
Abstract
Deposition of cadmium telluride (CdTe) from cadmium chloride (CdCl2) and tellurium oxide has been achieved by electroplating technique using two-electrode configuration. Cyclic voltammetry shows that near-stoichiometric CdTe is achievable between 1330 and 1400 mV deposition voltage range. The layers grown were characterised using X-ray diffraction (XRD), UV–Visible spectrophotometry, scanning electron microscopy (SEM), energy-dispersive X-ray analysis (EDX), photoelectrochemical (PEC) cell and DC conductivity measurements. The XRD shows that the electrodeposited CdTe layer is polycrystalline in nature. The UV–Visible spectrophotometry shows that the bandgap of both as-deposited and heat-treated CdTe films are in the range of (1.44–1.46) eV. The SEM shows grain growth after CdCl2 treatment, while, the EDX shows the effect of growth voltage on the atomic composition of CdTe layers. The PEC results show that both p- and n-type CdTe can be electrodeposited and the DC conductivity reveals that the high resistivity is at the inversion growth voltage (Vi) for the as-deposited and CdCl2 treated layers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Cadmium chloride post growth treatment of CdS/CdTe-based solar cell has been well explored due to its effect on the enhancement of photoelectrical, structural and morphological properties of CdTe layers [1]. This treatment also promotes grain boundary passivation and inter-diffusion at the CdS/CdTe heterojunction by reducing the lattice mismatch [2]. Different modes of application of CdCl2 treatment of CdS/CdTe-based solar cell structures have been explored [3] but there has been few literature on the in-situ treatment of CdTe with chlorine during growth. Although literature reveals that the incorporation of chlorine as a dopant during CdTe growth from cadmium sulphate precursor has been well explored [4–7], the use of CdCl2 as the main precursor for CdTe had been scarce. Preliminary investigation on the electrodeposition of CdTe using CdCl2 as cadmium precursor has been explored by Bonilla et al. [8] and Nor Abdul-Manaf et al. [9] using three-electrode configuration. The work presented in this paper focuses on the analysis and optimisation of the structural, morphological, optical and electrical properties of electrodeposited CdTe using cadmium chloride as cadmium precursor in a two-electrode electrodeposition configuration.
Electrodeposition as a semiconductor deposition technique was opted for due to its comparative advantages with respect to deposition process continuity, low-cost, simplicity, scalability and Cd-containing waste reduction amongst other advantages [10, 11]. The two-electrode electrodeposition configuration was utilised due to its industrial applicability, process simplification and also eliminate possible K and Ag ions doping [12, 13] which may emerge from the reference electrode.
2 Experimental details
2.1 Electrolytic bath preparation
CdTe thin films were electrodeposited cathodically on glass/FTO substrates by potentiostatic technique in which the anode was a high purity graphite rod. Cadmium chloride hydrate CdCl2·xH2O of 99.995% purity was utilised as cadmium precursor, while, tellurium oxide TeO2 of 5 N (99.999%) purity was used as the tellurium source. The electrolyte was prepared to make 1.5 M CdCl2·xH2O in 400 ml deionised (DI) water contained in a 500 ml polypropylene beaker. The polypropylene beaker was placed inside a 1000 ml glass beaker containing DI water. The glass beaker serves as the outer DI water jacket which helps in maintaining uniform heating of the electrolyte in the polypropylene beaker. Prior to the addition of TeO2, the CdCl2·xH2O aqueous solution was electro-purified for ~100 h by applying a cathodic potential just below the required potential for reduction of Cd2+. Afterwards, low level of TeO2 solution was added to the solution and stirred for ~5 h to achieve solution homogeneity. It should be noted that TeO2 is partially soluble in some acidic media but insoluble in water [14]. Therefore, 0.03 M of TeO2 solution was prepared by dissolving ~2 g of TeO2 in concentrated 30 ml hydrochloric acid. The acidic media was stirred for ~1 h to achieve homogeneity before the TeO2 is gently diluted with 400 ml of DI water in a polypropylene conical flask. All these steps were taken before TeO2 addition in minute quantity into the electro-purified CdCl2·xH2O electrolytic bath. An initial volume of 5 ml TeO2 solution was added to the CdCl2·xH2O electrolytic bath to give a total of 0.0023 M TeO2 concentration. The bath was continuously stirred and the temperature was maintained at ~85 °C. It should be noted that the Te concentration in the electrolytic bath was kept comparatively low due to its high positive reduction potential of Te as compared to Cd.
The pH at the start of the deposition was adjusted to 2.00 ± 0.02 at room temperature using diluted solutions of hydrochloric (HCl) and ammonium hydroxide (NH4OH). It should be noted that the 85 °C bath temperature used during the growth of CdTe layer was due to higher crystallinity achievable at higher growth temperature [7], but due to the aqueous solution utilised in this experiment, the growth temperature is limited. In this work, two-electrode configuration was utilised. This comprises of a high purity carbon anode and a 3 × 4 cm2 glass/FTO cathode with a sheet resistance of 7 Ω/sq.
The power supply source was a computerized Gill AC potentiostat. Prior to the deposition of CdTe layer, cyclic voltammetric test was performed to determine the cathodic potential range in which stoichiometric or near stoichiometric CdTe can be grown. It should be noted that, immediately after CdTe growth, the glass/FTO/CdTe was divided into two halves. One-half was left as-deposited while the other was CdCl2 treated (CCT) at 400 °C for 20 min in air. Experimentation on just heat treated CdTe layers were not performed due to improved material and electronic properties observed after CdCl2 treatment [1]. The CdCl2 treatment was carried out by dipping the as-grown CdTe layer into 0.1 M CdCl2 aqueous solution for 2 s, retracted and allowed to dry in air before heat treatment. The treatment with CdCl2 was necessitated due to its material and device quality improvement as reported by numerous independent researchers in the literature. All the substrates and chemicals used in this study were procured from Sigma-Aldrich Ltd, UK.
2.2 Substrate preparation
The glass/FTO substrates were cleaned using ultrasonic bath containing soap solution for ~15 min. The substrates were rinsed thoroughly afterwards in DI water, dried in a stream of nitrogen gas before it is been degreased using acetone and methanol. The substrates were immediately rinsed in DI water and attached to the cathode rod using polytetrafluoroethylene tape. Finally, the glass/FTO substrates were rinsed in DI water and transferred directly into the electrodeposition bath.
2.3 Experimental techniques
Cyclic voltammogram is the current–voltage characteristic of an electrolyte which provides information on the elemental/compound deposition taking place at different deposition potentials. The study helps in identifying the range in which stoichiometric or near-stoichiometric CdTe layers can be achieved. The level of crystallinity, crystalline structure and phase identification of the as-deposited and post-growth treated CdTe layers were obtained using X-ray diffraction (XRD) technique. This information was extracted using Philips PW 3710 X’pert diffractometer with Cu-Kα monochromator of wavelength λ = 1.54 Å. The X-ray generator tension was set to 40 kV while the current was adjusted to 40 mA for this set of experiments. Optical property such as the absorbance was studied using Carry 50 Scan UV–Vis spectrophotometer at room temperature between the wavelengths of 200–1000 nm. A thoroughly cleaned TEC7 glass/FTO substrate was setup as the baseline. Raman spectroscopy studies were performed on samples using Renishaw InVia Raman spectrometer using a 514 nm argon ion laser excitation source to determine the crystallinity, identify the phases present and also to determine the phonon modes of both the as-deposited and the CdCl2 treated samples. The Raman spectroscopy laser power and objective were set to 50% and ×100 respectively. The thickness of the CdTe layers was measured using UBM Microfocus optical depth profilometer (UBM, Messtechnik GmbH, Ettlingen, Germany). The surface morphology and compositional analysis of both the as-deposited and CdCl2 treated CdTe films were studied using FEI Nova 200 NanoSEM equipment. The electrical conductivity type of the CdTe layers was determined using photoelectrochemical (PEC) cell measurement as an alternative to the conventional Hall effect measurements. The Hall effect measurement of CdTe deposited on FTO are not possible due to the underlying conducting FTO substrate.
PEC measurement was conducted by dipping the glass/FTO/CdTe layer into an aqueous electrolyte containing 0.1 M of sodium thiosulphate to form a solid/liquid junction. The difference between the voltages measured under white light illuminated (V L ) and dark (V D ) conditions provides the open circuit voltage or the PEC signal. The sign of the PEC signal determines the electrical conductivity type of the CdTe layer [15].
The DC conductivity measurements on glass/FTO/CdTe/Ohmic contact structures were carried out on the electrodeposited CdTe layers to determine the electrical resistivity (ρ) and conductivity (σ) of the layers.
3 Results and discussion
3.1 Cyclic voltammetric study
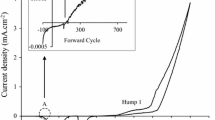
Figure 1 shows the cyclic voltammogram of an aqueous solution containing a mixture of 1.5 M CdCl2·xH2O and a low level of pre-prepared TeO2 solution in 400 ml of DI water during the forward and reverse cycle between 100 and −2000 mV cathodic voltage. The electrolytic bath pH was adjusted to 2.00 ± 0.02 using dilute solutions of HCl and NH4OH. The stirring rate and bath temperature were maintained at 300 rpm and ~85 °C respectively. Tellurium deposits first in the electrodeposition process due to its standard reduction potential value of +593 mV with respect to standard H2 electrode which is more positive than that of cadmium with standard reduction potential value of −403 mV.

A typical cyclic voltammogram for deposition electrolyte containing the mixture of 1.5 M CdCl2·xH2O and 0.0023 M TeO2 solution at ~85 °C and pH 2.00 ± 0.02. The scan rate was set to 3 mVs−1. The insets are the expanded sections of the forward cycle for both Te and Cd deposition initiation
It was observed from Fig. 1 that the deposition of Te starts in the forward cycle at cathodic potential of ~400 mV under the experimental conditions used (inset in Fig. 1) according to the following electrochemical reaction:
With an increase in cathodic potential, more Te is deposited. The deposition of Cd and formation of CdTe starts around ~1000 mV cathodic potential as depicted by the first hump shown in Fig. 1. Tellurium-rich CdTe is expected at the initial stage of deposition of cadmium, afterward, with an increase in deposition potential and there exists a narrow cathodic potential window in which stoichiometric CdTe compound can be deposited. Further increase in the cathodic potential above this point results in an increase in the cadmium richness of electrodeposited CdTe. The electrochemical equation for Cd deposition and the complete CdTe formation is given in Eq. (2) and (3) respectively.
It should be noted that the steep increase in current density (marked c) at a cathodic voltage above ~1400 mV might be due to either the formation of Cd dendrites on the working electrode or the electrolysis of water at any deposition potential above ~1230 mV [14]. Both scenarios have been reported to have a detrimental effect on the quality of the deposited CdTe layer [16]. However, the release of most active hydrogen atoms on the material surface also introduces an advantage of hydrogen-passivation during growth although the formation of hydrogen bubble could be a disadvantage due to a possible cause of material layer peeling. Therefore, selecting the growth voltage has a crucial effect on the growth of suitable CdTe layer. In the reverse cycle, the dissolution of elemental Cd and Cd from CdTe occurs in the voltage range of ~1500 and ~1000 mV, while the dissolution of Te from the glass/FTO substrate occurs below 750 mV as shown in Fig. 1.
Therefore, based on the information obtained from the voltammetric study, cathodic potential range between 1000 and 1400 mV was pre-characterised using XRD for as-deposited CdTe layers grown at a step size of 50 mV (results not presented in this paper). The highest peak intensity signalling highest crystallinity was observed at 1350 mV, therefore, surrounding cathodic potentials were scanned at 10 mV intervals and characterised to identify the best cathodic potential in which stoichiometric and near-stoichiometric CdTe can be achieved.
3.2 Material characterisation
3.2.1 X-ray diffraction study
The aim of this study is to identify the cathodic potential in which stoichiometric or near-stoichiometric CdTe can be grown. This was done by observing the level of crystallinity through XRD peak intensity. Figure 2a, b shows typical X-ray diffraction intensity plotted against 2θ angle for layers grown between 1330 and 1400 mV for both as-deposited and CdCl2 treated CdTe layers. For both the as-deposited and CdCl2 treated CdTe layers, only cubic CdTe phases were observed. CdTe (111)C peak corresponding to 2θ = ~23.8° is the dominant XRD peak and the preferred orientation of the electrodeposited CdTe at all growth voltages and conditions. Peaks attributed to CdTe, (220)C and (311)C corresponding to 2θ = ~38.6° and 2θ = ~45.8° were also observed asides the FTO peaks observed at 2θ = 20.6°, 33.8°, 37.9°, 51.6°, 60.7° and 65.6°. For better comparison, XRD patterns were shifted up in the graph as shown in Fig. 2a, b. As observed in both Fig. 2a, b, the highest XRD peak intensity of the CdTe (111)C for both as-deposited and CdCl2 treated CdTe layers was observed at 1360 mV. This suggests that highly crystalline CdTe corresponding to stoichiometric CdTe can be electrodeposited at 1360 mV cathodic voltage.

Typical XRD patterns of CdTe layers grown between 1330 and 1400 mV deposition potential for a as-deposited CdTe layers and b CdCl2 treated CdTe layers at 400 °C for 20 min in air
Figure 3a shows the comparison of CdTe (111)C peak intensity as a function of the cathodic potential at which the layers were grown while Fig. 3b shows the graph of crystallite size against cathodic voltage.

a Typical plot of CdTe (111) cubic peak intensity against cathodic voltage. b A typical plot of the crystallite sizes against cathodic voltage for the as-deposited and CdCl2 treated CdTe layers grown between 1330 and 1400 mV
It is clear from Fig. 3a that the treatment of the CdTe layer with CdCl2 improved the level of crystallinity of the CdTe layers grown at all the explored cathodic voltages. This improvement can be attributed to grain growth and recrystallisation. Furthermore, this improvement could also arise from the formation of CdTe from unreacted excess Te presented in the layer reacting with Cd from CdCl2 treatment. Full details of this recent understanding is reported by Dharmadasa et al. 2016 [17]. When the growth potential deviates from V i , the crystallinity suffers due to the presence of two phases; CdTe and Te at lower cathodic potential and CdTe and Cd at high cathodic voltage. The extracted XRD data from these CdTe work matches the JCPDS reference file No. 01-775-2086. The crystallite size, D, was calculated using the Scherrer’s formula:
where β is the full width at half maximum (FWHM) of the diffraction peak in radian, θ is the Bragg angle and λ is the wavelength of the X-rays used (1.54 Å). The summary of XRD data and obtained structural parameters of CdTe thin films grown at cathodic voltages between 1330 and 1400 mV using cubic (111) peak are tabulated in Table 1.
As observed in Table 1 and Fig. 3b, for the as-deposited CdTe layers, low crystallite size was observed at cathodic voltages of ±20 mV away from 1360 mV due to either Cd- or Te-richness of CdTe grown at these cathodic voltages. After CdCl2 treatment, improvement in crystallite size was observed for all layers. This observation is in accordance with the effect of CdCl2 treatment on CdTe as reported in the literature. However, it should be noted that there is a limitation of the use of Scherrer equation in determining the crystallite size. This equation is formalised to calculate smaller grains [18] of polycrystalline materials and may not be suitable for highly crystalline material. Therefore, as shown in Table 1 and Fig. 3b, the crystallite size saturates at ~65 nm. This must be due to the limitation of Scherrer equation and the XRD measurement system.
3.2.2 Thickness measurements
Figure 4 shows the graph of electrodeposited CdTe layer thickness estimated using both experimental and theoretical methods against cathodic voltage for CdTe layers grown for 120 min duration. The thickness of the layers grown between 1330 and 1400 mV were calculated theoretically using Faraday’s law of electrolysis as shown in equation (v). Where d is the density of CdTe, F = 96,485 Cmol−1 is the Faraday constant, n is the number of electrons transferred for deposition of 1 molecule of CdTe (n = 6 for CdTe), M is the molar mass of CdTe (240.01 gmol−1), t is the deposition time, J is the average deposition current density and T is the film thickness.

Graph of CdTe layer thickness (theoretical and experimental) against cathodic voltages for both as-deposited and CdCl2 treated CdS layers
As observed in Fig. 4, the value of the calculated thickness using Faraday’s law of electrolysis was higher than the measured thickness. It should be noted that the Faraday’s law of electrolysis assumes that all the electronic charges flowing through the electrolyte contribute to the deposition of the CdTe layers without considering the electronic charges involved in the decomposition of water into its constituent ions. It was further observed that an increase in the cathodic voltage results into increase in current density and hence affects deposited CdTe layer thickness. After CdCl2 treatment, a slight reduction in thickness was observed. This might be due to the sublimation of CdTe or excess elemental Cd or Te. This also can be due to the formation of a denser layer after CdCl2 treatment, between 1350 and 1390 mV cathodic voltages, a slightly uniform cathodic voltage which is an indication of the formation of CdTe layer with close deposition current density.
3.2.3 Optical absorption study
Using the data acquired through Carry 50 Scan UV–Vis spectrophotometer, the square of the absorbance (A 2) was plotted against the photon energy (hv) as shown in Fig. 5a, b for as-deposited and CdCl2 treated CdTe layers grown at different growth voltages respectively. The straight line segments were extrapolated from the straight line to A 2 = 0 in order to estimate the energy bandgaps of the CdTe layers tabulated in Table 2 for comparison. As observed in Table 2, the bandgap of both the as-deposited and the CdCl2 treated CdTe layers within the explored cathodic voltage range falls within 1.45 ± 0.01 eV range which is comparable with the bandgap of bulk CdTe [19]. As shown in Fig. 6, the growth of CdTe at 1360 mV shows the sharpest absorption edge which signifies superior CdTe layer [20], while the growth of CdTe layer away from 1360 mV shows a reduction in the slope of the optical absorption edge in both the as-deposited and CdCl2 treated CdTe layers due to Cd- or Te-richness in CdTe. It should be noted that CdCl2 treatment utilised in this work increases the sharpness of the absorption edge across all growth voltages explored. This further attests to the improvement in the optical absorption of CdTe layer after CdCl2 treatment as recorded in the literature.

Optical absorption spectra for electrodeposited CdTe thin-films grown between cathodic voltage range between 1330 and 1400 mV, a for as-deposited, and b for CdCl2 treated CdTe at 400 °C for 20 min in air

Graph of absorption edge slope against cathodic potential for as-deposited and CdCl2 treated CdTe thin films
3.2.4 Morphological and compositional analysis
For both the morphological and compositional experiments, CdTe layers were grown at cathodic voltages between 1330 and 1400 mV on glass/FTO for 120 min. Each glass/FTO/CdTe layer was divided into two halves; one-half was left as-deposited while the other was CdCl2 treated. Figure 7 shows the SEM images of as-deposited and CdCl2 treated CdTe grown at 1330, 1360 and 1400 mV. From topological observation, all the as-deposited CdTe layers show full coverage of the glass/FTO layer. The as-deposited CdTe layers show clear agglomeration of small crystallites forming into cauliflower-like clusters. After CdCl2 treatment, larger crystals were formed through recrystallization, coalescence of grains [16]. The presence of gaps was observable after CdCl2 treatment in all the layers. For CdCl2 treated CdTe layer grown at 1330 mV as shown in Fig. 7d, high density of pinholes was observed. This might be due to the Te-richness, as a result of the deviation from stoichiometric value and properties as explained by Dharmadasa et al. [16]. While the Cd-rich CdTe layer grown at 1400 mV shows less detrimental effect asides few pinholes along the grain boundaries. The major problem with the presence of pin-hole in between grain boundary is shunting due to contact between the back metal contact and the underlying substrate in solar cell structures. In comparison, larger grains are observed after CdCl2 treatment for layer grown at 1360 mV. The layers grown at 1400 mV shows better coverage of the substrate, although the grains are smaller.

SEM micrographs for CdTe layers grown at 1330, 1360 and 1400 mV, a–c for as-deposited and d–f for CdCl2 treated layers at 400 °C for 20 min in air
It should be noted that based on new material and device understanding in CdS/CdTe-based solar cell, the richness of Cd in CdTe layer has been demonstrated as more advantageous in high-efficiency device fabrication [21–24] as compared to Te-rich CdTe absorber layer.
Figure 8 shows the compositional analysis of as-deposited and CdCl2 treated CdTe layers using EDX results. As observed in Fig. 8, both the as-deposited and CdCl2 treated CdTe layers display Te-richness at a cathodic voltage lower than 1360 mV while CdTe layers grown at 1360 and above are rich in Cd. Stoichiometric CdTe was observed at ~1360 mV for both the as-deposited and the CdCl2 treated layers with the Cd/Te atomic ratio equal to 1.01. After CdCl2 treatment, a shift in the atomic composition ratio towards stoichiometry was observed. This might be due to the reaction between unreacted Te with Cd from CdCl2 and/or the sublimation of excess elemental Cd and Te from the layer. This result is in accord with the observations in Sect. 3.2.6.

Graphical representation of percentage compositions ratio of Cd to Te atoms in as-deposited and CdCl2 treated CdTe thin films at different deposition cathodic voltages
3.2.5 Photoelectrochemical (PEC) cell study
Figure 9 shows the PEC cell measurement results for CdTe layers grown at cathodic voltages between 1330 and 1480 mV in both the as-deposited and CdCl2 treated CdTe samples. Prior to the commencement of this experiment, the PEC cell was calibrated using a known n-type CdS layer. For the as-deposited CdTe layers shown in Fig. 9, cathodic voltages lower than 1360 mV shows p-type electrical conductivity, while layers grown at 1360 mV cathodic voltage and above were n-type in electrical conduction. This is due to the Te-richness in the CdTe layers at lower growth voltages and Cd-richness in CdTe grown at higher cathodic voltages. This observation can be related to the redox potential of both Cd and Te, and also on the cyclic voltammetric study as explained in Sect. 3.1. It can be deduced that stoichiometric or near-stoichiometric CdTe can be achieved at the cathodic voltage between n- and p-CdTe layer where the atomic ratio of Cd to Te is at 50:50. This observation is in line with the high crystallinity level observed at 1360 mV in Sect. 3.2.1. The presence of only one phase (CdTe) at this voltage increases the crystallinity of the layer.

PEC signals for layers grown at different cathodic voltages between 1330 and 1480 mV for both as-deposited and CdCl2 treated CdTe layers
After CdCl2 treatment, a shift in the PEC signal towards the p-type region was observed across the cathodic range explored. It should be noted that conductivity type change after CdCl2 treatment could depend on factors such as; the heat treatment temperature, duration of treatment, initial atomic composition of Cd and Te, the concentration of CdCl2 utilised in treatment, defect structure present in the starting material, and the material’s initial conductivity type as documented in the literature [1, 9, 25, 26]. It was interesting to see that CdTe layers grown at 1450 mV and above still retaining their initial n-type conductivity after CdCl2 treatment.
This observation is crucial to move towards understanding of CdCl2 treatment. It is clear that composition change is one of the factors determining the electrical conductivity of CdTe layer. Te-richness produces p-CdTe while Cd-richness produces n-CdTe. The addition of Cl into CdTe is a complex issue. Substitution of Cl into Te sites makes the material n-type by acting as a shallow donor [13]. However, there are experimental evidence of the formation of a defect level at 1.39 eV below the conduction band during CdCl2 treatment [27]. This shows that during the CdCl2 treatment, acceptor-like defect is also formed closer to the top of the valence band and this leads to the formation of p-type doping of CdTe. One explanation could be that Cl forms complex with currently unknown native defect. Therefore, Cl seems to act as an amphoteric dopant in CdTe.
In addition to the above doping effect, self-compensation can take place during heat treatment in the presence of CdCl2 due to the existence of numerous native defects in the material. Therefore, the final electrical conductivity depends on the most dominant process taking place during this treatment.
The experimental evidence in Fig. 9 shows the real situation; (i) in as-deposited layers Te-richness produces p-CdTe and Cd-richness produces n-CdTe (ii) CdCl2 treatment tend to change the material from n-properties towards p-properties. In other words, the Fermi level (FL) move from the upper half towards the lower half of the bandgap. The tendency of the FL crossing the mid-point depends on the initial nature of the material layer. If the Cd-richness is dominant in the layers, FL remains in the upper half of the bandgap keeping the material n-type in electrical conductivity. In this discussion, effects of external impurities have been neglected. If an external impurity with dominant doping is introduced in the CdTe layer, the above analogy can be changed.
3.3 DC resistivity
The DC resistivity experiment was performed on CdTe layers grown between the cathodic voltage of 1330 and 1400 mV. The CdTe layers were grown for 120 min each. 2 mm diameter of 100 nm thick gold contacts were evaporated at a high pressure of 10− 5 Nm−2 on the p-type CdTe layer for both the as-deposited and CdCl2 treated CdTe layers (glass/FTO/p-CdTe/Au), while 100 nm thick indium contacts were evaporated at a high pressure of 10−5 Nm−2 on the n-CdTe layers for both the as-deposited and CdCl2 treated CdTe layers (glass/FTO/n-CdTe/In) to achieve Ohmic contacts for the metal/semiconductor interfaces [28, 29]. The electrical resistivity (ρ) of the layers was calculated using equation (v) where R is the electrical resistance, A is the contact area and L is the film thickness. The average electrical resistance (R) was calculated using the I–V data extracted under dark condition using Rera Solution PV simulation system.
Figure 10 shows the plot of electrical resistivity (ρ) against the cathodic voltage in which the CdTe layers were grown. From observation in Fig. 10, the resistivity of CdTe layers reduces after CdCl2 treatment which might be due to defect and grain boundary passivation, reduction of grain boundaries due to grain growth during CdCl2 treatment, increase in CdTe crystallinity amongst other advantages of CdCl2 treatment as documented in the literature.

Typical graphs of electrical resistivity against cathodic voltage for CdTe layer grown within the cathodic voltage between 1330 and 1400 mV in both as-deposited and CdCl2 treated conditions
For both the as-deposited and CdCl2 treated CdTe layers grown at ~1360 mV cathodic voltage exhibit the high resistivity value. This is expected since the CdTe is stoichiometric and intrinsic in electrical conduction. The material grown at 1360 mV should, therefore, have the highest resistivity. However, cathodic voltages closer to the intrinsic voltage (V i ) would be favourable for the fabrication of devices due to high crystallinity, better optical and morphological properties achieved in stoichiometric or near-stoichiometric CdTe layers. It should be noted that based on the understanding as published by Dharmadasa et al. (2002) [21, 30, 31] and new results as published by independent researchers such as Reese et al. (2015) [23] and Burst et al. (2016) [22], fabricating devices with slightly Cd-rich CdTe is favourable due to increased carrier lifetime, defect reduction amongst other advantages.
4 Conclusion
In this work, CdTe layers were successfully electrodeposited using a two-electrode configuration from an acidic and aqueous solution containing cadmium chloride hydrate CdCl2·xH2O and tellurium oxide TeO2 as Cd and Te precursors respectively. XRD analysis shows cubic (111) CdTe diffraction as the preferred orientation of all the CdTe explored in this work, while, the highest diffraction intensity were observed at 1360 mV under both as-deposited and CdCl2 treated conditions. After CdCl2 treatment, an improvement in the absorption edge slope was observed with the highest slope signifying highest CdTe quality [20, 32] was observed at 1360 mV. Morphologically, better glass/FTO substrate coverage was observed at Cd-rich CdTe layer grown at 1400 mV while Te-rich layer grown at 1330 mV show high pinhole density. The best underlying substrate coverage, grain growth and size were observed at 1360 mV. PEC measurements show the ability to electroplate both n-, i- and p-type CdTe layers using precursors explored in this work. The incorporation of these layers into PV devices fabrication is on-going.
References
I.M. Dharmadasa, Review of the CdCl2 treatment used in CdS/CdTe thin film solar cell development and new evidence towards improved understanding. Coatings 4(2), 282–307 (2014)
X. Wu, High-efficiency polycrystalline CdTe thin-film solar cells. Sol. Energy 77(6), 803–814 (2004)
A. Abbas, G.D. West, J.W. Bowers, P. Isherwood, P.M. Kaminski, B. Maniscalco, P. Rowley, J.M. Walls, K. Barricklow, W.S. Sampath, K.L. Barth, The effect of cadmium chloride treatment on close-spaced sublimated cadmium telluride thin-film solar cells. IEEE J. Photovolta. 3(4), 1361–1366 (2013)
B.M. Basol, E.S. Tseng, D.S. Lo, S. Tseng, S. Dennis, Electrodeposition of thin film heterojunction photovoltaic devices that utilize Cd rich Hg1−x Cdx Te, Pat. US4548681, Oct 1985
V.D. Popovych, I.S. Virt, F.F. Sizov, V.V. Tetyorkin, Z.F. Tsybrii, L.O. Darchuk, O.A. Parfenjuk, M.I. Ilashchuk, The effect of chlorine doping concentration on the quality of CdTe single crystals grown by the modified physical vapor transport method. J. Cryst. Growth 308(1), 63–70 (2007)
L. Svob, A. Heurtel, Y. Marfaing, Deuteration effects in chlorine-doped CdTe. J. Cryst. Growth 101(1–4), 439–442 (1990)
M.P.R. Panicker, M. Knaster, F.A. Kroger, Cathodic deposition of CdTe from aqueous electrolytes. J. Electrochem. Soc. 125(4), 566 (1978)
S. Bonilla, E.A. Dalchiele, Electrochemical deposition and characterization of CdTe polycrystalline thin films. Thin Solid Films 204(2), 397–403 (1991)
N. Abdul-Manaf, H. Salim, M. Madugu, O. Olusola, I. Dharmadasa, “Electro-plating and characterisation of CdTe thin films using CdCl2 as the cadmium source. Energies 8(10), 10883–10903 (2015)
D. Lincot, Electrodeposition of semiconductors. Thin Solid Films 487(1–2), 40–48 (2005)
I.M. Dharmadasa, Advances in Thin-Film Solar Cells. (Pan Stanford, Singapore, 2013)
S. Dennison, Dopant and impurity effects in electrodeposited CdS/CdTe thin films for photovoltaic applications. J. Mater. Chem. 4(1), 41–46 (1994)
K. Zanio, Semiconductors and Semimetals, vol. 13 (Academic Press, New York, 1978)
C.G. Morris, Academic Press Dictionary of scieNce and Technology. (Academic Press, San Diego, 1991)
J. Nowotny, T. Bak, M. Nowotny, L. Sheppard, Titanium dioxide for solar-hydrogen I. Functional properties. Int. J. Hydrog Energy 32(14), 2609–2629 (2007)
I. Dharmadasa, P. Bingham, O. Echendu, H. Salim, T. Druffel, R. Dharmadasa, G. Sumanasekera, R. Dharmasena, M. Dergacheva, K. Mit, K. Urazov, L. Bowen, M. Walls, A. Abbas, Fabrication of CdS/CdTe-based thin film solar cells using an electrochemical technique. Coatings 4(3), 380–415 (2014)
I.M. Dharmadasa, O.K. Echendu, F. Fauzi, N.A. Abdul-Manaf, O.I. Olusola, H.I. Salim, M.L. Madugu, A.A. Ojo, Improvement of composition of CdTe thin films during heat treatment in the presence of CdCl2. J. Mater. Sci. (2016). doi:10.1007/s10854-016-5802-91-10
A. Monshi, Modified scherrer equation to estimate more accurately nano-crystallite size using XRD. World J. Nano Sci. Eng. 2(3), 154–160 (2012)
T.L. Chu, S.S. Chu, Thin film II–VI photovoltaics. Sol. State Electron. 38(3), 533–549 (1995)
A. Bosio, N. Romeo, S. Mazzamuto, V. Canevari, Polycrystalline CdTe thin films for photovoltaic applications. Prog. Cryst. Growth Charact. Mater. 52(4), 247–279 (2006)
I.M. Dharmadasa, W.G. Herrenden-Harker, R.H. Williams, Metals on cadmium telluride: Schottky barriers and interface reactions. Appl. Phys. Lett. 48(26), 1802 (1986)
J.M. Burst, J.N. Duenow, D.S. Albin, E. Colegrove, M.O. Reese, J.A. Aguiar, C.-S. Jiang, M.K. Patel, M.M. Al-Jassim, D. Kuciauskas, S. Swain, T. Ablekim, K.G. Lynn, W.K. Metzger, CdTe solar cells with open-circuit voltage breaking the 1 V barrier. Nat. Energy 1, 16015 (2016)
M.O. Reese, C.L. Perkins, J.M. Burst, S. Farrell, T.M. Barnes, S.W. Johnston, D. Kuciauskas, T.A. Gessert, W.K. Metzger, Intrinsic surface passivation of CdTe. J. Appl. Phys. 118(15), 155305 (2015)
D.G. Diso, F. Fauzi, O.K. Echendu, O.I. Olusola, I.M. Dharmadasa, Optimisation of CdTe electrodeposition voltage for development of CdS/CdTe solar cells. J. Mater. Sci. 27, 12464–12472 (2016)
H.I. Salim, V. Patel, A. Abbas, J.M. Walls, I.M. Dharmadasa, Electrodeposition of CdTe thin films using nitrate precursor for applications in solar cells. J. Mater. Sci. 26(5), 3119–3128 (2015)
B.M. Başol, Processing high efficiency CdTe solar cells. Int. J. Sol. Energy 12(1–4), 25–35 (1992)
I.M. Dharmadasa, O.K. Echendu, F. Fauzi, N.A. Abdul-Manaf, H.I. Salim, T. Druffel, R. Dharmadasa, B. Lavery, Effects of CdCl2 treatment on deep levels in CdTe and their implications on thin film solar cells: a comprehensive photoluminescence study. J. Mater. Sci. 26(7), 4571–4583 (2015)
M.K. Rabinal, I. Lyubomirsky, E. Pekarskaya, V. Lyakhovitskaya, D. Cahen, Low resistance contacts to p-CulnSe2 and p-CdTe Crystals. J. Electron. Mater. 26(8), 893–897 (1997)
S. Nozaki, A.G. Milnes, Specific contact resistivity of indium contacts to n-type CdTe. J. Electron. Mater. 14(2), 137–155 (1985)
I.M. Dharmadasa, A.P. Samantilleke, N.B. Chaure, J. Young, New ways of developing glass/conducting glass/CdS/CdTe/metal thin-film solar cells based on a new model. Semicond. Sci. Technol. 17(12), 1238–1248 (2002)
I.M. Dharmadasa, C.J. Blomfield, C.G. Scott, R. Coratger, F. Ajustron, J. Beauvillain, Metal/n-CdTe interfaces: a study of electrical contacts by deep level transient spectroscopy and ballistic electron emission microscopy. Sol. State Electron. 42(4), 595–604 (1998)
H. Metin, R. Esen, Annealing effects on optical and crystallographic properties of CBD grown CdS films. Semicond. Sci. Technol. 18(7), 647–654 (2003)
Acknowledgements
The authors would like to thank H. I. Salim, N.A. Abdul-Manaf, O. Olusola, M. Madugu, and K. Burak for their valuable contributions. The main author would also like to acknowledge the Sheffield Hallam University, Tertiary Education Trust Fund (TETFund), Nigeria and the University of Ado Ekiti for their support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ojo, A.A., Dharmadasa, I.M. Analysis of electrodeposited CdTe thin films grown using cadmium chloride precursor for applications in solar cells. J Mater Sci: Mater Electron 28, 14110–14120 (2017). https://doi.org/10.1007/s10854-017-7264-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-017-7264-0