
Abstract
Graphite oxide reduction is probably one of the best technique used to obtain large quantities of few-layer graphene. We developed a new method to produce reduced graphene oxide by using sodium metal as a reducing agent with subsequent dehydration in concentrated sulfuric acid. The resulting product was characterized using various analytical techniques with respect to the oxygen content and species of the residual oxygen-containing groups. The reduced graphene oxide prepared by this method was electrochemically tested as electrode in supercapacitors using two-electrode symmetric system and aqueous electrolyte. The product exhibits improved capacitance during cyclic voltammetry measurements. In comparison to parent graphite oxide specific capacity increased from 0.88 to 28 F/g after 10 cycles at scan rate 20 mV/s and dropped to 19.44 F/g after 100 cycles while at scan rate 5 mV/s specific capacity 46.41 F/g was recorded after first cycle and 39.90 F/g after 50 cycles.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Graphene, the parent of all graphitic forms, has been emerging as a material of great interest due to its remarkable physical, chemical and electrical properties. Since its discovery in 2004 [1] graphene has been thoroughly investigating in a physical and chemical point of view. At this moment a great challenge is an easy and scalable method to prepare this single layer of carbon atoms. The realization of potential applications is limited by the method of its production. Many different synthesis techniques have been developed including mechanical cleavage [2], chemical vapor deposition [3], epitaxial growth [4] and exfoliation of graphite oxide (GO) [5]. Mechanical exfoliation, repeated peeling of graphite using Scotch tape, is perfect for laboratory scale research but hard to control and optimize. In epitaxially-prepared graphene the resulting product cannot be used as it is and has to be transferred to appropriate substrate additionally some catalyst contaminations may appear. GO reduction is currently the only one that can be used to obtain large quantities of graphene without significant costs. Although such graphene suffers from defects of the residual active sites therefore is more often called reduced graphene oxide it has high surface area, good thermal and electrical conductivities and good catalytic activity. It has been widely studied as a promising candidate in various applications including electronic, optoelectronic and sensing devices as well as energy storage materials of supercapacitors or lithium-ion batteries. Residual oxygen species in reduced graphene oxide may introduce additional faradaic reactions and increase specific capacity of as-prepared supercapacitors electrode. GO can be converted to reduced graphene oxide using various chemical substances. Although corrosive and explosive most commonly hydrazine is used [6] because product with low oxygen content and high electrical conductivity can be obtained, however, metal hydrides mainly sodium borohydride [7] and lithium aluminumhydride [8] as well as some metals itself such as iron [9], zinc [10] and aluminum [11] are also often used as reducing agents. Other less popular reducing agents including glutathione [12], pyrogallol, vitamin C [13] and carrot roots [14] have been investigated too. GO can be also easily exfoliated by thermal treatment at high temperatures [15]. It is commonly known that efficient reduction of GO requires very strong reducing agent or high temperatures, however, neither strong chemical reducer nor very high temperature are able to reduce GO completely. Sufficiently strong reducing agent such as hydrazine or temperature close to 1,000 °C can generally reduce carboxyl groups located at the edges of graphene layers, hydroxyl groups and epoxy groups located at the interior [16]. It is much more difficult to remove carbonyl groups and some residual groups formed during transformation-reduction of the oxygen-containing species. At the temperature 800–1,000 °C most of the oxygen species are removed regarding production of the large number of defects [17]. At the higher temperature in the range 1,000–2,000 °C partial graphitization may occur resulting in decreasing in defects with simultaneous layer stacking and crystal growth. Perfect GO reducer has to efficiently reduce the oxygen species and prevent from sticking of graphene layers therefore we report here a method using violent and exothermic reaction of sodium metal in water. In fact this process utilizes hydrogen generated from metal–water reaction that can reduce and exfoliate GO.
In comparison to other metals used in GO reduction including transition metal such as zinc and iron there is no need to trigger the reaction by acid addition. Reduced GO is then dehydrated in boiled concentrated sulfuric acid to complete reduction and remove metal impurities.
2 Experimental
2.1 Synthesis of graphite oxide
Graphite oxide was prepared from synthetic graphite by the Staudenmaier method [18]. Briefly 20 g of commercial graphite powder (Fisher Scientific) was magnetically mixed with 350 mL 95–97 % sulfuric acid (Acros Organics) and 180 mL 100 % fuming nitric acid (Sigma-Aldrich) in a 1 L round-bottomed flask equipped with a reflux and thermometer and ice-cooled at the temperature close to 273 K. After an hour 220 g potassium chlorate was slowly added and reaction mixture was mixed for 330 h. The greenish GO slurry was transferred to a 10 L beaker and diluted with deionized water. After GO sedimentation almost 10 L of solution was decanted and a fresh portion of water was added. It was repeated 5 times till the solution was colorless. Brownish slurry was vacuum-filtrated on a fritted glass funnel and two hard filter papers (Munktel, 390) and washed with copious amount of deionized water. As-prepared GO was dried in desiccator with phosphorus pentoxide to prevent from desorption of some epoxy groups.
2.2 Graphite oxide reduction
0.5 g GO was ultrasonicated in 250 mL water and reduced with 10 g of sodium metal added in small pieces (Reachim). Next it was vacuum-filtrated, rinsed with 2.5 L of deionized water and dried. 0.3 g of reduced graphene oxide was mixed with 50 mL 95–97 % sulfuric acid and dehydrated at 150 °C for 3 h. Product was diluted, filtered, washed with 4 L of water and dried in an oven. The product was labeled Red Na SA.
2.3 Characterization
Graphite oxide and Red Na SA were characterized with powder X-ray diffraction (XRD, Seifert 3003) with a step size 0.02° and Cu Kα radiation to evaluate the interlayer distance (d002) and crystallite height (Lc).
The IR-spectra were measured on a Nicolet 6700 FT-IR spectrophotometer with attenuated total reflectance (ATR method). Raman spectroscopy was performed by using a Renishaw InVia with 514 nm laser excitation. Elemental analysis was performed on a Perkin Elmer 2400 to estimate mainly carbon and oxygen weight percent and C/O ratio.
Electrochemical experiments were carried out using two-electrode system. The working electrode materials composed of 90 wt% of active material and 10 wt% PTFE (Sigma-Aldrich, 35 μm) were pasted on electrochemical nickel current collectors to form films, 0.037 g of active material per electrode. The accurate weight of the electrodes was read by a high-precision balance (Metler Toledo AT 261 DeltaRange). Polypropylene separator (Fipro 25, 0.35 μm thickness) soaked with 6 M KOH aqueous electrolyte was used in all measurements. Electrodes, current collectors and separator were pressed by four screws in PMMA casing. Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements were performed with Autolab PGSTAT 30 workstation. EIS plots were tested in the frequency range from 100 kHz to 100 mHz with the amplitude of sinusoidal voltage signal equal 10 mV in potential range from 0 to 1 V and step potential 0.5 V.
3 Results
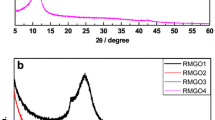
Structural change during the graphite oxidation and reduction of GO was studied by XRD. Figure 1 presents XRD patterns of the graphite precursor, GO and Red Na SA with an intense characteristic graphitic signal located around 26° corresponding to the interlayer distance 0.34 nm and crystallite height 24 nm. This peak shifts to about 12° for GO due to the increase in the interlayer distance to 0.74 nm and decrease in Lc to about 9 nm by intercalation of the oxygen-containing groups. After reduction and exfoliation GO signal broadens and flattens additionally small peak in graphitic region is created that may indicate the loss of the long-range order in graphenes [19]. Reduced graphene oxide has the interlayer spacing very close to that of graphite 0.37 nm and height of crystallite about 4 nm.

XRD patterns of graphite, GO and Red Na SA
Raman spectroscopy is a useful method to characterize graphite-based materials. It can be used to investigate crystallite size, hybridization of carbon atoms, defects, crystal disorder and number of layers. Figure 2 presents the Raman spectra obtained for graphite, GO and Red Na SA. There are observed two vibration bands D around 1,355 cm−1 and G around 1,600 cm−1. The D band corresponding to disorder is very small in highly-ordered graphite structure but increases during oxidation and reduction. The G band that corresponds to sp 2 domains is broadened after oxidation due to defects formation and small crystallite size.

Raman spectra of graphite, GO and Red Na SA
Intensity ratio ID/IG is a convenient measure of disordered carbon that simply corresponds to amount of sp 3 and sp 2 carbon atoms. For graphite, possessing ordered and layered structure with high crystallite size, ID/IG ratio is 0.36 and rises to 0.9 for GO due to increase in disorder of turbostratically aligned graphene layers and diminishing of randomly-located crystallite size. Upon reduction there is increase in sp 2 domains due to retrieving of ordered structure but also decrease in the average size of crystallites therefore ID/IG ratio is slightly higher than that for GO and equals 1.04.
Qualitative analysis of GO and sodium-reduced graphene oxide was performed using IR spectroscopy. Figure 3 shows FTIR spectra recorded for GO and Red Na SA. GO spectra showed a broad signal located at 3,700–2,800 cm−1 attributed to the stretching vibration in hydroxyl groups and water molecules, as well as signal at 1,700 cm−1 belonging to the carboxylic groups, 1,600 cm−1 to OH groups, 1,350 cm−1 due to deformation vibration in C–OH, 1,024 cm−1 represented by C–O groups sometimes designated to C–C and finally peak around 940 cm−1 from epoxy groups.

FTIR ATR spectra of GO and Red Na SA
Pumera et al. [20] suggests that large peak at 1,600 cm−1 corresponds to the unoxidized sp 2 regions but in fact signal slightly above 1,600 cm−1 is more often attributed to OH groups while signal around 1,580 cm−1, very common in less-oxidized graphite, may result from sp 2 domains [21]. After reduction most of the signals disappeared there were observed only very small peaks around 1,660 cm−1 that may be related to some residual C=O groups [22] and unexpectedly around 1,330 cm−1 due to hydroxyl groups.
Elemental analysis was used to evaluate amount of oxygen before and after reduction. Graphite oxidized using Staudenmaier method contained 51.5 wt% C and 47.32 wt% O with about 1 wt% H and 0.1 wt% N from solvent. It should be noted that there is also some amount of sulfur, based on XPS results generally GO contains about 0.3 wt% of sulfur. GO reduced with sodium and dehydrated in sulfuric acid results in diminishing of the oxygen content to about 18 wt% and increase in carbon amount to 80.5 wt%.
The capacitive behavior of Red Na SA was performed in a two-electrode symmetric cell using polypropylene separator soaked with 6 M KOH electrolyte. Figure 4 presents cyclic voltammograms after 10, 50, 60, 70, 80, 90 and 100 cycles at scan rate 20 mV/s in potential window 0–1 V, the specific capacities per mass of one electrode calculated from CV curves [23] were 28.05, 20.14, 19.85, 19.56, 19.31, 19.09 and 19.44 F/g respectively and remained close to 19 F/g even after 200 cycles that is higher than 0.88 F/g obtained for GO after 10 cycles.

CV curves for Red Na SA during 100 cycles at scan rate 20 mV/s, the biggest loop (grey) was recorded after 10 cycles
Measurements at scan rate 5 mV/s presented in Fig. 5 exhibit better cycling and higher capacity. There is smaller difference between curves after tenth and fiftieth cycle than observed for 20 mV/s. Consequently similar specific capacities were obtained that was 46.41 F/g after the first cycle, 44.49 F/g after ten cycles and 39.90 F/g after 50 cycles. This is much higher value than recorded at the same scan rate for GO, 5.54 F/g after the first cycle and only 2.32 F/g after 10 cycles.

CV curves for Red Na SA recorded at scan rate 5 mV/s after 1, 10, 20, 30, 40 and 50 cycles
Electrochemical impedance spectroscopy can be used to understand the resistive components involved with electrochemical systems that is very important for supercapacitors characterization.
The Nyquist plots obtained at an applied potential of 0 V and a frequency range between 100 kHz and 100 mHz are presented in Fig. 6 and 7 for GO and Red Na SA respectively.

Nyquist plot for GO

Nyquist plot for sodium-reduced GO dehydrated in sulfuric acid
Electrochemical impedance spectroscopy plots present quasi-reversible behavior of GO and reduced graphene oxide. Two regions can be distinguished including the semicircle corresponding to the faradaic charge-transfer resistance [24] and straight line in the low-frequency region indicating a pure capacitive behavior and representing the ion diffusion in the electrode structure. More steep curve in the low-frequency region for Red Na SA is attributed to better Warburg diffusion that is more effective mass transfer from the electrolyte to the electrode interface. Although GO has higher wettability [25] towards aqueous electrolyte it is a poor conductor with very high real resistance therefore its specific capacity is relatively low. Red Na SA has some residual oxygen-containing groups that may provide additional pseudo-capacitance and better wettability but exhibits much lower resistance than GO so transport of electrolyte ions to graphene surface is more favored.
4 Conclusions
We have demonstrated that sodium metal is a powerful reducing agent for graphene oxide. Although its reaction with water is very explosive tiny amounts slowly added during ultrasonic treatment limit exothermic effects. Subsequent dehydration of as-prepared sample in concentrated sulfuric acid may result well-reduced graphene oxide with some residual oxygen-containing groups. This is quite fast reduction method and in comparison to hydrazine there is no need to use reducing agent in a huge excess. This material shows possibility to be used as electrode in supercapacitors because of higher specific capacity than parent GO and lower charge and mass transfer resistance.
References
K.S. Novoselov, A.K. Geim, S.V. Morozov, D. Jiang, Y. Zhang, S.V. Dubonos, I.V. Grigorieva, A.A. Firsov, Science 306, 666 (2004)
P. Kun, F. Weber, C. Balazsi, Cent. Eur. J. Chem. 9, 47 (2011)
X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S.K. Banerjee, L. Colombo, R.S. Ruoff, Science 324, 1312 (2009)
J. Hass, R. Feng, T. Li, X. Li, Z. Zong, W.A. deHeer, P.N. First, E.H. Conrad, C.A. Jeffrey, C. Berger, Appl. Phys. Lett. 89, 143106 (2006)
S. Stankovich, D.A. Dikin, G.H.B. Dommett, K.M. Kohlhaas, E.J. Zimney, E.A. Stach, R.D. Piner, S.B.T. Nguyen, R.S. Ruoff, Nature 442, 282 (2006)
S. Stankovich, D.A. Dikin, R.D. Piner, K.A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S.B.T. Nguyen, R.S. Ruoff, Carbon 45, 1558 (2007)
J. Li, H. Lin, Z. Yang, J. Li, Carbon 49, 3024 (2011)
A. Ambrosi, C.K. Chua, A. Bonanni, M. Pumera, Chem. Mater. 24, 2292 (2012)
Z.J. Fan, W. Kai, J. Yan, T. Wei, L.J. Zhi, J. Feng, Y. Ren, L.P. Song, F. Wei, ACS Nano 5, 191 (2011)
X. Mei, J. Ouyang, Carbon 49, 5389 (2011)
Z. Fan, K. Wang, T. Wei, J. Yan, L. Song, B. Shao, Carbon 48, 1670 (2010)
T.A. Pham, J.S. Kim, J.S. Kim, Y.T. Jeong, Coll. Surf. A 384, 543 (2011)
M.J. Fernandez-Merino, L. Guardia, J.I. Paredes, S. Villar-Rodil, P. Solis-Fernandez, A. Martınez-Alonso, J.M.D. Tascon, J. Phys. Chem. C 114, 6426 (2010)
T. Kuila, S. Bose, P. Khanra, A.K. Mishra, N.H. Kim, J.H. Lee, Carbon 50, 914 (2012)
H.M. Ju, S.H. Huh, S.H. Choi, H.L. Lee, Mater. Lett. 64, 357 (2010)
S. Pei, H.M. Cheng, Carbon 50, 3210 (2012)
S.H. Huh, in Physics and Applications of Graphene—Experiments, ed. by S. Mikhailov (InTech, 2011), p. 73
L. Staudenmaier, Ber. Dtsch. Chem. Ges. 31, 1481 (1898)
A. Kaniyoor, T.T. Baby, T. Arockiadoss, N. Rajalakshmi, S. Ramaprabhu, J. Phys. Chem. C 115, 17660 (2011)
C.K. Chua, Z. Sofer, M. Pumera, Chem. Eur. J. 18, 13453 (2012)
Y.M. Shulga, V.M. Martynenko, V.E. Muradyan, S.A. Baskakov, V.A. Smirnov, G.L. Gutsev, Chem. Phys. Lett. 498, 287 (2010)
M. Acik, G. Lee, C. Mattevi, A. Pirkle, R.M. Wallace, M. Chhowalla, K. Cho, Y. Chabal, J. Phys. Chem. C 115, 19761 (2011)
E. Frackowiak, V. Khomenko, K. Jurewicz, K. Lota, F. Béguin, J. Power Sources 153, 413 (2006)
Y. Chen, X. Zhang, D. Zhang, P. Yu, Y. Ma, Carbon 49, 573 (2011)
B. Xu, S. Yue, Z. Sui, X. Zhang, S. Hou, G. Cao, Y. Yang, Energy Environ. Sci. 4, 2826 (2011)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Ciszewski, M., Mianowski, A. & Nawrat, G. Preparation and electrochemical properties of sodium-reduced graphene oxide. J Mater Sci: Mater Electron 24, 3382–3386 (2013). https://doi.org/10.1007/s10854-013-1259-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-013-1259-2