
Abstract
A nuclear magnetic resonance-based ligand screening strategy utilizing a paramagnetic lanthanide probe is presented. By fixing a paramagnetic lanthanide ion to a target protein, a pseudo-contact shift (PCS) and a paramagnetic relaxation enhancement (PRE) can be observed for both the target protein and its bound ligand. Based on PRE and PCS information, the bound ligand is then screened from the compound library and the structure of the ligand–protein complex is determined. PRE is an isotropic paramagnetic effect observed within 30 Å from the lanthanide ion, and is utilized for the ligand screening in the present study. PCS is an anisotropic paramagnetic effect providing long-range (~40 Å) distance and angular information on the observed nuclei relative to the paramagnetic lanthanide ion, and utilized for the structure determination of the ligand–protein complex. Since a two-point anchored lanthanide-binding peptide tag is utilized for fixing the lanthanide ion to the target protein, this screening method can be generally applied to non-metal-binding proteins. The usefulness of this strategy was demonstrated in the case of the growth factor receptor-bound protein 2 (Grb2) Src homology 2 (SH2) domain and its low- and high-affinity ligands.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In fragment-based drug design (FBDD), small simple compounds (fragments) are screened for binding to a target protein, and the hit compounds are then optimized to increase their affinity. While the fragments found in the FBDD screening often show weak affinity (K d~30 μM), their affinity can be improved through structure-based optimization (Rees et al. 2004). If several fragments are identified as binding to different sites of the target protein, they can be linked (linking) (Shuker et al. 1996). If there is extra space around the binding pocket, it can be filled by modifying the initially identified fragment (growing) (Boehm et al. 2000). In contrast to the high-throughput screening (HTS), where the identified hits are often hydrophobic and possess relatively higher molecular weights, FBDD carries a higher potential to produce drug candidates with more efficient binding properties, lower molecular mass, and higher solubility (Klages et al. 2006).
For efficient FBDD, it is important to obtain structural information on the ligand-target protein complex, even for weakly bound ligands. Two of the most powerful techniques for obtaining structural information on the ligand–protein complex are nuclear magnetic resonance (NMR) and X-ray crystallography. X-ray crystallography has the ability to determine the precise three-dimensional structure of a complex. However, the preparation of crystals of sufficient quality for structure-based ligand optimization remains difficult. Moreover, X-ray crystallography is not free from crystallization artifacts. In contrast, NMR can rapidly characterize molecular interactions, at atomic resolutions and is applicable even for weakly bound molecules. It also has a lower risk of false positives.
Several NMR-based screening techniques have been reported which are classified into two types: methods that observe the NMR signals of small ligands (the ligand-based approach) and those that focus on the protein NMR signals (the protein-based approach). In a protein-based approach, the protein NMR spectra such as 1H–15N HSQC spectra are measured by adding candidate ligands, thus identifying the protein surface area that interacts with the ligands (Shuker et al. 1996). However, this method requires a lot of protein samples and much NMR measurement time, both of which are an obstacle for high-throughput screening. Several techniques have been reported for the ligand-based approach: saturation transfer difference (STD) NMR (Mayer and Meyer 1999; Klein et al. 1999), water-ligand observed via gradient spectroscopy (WaterLOGSY) (Dalvit et al. 2000, 2001, 2002; Hu et al. 2010), spin labels attached to protein side chains as a tool to identify interacting compounds (SLAPSTIC) (Jahnke et al. 2000, 2001), inter-ligand NOE for pharmacophore mapping (INPHARMA) (Sánchez-Pedregal et al. 2005; Orts et al. 2009), structural information using Overhauser effects and selective labeling (SOS)-NMR (Hajduk et al. 2004), and structure–activity relationship (SAR) by interligand nuclear Overhauser effects (ILOEs) (Becattini and Pellecchia 2006). Since most of these techniques are based on the 1H 1D spectra of the ligands, they require less measurement time and are free from signal assignment for the target protein. However, despite their high-throughput capabilities, these methods can’t provide structural information on the ligand–protein complex. The exceptions are SOS-NMR (Hajduk et al. 2004) and INPHARMA (Sánchez-Pedregal et al. 2005; Orts et al. 2009) that give structural information on the ligand–protein complex. SOS-NMR utilizes STD NMR experiments performed on a ligand complexed to a series of protein samples that are deuterated except for specific amino acid types. Analysis of the signal decaying properties for each sample enables the structure of the ligand–protein complex to be modeled. The INPHARMA method exploits protein-mediated inter-ligand nuclear Overhauser effects (NOEs) observed between two competitive ligands in the presence of a target protein. If the structure of the ligand–protein complex is known for one ligand, the complex structure for the other ligand can be estimated based on the intermolecular NOEs. However, these two methods are not widely used. SOS-NMR requires several protein samples with different deuterium labeling patterns, which is both resource intensive and time consuming. In order to apply INPHARMA, the two ligands should bind weakly to the same binding pocket, with similar exchange rates, and the spin diffusion among protein protons should be considered. Though the structure of the ligand–protein complex can be determined by a standard NOE-based NMR method, it is time-consuming because almost all proton resonances and NOE signals have to be assigned.
Here we demonstrate an NMR-based ligand screening strategy, which can identify the ligand binding to a target protein and also determine the complex structure, based on the proton resonances of the ligand. In this method, a paramagnetic lanthanide probe is utilized. Paramagnetic lanthanide ions fixed in a protein frame induce several paramagnetic effects in the NMR spectra of the proteins, such as a pseudo-contact shift (PCS) and a paramagnetic relaxation enhancement (PRE) (Otting 2008). PCS is a chemical shift change induced by the paramagnetism of the lanthanide ion and provides long-range (~40 Å) distance and angular information on the observed nuclei. The NMR signals derived from the nuclei close to the lanthanide ion are broadened because of paramagnetic relaxation enhancement (PRE), which provides long-range (~30 Å) distance information of the nuclei. In contrast to the conventional approaches, most of which utilize NOE-based short-range distance information, the paramagnetic lanthanide probe provides long-range quantitative information and thus is useful for the rapid structural determination of protein–protein or protein–ligand complex structures (Saio et al. 2010; Pintacuda et al. 2007). Lanthanide probes can be applied to any desired protein by the use of a two-point anchored lanthanide-binding peptide tag (LBT) (Saio et al. 2009). Once the lanthanide ion is fixed on the target protein, the paramagnetic effects such as PCS and PRE can be observed both for the target protein and the bound ligand. These paramagnetic effects can be rapidly translated into the structural information on the ligand–protein complex. This strategy was demonstrated on the growth factor receptor-bound protein 2 (Grb2) Src homology 2 (SH2) domain and its two ligands: 4-[(10S,14S,18S)-18-(2-amino-2-oxoethyl)-14-(1-naphthylmethyl)-8,17,20-trioxo-7,16,19-triazaspiro[5.14]icos-11-en-10-yl]benzylphosphonic acid (macrocyclic high-affinity inhibitor) (Gao et al. 2001) as a high affinity inhibitor and pYTN tripeptide as a low-affinity ligand.
Materials and methods
Construct design
In order to fix a lanthanide ion on the Grb2 SH2 domain (60–159), a two-point anchored lanthanide-binding tag (LBT) was utilized (Saio et al. 2009, 2010). The LBT sequence (CYVDTNNDGAYEGDEL) (Nitz et al. 2003, 2004; Su et al. 2006, 2008) was fused to the N-terminus of Grb2 SH2 domain and an M73C mutation was introduced: the product is hereafter referred to as LBT-Grb2 (supporting information Figure S-1A). The position of the Cys mutation was designed based on the structure of the LBT (1tjb.pdb, Nitz et al. 2004) and the Grb2 SH2 domain (1x0n.pdb, Ogura et al. 2008). LBT-Grb2 was subcloned, together with a Glutathione S-transferase (GST) tag and a tobacco etch virus (TEV) protease cleavage site, into a pGSTV vector (Saio et al. 2010) derived from the pET-21 plasmid (Novagen, USA).
Preparation of protein samples
LBT-Grb2 was expressed in E. coli strain BL21 (DE3) cells. For the unlabeled sample, cells were grown in Luria–Bertani media. For the uniformly 15N-labeled sample, cells were grown in M9 media containing 15NH4Cl (1 g/L), Celtone-N powder (0.2 g/L) (Cambridge Isotope Laboratories, USA) and unlabeled glucose (10 g/L). The uniformly 2H/15N-labeled sample was prepared by culturing cells in 100% 2H2O M9 medium containing 15NH4Cl (1 g/L), [U–2H] glucose (2 g/L), and Celtone-dN powder (0.2 g/L). Cells were grown at 37°C to A600 of 0.8, and protein expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside to a final concentration of 0.5 mM for 16 h at 25°C.
LBT-Grb2 was purified using glutathione-Sepharose 4B resin (GE Healthcare, UK). The GST tag was removed by incubation for 4 h at room temperature with TEV protease. The isolated protein was then further purified by gel filtration chromatography on a Superdex 75 column (GE Healthcare).
After gel filtration, LBT-Grb2 was incubated with 1 mM 5,5′-ditiobis(2-nitrobenzoic acid) (DTNB) for 2 h at room temperature, which linked the N-terminal Cys of LBT and the Cys73 on Grb2 via an intramolecular disulfide bond (Saio et al. 2009, 2010). After incubation, the DTNB was removed by dialysis.
Solid-phase synthesis of the pYTN peptide
All commercially available solvents and reagents were used without further purification. Tentagel S RAM (Bayer and Rapp 1986) was purchased from Hipep Laboratories. Fmoc amino acid derivatives were purchased from NOVA Biochem. Fmoc-protected amino acids (Asn, Thr and Tyr were employed as Asn(Trt), Thr(OtBut), Tyr(PO(OBzl)OH). All solid-phase reactions were performed manually in a polypropylene tube equipped with a filter (LibraTube; Hipep Laboratories).
Tentagel S RAM resin (0.035 mmol) functionalized with a Rinkamide linker (0.25 mmol/g) was stirred with 20% piperidine/DMF in a polypropylene tube for 10 min using a vortex mixer to remove the Fmoc group. Then the resin was washed with dichloromethane and DMF repeatedly and each amino acid (0.105 mmol) was coupled with the resin in the presence of HBTU (0.105 mmol), HOBt (0.105 mmol), and DIEA (0.21 mmol) in DMF for 1 h at room temperature. After washing the resin with DMF, removal of N Fmoc protection and the coupling reaction were repeated until the N-terminal amino acid residue was coupled. Upon completion of the synthesis, the peptide resin was treated with a mixture of (TFA/H2O/TIS) 95:2.5:2.5 for 2 h at room temperature. The solution was filtered and concentrated by a flow of nitrogen gas, and the crude peptide was precipitated using cold tert-butylmethylether. The crude material was purified by preparative RP-HPLC.
The resulting crude precipitate was purified using a preparative C-18 reversed phase column (Inertsil ODS-3 20 mm × 250 mm) on a SHIMADZU liquid chromatography system (HPLC) with an LC-6AD pump, at a flow rate of 5 mL/min. The column temperature was 25°C, and UV monitoring was carried out at 220 nm. Solvent A was distilled water containing 0.1% TFA, and solvent B was acetonitrile containing 0.1% TFA. A linear gradient of 5–15% of B over 45 min was used unless otherwise stated.
NMR spectroscopy
For the LBT-Grb2 NMR experiments, the samples were prepared in 20 mM tris buffer (pH 7.2) with 100 mM NaCl. For the 1H 1D NMR experiments observing the ligand 1H signals, the samples were prepared in a deuterium buffer consisting of 20 mM [U-2H] tris buffer (pD 7.2) with 100 mM NaCl in 100% 2H2O. All NMR experiments were run on UNITY inova 800 or 500 MHz NMR spectrometers (Varian, USA) at 25°C. Spectra were processed using the NMRPipe program (Delaglio et al. 1995) and data analysis was performed with the help of the Olivia program developed in our laboratory (Yokochi et al. http://fermi.pharm.hokudai.ac.jp/olivia/). For the measurement of PCS and PRE, the NMR spectra were acquired in complex with 1 equivalent of lanthanide ions. Aliquots of 5 mM LnCl3 stock solution were added to the NMR sample.
Δχ-tensor calculation
Δχ-tensors and the position of the lanthanide ion for LBT-Grb2 were calculated from the PCS values and the structure of Grb2 SH2, using the Numbat program (Schmitz et al. 2008), from (1),
where Δδ PCS is the pseudo-contact shift, r, ∂ and φ are polar coordinates of the nucleus with respect to the principal axes of the magnetic susceptibility tensor, and Δχ ax and Δχ rh are the axial and rhombic components, respectively, of the anisotropic magnetic susceptibility tensor. Conformer 1 of the family of NMR structures of Grb2 SH2 (1x0n.pdb, Ogura et al. 2008) was used for the tensor fit.
Estimation of the binding affinity of the pYTN tripeptide and Grb2 SH2
The binding affinity of the pYTN tripeptide and Grb2 SH2 was estimated using a 1H–15N HSQC-based titration experiment, where the non-labeled tripeptide was titrated into 0.19 mM 15N-labeled Grb2 SH2. Titration curves were obtained by plotting absolute value of chemical shift perturbations (Δδppm) against the concentration of the tripeptide. Non-linear least square fitting calculations with a 1:1 binding model were performed in GraphPad Prism (GraphPad Software, USA), using (2),
where [L] and [P] are the concentrations of the peptide ligand and protein, respectively, and K d is the dissociation constant.
PCSbound calculation
In the case of low-affinity ligands indicating fast-exchange on the NMR timescale, the chemical shifts are average of the bound and free states when the ligands and the protein are mixed. Thus the chemical shifts of the ligand signals are gradually changed by the titration of the protein. The chemical shift differences between the free and bound states (\( \Updelta \delta_{\text{ppm}}^{\text{bound}} \)) of the low-affinity ligands were calculated from the observed chemical shift change (Δδppm), using (3),
where [L] and [P] are the concentrations of the ligand and protein, respectively, and K d is the dissociation constant. PCS of the bound state, PCSbound, was calculated by (4),
where \( \Updelta \delta_{\text{ppm}}^{\text{bound}} \)(Ln) and \( \Updelta \delta_{\text{ppm}}^{\text{bound}} \)(apo) are the chemical shift difference upon the binding to the protein with and without a paramagnetic lanthanide ion, respectively.
Ligand–protein docking calculation
The high-affinity inhibitor and Grb2 SH2 were docked based on the PCS as described previously (Saio et al. 2010). PCS-based rigid body docking was carried out using the Xplor-NIH program (Schwieters et al. 2003, 2006) equipped with PARA restraints for Xplor-NIH (Banci et al. 2004). The coordinates of Grb2 SH2 and the metal were fixed, whereas the high-affinity inhibitor was treated as a rigid body and allowed to rotate and translate. First, the coordinates of Grb2 SH2 and the high-affinity inhibitor were both extracted from their complex structure as determined by NOE-based NMR (1x0n.pdb, Ogura et al. 2008), and their relative orientation and position were randomized to generate 100 starting structures. The starting positions of the high-affinity inhibitor were located within 100 Å of Grb2 SH2. The coordinates of Grb2 SH2 and the metal, on the other hand, were fixed. Next, the rigid body docking calculation was performed based on the PCS and binding restraints. For the PCS restraints, pseudo atoms representing the tensor axes were introduced. The atom representing the origin of the axis was restrained within 0.02 Å of the metal, while the coordinates of the tensor were allowed to rotate around the origin. The position of the metal and values of Δχ ax and Δχ rh were fixed to those determined in the Δχ-tensor calculation for Grb2 SH2 using Numbat. Though the directions of the principal axes were also determined in the calculation by Numbat, the directions were recalculated during the docking calculation based on the PCSs since it was difficult to fix the tensor directions in the Xplor-NIH calculation. Note that the directions of the principal axes determined in the docking calculation using Xplor-NIH were almost identical to those determined by Numbat. In PCS-based structure calculations, the symmetry of the PCS isosurface causes several degenerated solutions that include false structures lacking any intermolecular interaction (Saio et al. 2010). To avoid these artifacts, a binding restraint was added to the calculation. The binding restraint was set up as a loose distance restraint between the two molecules, the high-affinity inhibitor and Grb2 SH2, using the r −6 averaging option, which means the two molecules locate close to each other (Saio et al. 2010). The Xplor-NIH script for the docking calculation is provided in Script 1 (supporting information).
The docking procedure for the pYTN tripeptide and Grb2 SH2 was almost the same as that for the high-affinity inhibitor, except that each of the three residues in the peptide was treated as a separate rigid body. The coordinates of pYTN peptide were first extracted from the crystal structure of Grb2 SH2 complexed with the PSpYVNVQN peptide (Nioche et al. 2002, 1jyr.pdb), with modification of Val to Thr using PyMOL software (DeLano 2002). Second, in order to generate starting coordinates, the relative orientations between the three residues were randomized in two-steps as follows. (i) Each of the three residues was treated as a rigid body, and was randomly translated and rotated against Grb2 SH2, generating the coordinates where the three residues randomly and separately located. (ii) The carbonyl carbon of pTyr and the amide nitrogen of Thr, and the carbonyl carbon of Thr and the amide nitrogen of Asn were tethered to each other, thus generating the coordinates of the three-residue peptide linked by C′ and N. These processes were repeated 100 times to generate 100 sets of the starting coordinates. Finally, the pYTN peptide and Grb2 SH2 were docked based on PCS restrains and binding restraints. During the docking calculation, each of the three residues of the peptide was treated as a rigid body, while the relative orientations between them were variable. The Xplor-NIH script for the docking calculation is provided in Script 2 (supporting information).
Results and discussion
Screening strategy utilizing a paramagnetic lanthanide probe
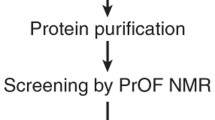
The ligand screening strategy utilizing the paramagnetic lanthanide probe is described as follows (Fig. 1). (A) Screening step: Ligand binding to the target protein can be identified from compound mixture based on Gd3+-induced PRE. Gd3+ induces strong PRE due to long electron-spin relaxation time but does not induce PCS due to the isotropic paramagnetic susceptibility tensor. A T1ρ relaxation experiment is carried out on a mixture of ligand candidates in the presence of the target protein containing Gd3+. A two-point anchored LBT enables Gd3+ to be introduced into any target proteins (Saio et al. 2009). The 1H 1D NMR spectra with short and long spinlock periods are measured. If any ligands bind to the target protein, the NMR signals of the ligand are affected by PRE, which is reflected by the reduction in the signal intensity at the long spinlock period. Thus, efficient ligand screening can be achieved by T1ρ measurements. (B) Structural analysis: Ligands hit in the screening step are further analyzed, where the structure of the ligand–protein complex can be rapidly determined based on PCSs. PCS restraints can be readily collected by replacing the Gd3+ ion with the paramagnetic lanthanide ions that have anisotropic magnetic susceptibility tensors such as Tb3+, Tm3+, and Dy3+. Once anisotropic magnetic susceptibility tensor (Δχ-tensor) parameters are determined for each lanthanide ion based on the PCSs observed for the protein, ligand PCSs can be translated into quantitative structural information on the complex. Since the Δχ-tensor parameter determination requires at least 8 PCSs, a sufficient number of PCSs can be collected based on the limited number of NMR signals, such as backbone amide signals from 1H–15N HSQC spectra.

Schematic representation of the NMR ligand screening method utilizing a paramagnetic lanthanide probe
In this study, the above strategy was applied to Grb2 SH2 and its two ligands: 4-[(10S,14S,18S)-18-(2-amino-2-oxoethyl)-14-(1-naphthylmethyl)-8,17,20-trioxo-7,16,19-triazaspiro[5.14]icos-11-en-10-yl]benzylphosphonic acid (macrocyclic high-affinity inhibitor) (Gao et al. 2001) and pYTN tripeptide (low-affinity ligand).
A lanthanide ion was fixed on a target protein using a two-point anchored LBT
For the application of the paramagnetic lanthanide probe, the lanthanide ion has to be rigidly fixed in a protein frame. Although some metal-binding proteins can bind lanthanide ions, many proteins lack metal-binding sites and require a lanthanide-binding tag for the application of the lanthanide probe method. In our previous work, we proposed a two-point anchored LBT that is attached to the target protein via N-terminal fusion and disulfide bond, thus holding the lanthanide ion in a rigid position (Saio et al. 2009). In this study we utilized this tagging method, by which a lanthanide ion was attached to Grb2 SH2. The LBT sequence (CYVDTNNDGAYEGDEL) was fused to the N-terminus of Grb2 SH2, and an M73C mutation was introduced on the surface of Grb2 SH2. The position of the Cys mutation was designed based on the structures of LBT (1tjb.pdb, Nitz et al. 2004) and Grb2 SH2 (1x0n.pdb, Ogura et al. 2008), according to the following considerations. In the LBT crystal structure, the distance between the Cα atoms of the N- and C-terminal residues is around 7 Å. We searched for a residue about 7 Å in distance from N-terminal residue (Trp60) of Grb2 SH2, and found Met73 (supporting information Figure S-1A). The Cα distance between Trp60 and Met73 was 9.9 Å. A linker sequence was introduced to the fusion point between LBT and Grb2 SH2, in order to avoid structural distortion and steric hindrance. The length of the linker was optimized. 15N-labeled LBT-Grb2 containing three- (HMA), four- (HMAG), or five-residue (HMAGS) linkers were prepared and the 1H–15N HSQC spectra were acquired in the presence of 1 equivalent of terbium ion. As shown in Figure S-1B (supporting information), the construct with the three-residue linker showed broad signals, whereas the constructs with four- and five-residue linkers showed sharp well-dispersed single peaks. Thus we concluded that the three-residue linker is too short, and that the four- and five-residue linkers match the distance requirements. The construct with the four-residue linker, hereafter referred to as LBT-Grb2, was used for the following experiments.
PCS observation and tensor determination for LBT-Grb2
Pseudo-contact shift values were measured as the difference in the backbone amide proton chemical sifts; i.e., the chemical shifts observed in complex with paramagnetic lanthanide ions minus those observed in complex with the diamagnetic Lu3+ ion. The backbone amide signals of LBT-Grb2 containing Lu3+ were assigned based on the assignments reported previously (Wang et al. 1996; Thornton et al. 1996; Ogura et al. 2008). 1H–15N HSQC spectra of the 15N-labeled LBT-Grb2 were recorded in the presence of 1 equivalent of lanthanide ions: Lu3+, Tb3+, Dy3+, Er3+ and Tm3+ (Fig. 2). Since the 1H and 15N of each amide group are spatially close, the PCS has similar ppm values in both 1H and 15N dimensions (Saio et al. 2009, 2010). Thus, by overlaying the spectra recorded with different lanthanide ions, the signals were aligned along a straight line. Based on this behavior, the 1H–15N HSQC cross-peaks of the paramagnetic samples were readily assigned using the assignment of the diamagnetic samples. Finally a total of 224 PCSs were observed for amide proton resonances of LBT-Grb2 (55, 56, 57, and 56 PCSs for Dy3+, Tb3+, Er3+ and Tm3+, respectively) (Supporting information Table S-1).

1H–15N HSQC spectra of 15N LBT-Grb2 in complex with Lu3+ (gray), Dy3+ (red), Tb3+ (orange), Er3+ (green), and Tm3+ (blue). Spectra were acquired using an 800 MHz NMR spectrometer at 25°C
Based on the PCSs observed for the backbone amide signals of LBT-Grb2, Δχ-tensor was determined for each lanthanide ion using the Numbat program (Schmitz et al. 2008). In the tensor calculation, tensor parameters for Dy3+, Tb3+, Er3+ and Tm3+ were simultaneously fitted with each lanthanide having a shared metal position because of the isomorphous nature of the lanthanide ions (Saio et al. 2010). The Δχ-tensor parameters were well defined with the principal axes for the four lanthanides oriented in similar directions (Table 1; Fig. 3a, supporting information Figure S-2). The correlations between the experimental and back-calculated values were good (Fig. 3b–e). Thus we concluded that the lanthanide ion was fixed in the protein frame and the Δχ-tensor parameters including metal position were well determined.

Δχ-tensor determination for LBT-Grb2. a Orientation of the principal axes of the Δχ-tensors of Dy3+, Tb3+, Er3+, and Tm3+ in complex with LBT-Grb2, visualized in Sanson-Flamsteed projection. The plots show the points where the principal axes of the Δχ-tensor penetrate the sphere. The convention |z| > |y| > |x| was used to name the axes, which occasionally caused swapping between the z- and y-axes of the tensors when their magnitudes were similar. One hundred sets of plots represent the results of the Monte-Carlo analysis using the 100 partial PCS data sets in which 30% of the input data were randomly deleted. b–e Comparison between experimental and back-calculated PCSs of backbone amide protons observed in LBT-Grb2 in the presence of Dy3+ (b), Tb3+ (c), Er3+ (d), and Tm3+ (e). The ideal correlations are indicated
Ligands bound to the target protein can be identified based on PREs
Once the LBT is introduced to the target protein, several paramagnetic effects can be exploited simply by changing the lanthanide ion. First we examined PRE-based ligand screening using Gd3+, which induces strong PRE but without any PCS. Jahnke et al. (2000, 2001) reported a PRE-based screening method utilizing a 2,2,6,6-tetramethyl-piperidine-1-oxyl (TEMPO) spin-label as a paramagnetic center (SLAPSTIC). For the lanthanide probe-based screening strategy, we utilized Gd3+ as a fixed paramagnetic center and carried out a T1ρ relaxation experiment. A 0.2 equivalent of LBT-Grb2 containing Gd3+ was added to a solution of the pYTN tripeptide (840 μM) mixed with the other six compounds (840 μM for each) as a model study and 1H spinlock 1D NMR spectra were acquired with spinlock period of 10 or 200 ms (Fig. 4). The signals of the pYTN tripeptide were specifically attenuated in the spectra with long spinlock period. The NMR signals of the ligand binding to the target protein were affected by the Gd3+ PRE, which was reflected by the signal decay at the long spinlock period. In contrast, the signals derived from the compounds other than pYTN showed no significant signal decay. Thus the ligand binding to the target protein can be identified in the compound mixture based on the PRE. This screening experiment requires only two 1H spinlock 1D NMR spectra measured on a single NMR sample, which is advantageous to rapid screening. Although the signals of the pYTN tripeptide were also attenuated in the experiment using LBT-Grb2 lacking Gd3+ (Fig. 4b, d), the relaxation effects were much smaller. This means that use of Gd3+ allows a reduction in the amount of protein used for the experiments, and also allows more weakly bound ligands to be detected.

1H spinlock 1D NMR spectra of the pYTN tripeptide in the presence of the other six compounds including adenine, thymine, uracil, glycine, serine, and DMSO. The signals derived from adenine (8.23, 8.18 ppm), thymine (7.35 ppm) and uracil (7.53, 7.51, 5.80, 5.78 ppm) are observed in the selected region of the spectra. The spectra were acquired with a spinlock period of 10 ms (a, b) or 200 ms (c, d). A 0.2 eq of LBT-Grb2 with (a, c) and without (b, d) Gd3+ were added. Asterisks indicate the resonances derived from the pYTN tripeptide. Spectra were acquired using a 500 MHz NMR spectrometer at 25°C
Structure of the ligand–protein complex is rapidly determined based on the ligand PCSs: the case of the low-affinity ligand
The ligand identified in the above PRE-based screening is further analyzed based on PCSs. Quantitative analysis of the PCSs observed for the ligand enables structure determination of the ligand–protein complex, from PCS-based ligand–protein docking calculation. To demonstrate the PCS-based structure determination of the ligand–protein complex, we first carried out the analysis on the low affinity pYTN peptide (Fig. 5c) (Gay et al. 1999; Kessels et al. 2002). Prior to the structural analysis, the binding affinity of the tripeptide and Grb2 SH2 was estimated from an NMR titration experiment, where the non-labeled tripeptide was titrated into a solution of 15N-labeled Grb2 SH2. Shifts in the resonances during the titration indicated that the binding and dissociation were fast on the NMR timescale (supporting information Figure S-3). The dissociation constant was calculated based on the signals of A91, F95, S96, and W121 (supporting information Figure S-4). A non-linear least square fitting of the data to a 1:1 binding model yielded a dissociation constant (K d) of 70.4 ± 7.8 μM.

Calculation of the PCSs for the bound state of the pYTN tripeptide. a Selected region of the 1H NMR spectra of the pYTN tripeptide, acquired during titration of LBT-Grb2 containing Tm3+. The spectra of the tripeptide in the presence of 0 equivalent (blue) to 0.7 equivalent (purple) LBT-Grb2-Tm3+ are overlaid. b Chemical shift changes of the signals of the pYTN tripeptide during titration with increasing amounts LBT-Grb2 containing Tm3+. c The chemical structure of the pYTN tripeptide on which the observed PCS values are indicated
In the case of the lower affinity ligands, which are often encountered in FBDD screening, the observed PCSs are weighted averages of the free and bound states because of the fast exchange process. In such cases, the structure of the ligand–protein complex can be determined based on the PCSbound (the PCS values for the bound state), which is obtained from titration experiments and curve fitting. 1H 1D NMR spectra of the pYTN tripeptide were measured during the titration with non-labeled LBT-Grb2. Two sets of data were collected: one for titration with Tm3+-bound LBT-Grb2, and the other for titration with LBT-Grb2 lacking the lanthanide ion (apo-state). Although the signals of the tripeptide were gradually obscured by the signals derived from LBT-Grb2, the signal shifts could be observed during the early stage of the titration experiment (Fig. 5a). Chemical shift changes of the pYTN tripeptide were plotted against the amount of LBT-Grb2 added to the solution (Fig. 5b). The titration curves were fitted using (3) to determine the chemical shift difference between the free and bound state of the pYTN tripeptide, as described in the “Materials and methods” section. The K d value was fixed to 70.4 μM as determined by the titration experiment with 15N-labeled Grb2 SH2 and the non-labeled pYTN tripeptide. Chemical shift changes were determined both for LBT-Grb2(Tm3+) and LBT-Grb2(apo), and the differences in these values were defined as the PCSs for the bound state (4). In addition to Tm3+, PCSs were observed using Tb3+ and Dy3+, and a total of 16 PCS values were obtained for Tm3+, Tb3+ and Dy3+ (Fig. 5c).
Pseudo-contact shift-based docking calculation was performed for the pYTN peptide and Grb2 SH2, using the Xplor-NIH program (Schwieters et al. 2003, 2006). Each of the three residues in the peptide was separately treated as a rigid body and their relative orientations were set to be variable during the calculation (see the “Materials and methods” section for details). One hundred calculations were performed and the 10 lowest energy structures were selected according to the two-step selection: the 20 structures were selected based on the PCS energy, then the final 10 structures were selected based on the total energy.
The lowest energy structure and the ensemble of the 10 lowest energy structures are shown in Fig. 6a and b, respectively. The RMSD value calculated for all atoms of pYTN peptide was 2.96 Å. The structure of the pYTN peptide roughly corresponds to that determined by X-ray crystallography (Fig. 6c), and the correlations between the experimental and back-calculated PCS values were good (Fig. 6d). Although the convergence of pTyr and Thr was reasonably good, the direction of the Asn residue was not defined: some conformers indicated Asn to be oriented in the direction of the protein, whereas others indicated it to be oriented in the opposite direction. This is presumably because of the distribution of the PCS isosurface (supporting information Figure S-2), on which the Asn residue slides away. Though this ambiguity could be eliminated by the use of the additional PCSs induced by the lanthanide ion attached at the different positions on the protein, the interaction between the Asn residue and Grb2 SH2 is well implied by the present structure.

The docking structure of Grb2 SH2 and the pYTN tripeptide. a The lowest energy structure. Grb2 SH2 and the tripeptide are represented as ribbon and stick models, respectively. The position of the lanthanide ion is represented as a yellow sphere. b Ensemble of the 10 lowest energy structures. Grb2 SH2 moieties are superimposed. pTyr, Thr, and Asn are colored blue, green, and orange, respectively. c X-ray crystal structure of Grb2 SH2 complexed with a phosphorylated peptide (PSpYVNVQN) (Nioche et al. 2002, 1jyr.pdb). The corresponding residues of the peptide are displayed as a stick model. The structures were drawn using the program PyMOL (DeLano 2002). d Comparison of experimental and back-calculated PCSs of proton signals of the low-affinity peptide (filled circles) and backbone amide proton signals of Grb2 SH2 (open squares) in complex with Tm3+. The PCS calculation was carried out for the lowest energy structure of the complex
Though structural information on the ligand–protein complex is inevitable for FBDD, it is often difficult to obtain the structural information in case of low-affinity, fast-exchanging ligands. We here demonstrated that the structure of the low-affinity ligand in complex with the target protein could be determined based on the PCSs, based on which the binding region for the protein and the orientation of the ligand could be estimated. The binding position and orientation of each of the fragments, “residues” in this case, can also be estimated. Despite the simplified treatment of the present fragment approach, the PCS data of both Grb2 and the peptide can define the structure of the peptide bound to Grb2 in a reasonable quality.
Structure determination of the complex: the case of the high-affinity inhibitor
In order to demonstrate the PCS-based structure determination for high-affinity ligands, we conducted an analysis on the high-affinity inhibitor, 4-[(10S,14S,18S)-18-(2-amino-2- oxoethyl)-14-(1-naphthylmethyl)-8,17,20-trioxo-7,16,19-triazaspiro[5.14]icos-11-en-10-yl]benzylphosphonic acid (Fig. 7a) (Gao et al. 2001), in complex with LBT-Grb2. The high-affinity inhibitor strongly binds to Grb2 SH2, and their binding and dissociation are slow on the NMR timescale (Ogura et al. 2008). Thus, the chemical shift changes of the high affinity inhibitor, upon the addition of Grb2 SH2, are equal to those between the free and bound states.

Analysis on the high-affinity inhibitor and LBT-Grb2. a The chemical structure of the inhibitor in which the protons were labeled. Proton signals were assigned according to the previous work (Ogura et al. 2008). Observed PCS values are indicated. b Overlay of the 1H 1D spectra of the high-affinity inhibitor complexed with 2H15N-labeled LBT-Grb2. The spectra measured in the presence and absence of the paramagnetic lanthanide ion Tm3+ are shown in blue and gray, respectively. An asterisk indicates an impurity. Spectra were acquired using an 800 MHz NMR spectrometer at 25°C
We prepared the high-affinity inhibitor in complex with 2H/15N-labeled LBT-Grb2 and measured 1H 1D NMR spectra in the presence and absence of the paramagnetic lanthanide ion Tm3+. Based on these 1D spectra, 11 Tm3+-induced PCSs were observed for the high-affinity inhibitor (Fig. 7b). The PCSs were further assigned with the help of 2D TOCSY experiment. In addition to Tm3+, PCSs were observed using Tb3+ and Dy3+, and finally 56 PCS values were assigned for the high-affinity inhibitor (Fig. 7a). Although PCS values are usually measured with reference to the diamagnetic lanthanide ion, such as Lu3+, here we used an apo-state sample as the reference as we were aiming for a rapid and simple experiment based on the fact that the 1H 1D spectrum of the inhibitor in complex with apo-state 2H/15N-labeled LBT-Grb2 was identical to that with Lu3+-bound LBT-Grb2 (data not shown).
Next, PCS-based docking calculation was carried out using the Xplor-NIH program (Schwieters et al. 2003, 2006) with a rigid body minimization protocol (Clore 2000; Tang and Clore 2006; Saio et al. 2010). For the docking calculation, PCSs derived from Tm3+, Tb3+ and Dy3+ were used: 125 PCSs for Grb2 SH2 and 56 PCSs for the high-affinity inhibitor. The position of the lanthanide ion against Grb2 SH2 was fixed at the position determined by the tensor calculation using Numbat. Grb2 SH2 and the inhibitor were treated as rigid bodies, and docked based on the PCS restraints and binding restraints (see “Materials and method” section for details). One hundred calculations were performed and the 10 lowest energy structures were selected according to the two-step selection procedure described above.
The lowest energy structure is shown in Fig. 8a. The position and orientation of the high-affinity inhibitor correspond well to those of the structure determined by an NOE-based NMR method (Fig. 8c). Furthermore, the 10 lowest energy structures are well defined as shown in Fig. 8b. The RMSD value calculated for all atoms of the high-affinity inhibitor was 1.91 Å. It should be also noted that the correlations between the experimental and back-calculated PCS values were good, which supports the compatibility of the docking structure (Fig. 8d).

The docking structure of Grb2 SH2 and the high-affinity inhibitor. a The lowest energy structure. Grb2 SH2 and the high-affinity inhibitor are represented as ribbon and stick models, respectively. The position of the lanthanide ion is represented as a yellow sphere. b Ensemble of the 10 lowest energy structures. The structure of Grb2 SH2 moiety is superimposed. c The structure of the complex determined by the NOE-based NMR method (Ogura et al. 2008, 1 x 0n.pdb). The lowest energy structure is represented as ribbon and stick models. The structures were drawn using the program PyMOL (DeLano 2002). d Comparison of experimental and back-calculated PCSs of proton signals of the high-affinity inhibitor (filled circles) and backbone amide proton signals of Grb2 SH2 (open squares), in complex with Tm3+. The PCS calculation was carried out for the lowest energy structure of the complex
Advantages against conventional methods
As shown in Figs. 6 and 8, the PCS-based structures of the ligand–protein complexes were less precise than those determined by X-ray crystallography or an NOE-based conventional NMR method. However, a high-resolution structure is not necessarily required at the early stage of FBDD (Jahnke 2007). One of the advantages of our strategy is its rapidness.
In our PCS-based strategy, no time-consuming side-chain assignment or NOE analysis is required. Δχ-tensor parameters, required for PCS-based structure determination, can be determined based on at least 8 PCSs of the protein: thus a limited number of signal assignments are sufficient for tensor determination. Several assignment techniques, that are free from time-consuming, low-sensitive three-dimensional NMR experiments, have been developed: signal assignment based on the amino acid selective labeling (Trbovic et al. 2005; Ozawa et al. 2006), site-specific labeling of unnatural amino acids (Jones et al. 2010), site-directed mutagenesis (Religa et al. 2010), and the lanthanide probe method (Pintacuda et al. 2004; John et al. 2007). Although it is difficult to assign all of the resonances of the protein using the above methods, these techniques are suitable for rapid assignments and are applicable for even large proteins or membrane proteins. Once the Δχ-tensor is determined for the target protein, PCSs observed for the ligand can be translated into quantitative structural information on the ligand–protein complex. In the conventional NMR approach, it is difficult to discuss the ligand–protein complex structure based only on limited NMR signals.
The PCS-based structure determinations of protein–ligand complexes were previously reported for a metalloprotein (John et al. 2006) and a non-metalloprotein with the LBT attached via a single fusion point (Zhuang et al. 2008). Although the ligand–protein structure was precisely determined in the case of the metalloprotein, the method was not applicable to general proteins lacking a metal-binding site. In case of the analysis utilizing the one-point anchored LBT, an inter-molecular NOE restraint was required for accurate structure determination, which would be a disadvantage for rapid structural analysis. In this study we determined more precise ligand–protein structures without specific inter-molecular NOE restraints, by exploiting the two-point anchored LBT that can hold the lanthanide ion more rigidly (Saio et al. 2009).
Conclusion
Although the use of the lanthanide probe has been limited to some metalloproteins, here we exploited the two-point anchored LBT and demonstrated that a lanthanide probe-based ligand screening method can be applied to non-metalloproteins. Since lanthanide ions have similar chemical properties but diverse magnetic properties, the distinct paramagnetic effects can be utilized simply by replacing the lanthanide ion added to the sample. Gd3+ induces strong PRE but no PCS. Thus it is useful for rapid identification of the hit ligand. Paramagnetic lanthanide ions other than Gd3+ induce PCSs that are useful for rapid structure determination of the ligand–protein complex. We proposed a hybrid strategy exploiting the advantages of both paramagnetic effects, by which the experiments from screening to structural analysis can be completed in a single system. The method was successfully applied to ligand screening and structural analysis of a protein–ligand complex with both low- and high-affinity ligands.
References
Banci L, Bertini I, Cavallaro G, Giachetti A, Luchinat C, Parigi G (2004) Paramagnetism-based restraints for Xplor-NIH. J Biomol NMR 28:249–261
Bayer E, Rapp W (1986) New polymer supports Jor solid-liquid phase peptide synthesis. In: Voelter W, Bayer E, Ovchinnikov YA, Ivanov VT (eds) Chemistry of peptides and proteins, vol 3. Waltler de Gruyter & Co, Berlin, pp 3–8
Becattini B, Pellecchia M (2006) SAR by ILOEs: an NMR-based approach to reverse chemical genetics. Chemistry 12:2658–2662
Boehm HJ, Boehringer M, Bur D, Gmuender H, Huber W, Klaus W, Kostrewa D, Kuehne H, Luebbers T, Meunier-Keller N, Mueller F (2000) Novel inhibitors of DNA gyrase: 3D structure based biased needle screening, hit validation by biophysical methods, and 3D guided optimization. A promising alternative to random screening. J Med Chem 43:2664–2674
Clore GM (2000) Accurate and rapid docking of protein–protein complexes on the basis of intermolecular nuclear overhauser enhancement data and dipolar couplings by rigid body minimization. Proc Natl Acad Sci 97:9021–9025
Dalvit C, Pevarello P, Tatò M, Veronesi M, Vulpetti A, Sundström M (2000) Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water. J Biomol NMR 18:65–68
Dalvit C, Fogliatto G, Stewart A, Veronesi M, Stockman B (2001) WaterLOGSY as a method for primary NMR screening: practical aspects and range of applicability. J Biomol NMR 21:349–359
Dalvit C, Fasolini M, Flocco M, Knapp S, Pevarello P, Veronesi M (2002) NMR-Based screening with competition water-ligand observed via gradient spectroscopy experiments: detection of high-affinity ligands. J Med Chem 45:2610–2614
Delaglio F, Grzesiek S, Vuister G, Zhu W, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293
DeLano WL (2002) The PyMOL molecular graphics system. Palo Alto, CA
Gao Y, Voigt J, Wu JX, Yang D, Burke TR Jr (2001) Macrocyclization in the design of a conformationally constrained Grb2 SH2 domain inhibitor. Bioorg Med Chem Lett 11:1889–1892
Gay B, Suarez S, Caravatti G, Furet P, Meyer T, Schoepfer J (1999) Selective GRB2 SH2 inhibitors as anti-Ras therapy. Int J Cancer 83:235–241
Hajduk PJ, Mack JC, Olejniczak ET, Park C, Dandliker PJ, Beutel BA (2004) SOS-NMR: a saturation transfer NMR-based method for determining the structures of protein-ligand complexes. J Am Chem Soc 126:2390–2398
Hu J, Eriksson PO, Kern G (2010) Aroma WaterLOGSY: a fast and sensitive screening tool for drug discovery. Magn Reson Chem 48:909–911
Jahnke W (2007) Perspectives of biomolecular NMR in drug discovery: the blessing and curse of versatility. J Biomol NMR 39:87–90
Jahnke W, Perez LB, Paris CG, Strauss A, Fendrich G, Nalin CM (2000) Second-Site NMR Screening with a Spin-Labeled First Ligand. J Am Chem Soc 122:7394–7395
Jahnke W, Rüdisser S, Zurini M (2001) Spin label enhanced NMR screening. J Am Chem Soc 123:3149–3150
John M, Pintacuda G, Park AY, Dixon NE, Otting G (2006) Structure determination of protein-ligand complexes by transferred paramagnetic shifts. J Am Chem Soc 128:12910–12916
John M, Schmitz C, Park AY, Dixon NE, Huber T, Otting G (2007) Sequence-specific and stereospecific assignment of methyl groups using paramagnetic lanthanides. J Am Chem Soc 129:13749–13757
Jones DH, Cellitti SE, Hao X, Zhang Q, Jahnz M, Summerer D, Schultz PG, Uno T, Geierstanger BH (2010) Site-specific labeling of proteins with NMR-active unnatural amino acids. J Biomol NMR 46:89–100
Kessels HW, Ward AC, Schumacher TN (2002) Specificity and affinity motifs for Grb2 SH2-ligand interactions. Proc Natl Acad Sci U S A 99:8524–8529
Klages J, Coles M, Kessler H (2006) NMR-based screening: a powerful tool in fragment-based drug discovery. Mol BioSyst 2:318–332
Klein J, Meinecke R, Mayer M, Meyer B (1999) Detecting binding affinity to immobilized receptor proteins in compound libraries by HR-MAS STD NMR. J Am Chem Soc 121:5336–5337
Mayer M, Meyer B (1999) Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Chem Int Ed 38:1784–1788
Nioche P, Liu WQ, Broutin I, Charbonnier F, Latreille MT, Vidal M, Roques B, Garbay C, Ducruix A (2002) Crystal structures of the SH2 domain of Grb2: highlight on the binding of a new high-affinity inhibitor. J Mol Biol 315:1167–1177
Nitz M, Franz KJ, Maglathlin RL, Imperiali B (2003) A powerful combinatorial screen to identify high-affinity terbium(III)-binding peptides. Chembiochem 4:272–276
Nitz M, Sherawat M, Franz KJ, Peisach E, Allen KN, Imperiali B (2004) Structural origin of the high affinity of a chemically evolved lanthanide-binding peptide. Angew Chem Int Ed Engl 12:3682–3685
Ogura K, Shiga T, Yokochi M, Yuzawa S, Burke TR Jr, Inagaki F (2008) Solution structure of the Grb2 SH2 domain complexed with a high-affinity inhibitor. J Biomol NMR 42:197–207
Orts J, Griesinger C, Carlomagno T (2009) The INPHARMA technique for pharmacophore mapping: a theoretical guide to the method. J Magn Reson 200:64–73
Otting G (2008) Prospects for lanthanides in structural biology by NMR. J Biomol NMR 42:1–9
Ozawa K, Wu PS, Dixon NE, Otting G (2006) 15N-Labelled proteins by cell-free protein synthesis. Strategies for high-throughput NMR studies of proteins and protein-ligand complexes. FEBS J 273:4154–4159
Pintacuda G, Keniry MA, Huber T, Park AY, Dixon NE, Otting G (2004) Fast structure-based assignment of 15N HSQC spectra of selectively 15N-labeled paramagnetic proteins. J Am Chem Soc 126:2963–2970
Pintacuda G, John M, Su XC, Otting G (2007) NMR structure of protein-ligand complexes by lanthanide labeling. Acc Chem Res 40:206–212
Rees DC, Congreve M, Murray CW, Carr R (2004) Fragment-based lead discovery. Nat Rev Drug Discov 3:660–672
Religa TL, Sprangers R, Kay LE (2010) Dynamic regulation of archaeal proteasome gate opening as studied by TROSY NMR. Science 328:98–102
Saio T, Ogura K, Yokochi M, Kobashigawa Y, Inagaki F (2009) Two-point anchoring of a lanthanide-binding peptide to a target protein enhances the paramagnetic anisotropic effect. J Biomol NMR 44:157–166
Saio T, Yokochi M, Kumeta H, Inagaki F (2010) PCS-based structure determination of protein–protein complexes. J Biomol NMR 46:271–280
Sánchez-Pedregal VM, Reese M, Meiler J, Blommers MJ, Griesinger C, Carlomagno T (2005) The INPHARMA method: protein-mediated interligand NOEs for pharmacophore mapping. Angew Chem Int Ed Engl 44:4172–4175
Schmitz C, Stanton-Cook MJ, Su XC, Otting G, Huber T (2008) Numbat: an interactive software tool for fitting Deltachi-tensors to molecular coordinates using pseudocontact shifts. J Biomol NMR 41:179–189
Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM (2003) The Xplor-NIH NMR molecular structure determination package. J Magn Reson 160:65–73
Schwieters CD, Kuszewski JJ, Clore GM (2006) Using Xplor–NIH for NMR molecular structure determination. Progr Nucl Magn Resn Spectr 48:47–62
Shuker SB, Hajduk PJ, Meadows RP, Fesik SW (1996) Discovering high-affinity ligands for proteins: SAR by NMR. Science 274:1531–1534
Su XC, Huber T, Dixon NE, Otting G (2006) Site-specific labelling of proteins with a rigid lanthanide-binding tag. ChemBioChem 7:1599–1604
Su XC, McAndrew K, Huber T, Otting G (2008) Lanthanide-binding peptides for NMR measurements of residual dipolar couplings and paramagnetic effects from multiple angles. J Am Chem Soc 130:1681–1687
Tang C, Clore GM (2006) A simple and reliable approach to docking protein–protein complexes from very sparse NOE-derived intermolecular distance restraints. J Biomol NMR 36:37–44
Thornton KH, Mueller WT, McConnell P, Zhu G, Saltiel AR, Thanabal V (1996) Nuclear magnetic resonance solution structure of the growth factor receptor-bound protein 2 Src homology 2 domain. Biochemistry 35:11852–11864
Trbovic N, Klammt C, Koglin A, Löhr F, Bernhard F, Dötsch V (2005) Efficient strategy for the rapid backbone assignment of membrane proteins. J Am Chem Soc 127:13504–13505
Wang YS, Frederick AF, Senior MM, Lyons BA, Black S, Kirschmeier P, Perkins LM, Wilson O (1996) Chemical shift assignments and secondary structure of the Grb2 SH2 domain by heteronuclear NMR spectroscopy. J Biomol NMR 7:89–98
Zhuang T, Lee HS, Imperiali B, Prestegard JH (2008) Structure determination of a Galectin-3-carbohydrate complex using paramagnetism-based NMR constraints. Protein Sci 17:1220–1231
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Saio, T., Ogura, K., Shimizu, K. et al. An NMR strategy for fragment-based ligand screening utilizing a paramagnetic lanthanide probe. J Biomol NMR 51, 395–408 (2011). https://doi.org/10.1007/s10858-011-9566-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10858-011-9566-5