D HSQC-TOCSY experiments. J Magn Reson Ser B 106:170–177
Mourao PAS et al (1996) Structure and anticoagulant activity of a fucosylated chondroitin sulfate from echinoderm: sulfated fucose branches on the polysaccharide account for its high anticoagulant action. J Biol Chem 271:23973–23984
Nagase H et al (1995) Depolymerized holothurian glycosaminoglycan with novel anticoagulant actions: antithrombin III- and heparin cofactor II-independent inhibition of factor X activation by factor IXa-factor VIIIa complex and heparin cofactor II-dependent inhibition of thrombin. Blood 85:1527–1534
Palmer AG, Cavanagh J, Wright PE, Rance M (1991) Sensitivity improvement in proton-detected 2-dimensional heteronuclear correlation NMR spectroscopy. J Magn Reson 93:151–170
Panagos C, Thomson D, Bavington CD, Uhrin D (2012) Structural characterisation of oligosaccharides obtained by Fenton-type radical depolymerisation of dermatan sulfate. Carbohyd Polym 87:2086–2092
Panagos CG et al (2014) Fucosylated chondroitin sulfates from the body wall of the sea cucumber Holothuria forskali. Conformation, selectin binding, and biological activity. J Biol Chem 289:28284–28298
Pudakalakatti SM et al (2014) A fast NMR method for resonance assignments: application to metabolomics. J Biomol NMR 58:165–173
Rodriguez-Carvajal MA, du Penhoat CH, Mazeau K, Doco T, Perez S (2003) The three-dimensional structure of the mega-oligosaccharide rhamnogalacturonan II monomer: a combined molecular modeling and NMR investigation. Carbohyd Res 338:651–671
Rucker SP, Shaka AJ (1989) Broad-band homonuclear cross polarization in 2D NMR using DIPSI-2. Mol Phys 68:509–517
Sato H, Fukae K, Kaiihara Y (2008) 2D Selective-TOCSY-DQFCOSY and HSQC-TOCSY NMR experiments for assignment of a homogeneous asparagine-linked triantennary complex type undecasaccharide. Carbohyd Res 343:1333–1345
Schleucher J et al (1994) A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed-field gradients. J Biomol NMR 4:301–306
Sonti R, Rai R, Ragothama S, Balaram, P (2012) NMR Analysis of cross strand aromatic interactions in an 8 residue hairpin and a 14 residue three stranded beta sheet peptide. JPhys Chem B 116:14207–14215
Uhrin D, Liptaj T, Kover KE (1993) Modified BIRD pulses and design of heteronuclear pulse sequences. J Magn Reson A 101:41–46
Uhrin D, Brisson JR, Maclean LL, Richards JC, Perry MB (1994) Application of 1D and 2D NMR techniques to the structure elucidation of the O-polysaccharide from Proteus mirabilis O-57. J Biomol NMR 4:615–630
Ustyuzhanina NE et al (2016) Structure and biological activity of a fucosylated chondroitin sulfate from the sea cucumber Cucumaria japonica. Glycobiology 26:449–459
Ustyuzhanina NE et al (2017) A highly regular fucosylated chondroitin sulfate from the sea cucumber Massinium magnum: structure and effects on coagulation. Carbohyd Polym 167:20–26
Wei FF, Furihata K, Hu FY, Miyakawa T, Tanokura M (2011) Two-dimensional H-1-C-13 nuclear magnetic resonance (NMR)-based comprehensive analysis of roasted coffee bean extract. J Agric Food Chem 59:9065–9073
Willker W, Leibfritz D (1998) Assignment of mono- and polyunsaturated fatty acids in lipids of tissues and body fluids. Magn Reson Chem 36:S79–S84
Acknowledgements
The authors would like to thank Juraj Bella and Lorna Murray for their maintenance of the Edinburgh NMR facility, and Mr Will Kew for writing an AU program for processing of the spectra, GlycoMar Ltd for providing purified fCS and determining the Mw of the depolymerised products. Funding was provided by the EastBio (NB), IBioICExemplar grant 2015-1-9 (ZK) and GlycoMar Ltd (NB, ZK).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Brodaczewska, N., Košťálová, Z. & Uhrín, D. (3, 2)D 1H, 13C BIRDr,X-HSQC-TOCSY for NMR structure elucidation of mixtures: application to complex carbohydrates. J Biomol NMR 70, 115–122 (2018). https://doi.org/10.1007/s10858-018-0163-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-018-0163-8