
Abstract
Layered double hydroxides (LDHs) of various compositions, i.e. Mg–Al, Mg–Mn–Al, are applied as the precursors of metal oxides for the preparation of N-doped carbon materials via chemical vapour deposition (CVD) with acetonitrile (as carbon and nitrogen source) at 600 and 700 °C. The use of Mn-containing LDHs for the preparation of the carbon materials is a novelty. The impact of transition metal species, i.e. MnxOy, in a blend of metal oxides derived from LDHs on the amount of carbon deposit and its composition, morphology, textural and capacitive properties is investigated. Mn-containing species occurring in a mixture of metal oxides enhance the quantity of carbonaceous product compared to those derived from Mg–Al LDHs. Thermally heated Mg–Mn–Al LDHs contain structural defects due to manganese oxides, which promote the formation of carbon deposit, especially higher production of amorphous carbons. The addition of Mn into Mg–Al LDHs matrix leads to carbon particles with increased N-doping and enhanced volume of mesopores. Furthermore, graphitic domains occurring in the carbon materials obtained with Mg–Mn–Al LDHs are thicker than those in the corresponding samples obtained with Mg–Al LDHs as Mn-containing species influence the concentration and location of N-containing groups in graphitic array. The specific capacitance of the carbon materials produced by CVD with the compounds derived from Mg–Al LDHs or Mg–Mn–Al LDHs is comparable (20–25 μF cm−2). The formation of electrical double layer at electrode/electrolyte interface is easier for the carbon materials prepared at 700 °C than for the carbon materials prepared at 600 °C. The maximum charge is stored either in the shallow parts of carbon particles for the former, as they contain bottleneck mesopores, or in the deep parts of carbon particles for the latter, as they contain slit-shaped mesopores.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chemical vapour deposition (CVD) is a commonly used method for the preparation of the carbon materials via thermal decomposition of gaseous organic compounds at high temperature (600–1000 °C) in the inert atmosphere in the presence of porous matrixes (e.g. hard templates). CVD experiments are also carried out in the presence of the catalyst, and then, the process is called catalytic chemical vapour deposition (CCVD). Depending on the synthesis conditions, a wide range of carbon materials differing in chemical composition, carbon particle morphology and textural properties can be obtained by means of CVD/CCVD method.
This work describes the preparation of the carbon materials via CVD using acetonitrile (CH3CN) as a source of carbon and nitrogen and layered double hydroxides (LDHs) as metal oxide precursors. LDHs consist of positively charged layers [(M2+)1−x(M3+)x(OH−)2)]x+, containing di- and trivalent metal cations and negatively charged interlayers, consisting of anions such as nitrates, carbonates and water molecules. The composition of LDHs can be appropriately modified by changing type and ratio of di- and/or trivalent metal cations in the layers and/or by changing type and/or concentration of anions in the interlayer space. Thermal decomposition of LDHs results in a mixture of metal oxides. Such a blend of metal oxides of various phase compositions can serve as supports or catalysts during carbonization.
LDHs were used previously for the preparation of carbon materials according to different procedures. In the 1990s MgAl LDHs with organic, aromatic anions between layers were used to prepare microporous carbon materials with specific surface area (SBET) ~ 1000 m2 g−1 [1,2,3,4,5]. In the 2000s, LDHs with organic, aliphatic anions between layers were used to prepare microporous carbon materials with higher SBET ~ 1700 m2 g−1 [6,7,8,9,10]. LDHs containing transition metal ions in the layers, i.e. Fe2+ or Ni2+, were applied to obtain transition metal oxides, then followed by transition metal nanoparticles, i.e. Fe or Ni, which were next utilized for carbon nanotubes (CNT) synthesis [11,12,13].
In a very recent review article titled “Layered double hydroxides functionalized by carbonaceous materials: from preparation to energy and environmental applications” one section is dedicated to the formation of carbonaceous materials (CMs) with the application of LDHs [14]. According to the information provided in that paper and elsewhere, LDHs can be used for the preparation of metal oxide/CMs composites by CVD via one- (without additional reduction) or two-step-procedure, especially Co-containing LDHs, i.e. Mg–Co–Al, Co–Al, Co–Fe–Mg–Al [15,16,17]. In addition, Mg–Al LDHs can be utilized as precursors of microspheres or flakes to obtain graphene microspheres (GMSs) or unstacked graphene, respectively, by means of CVD with CH4 [18, 19].
This work describes the application of Mg–Al LDHs, in which Mg2+ was partially substituted by Mn2+ (Mg–Mn–Al LDHs). We systematically studied the use of Mg–Mn–Al LDHs of various contributions of Mn, i.e. Mg:Mn:Al atomic ratios equal 2.8:0.2:1.0 or 2.6:0.4:1.0, for the preparation of carbon materials at 600 or 700 °C. Our intention was to check how the presence of Mn-containing species in a mixture of metal oxides derived from LDHs affects the yield of carbon deposit and its features. We focused on this issue because there is no report in the literature on the application of Mn-containing LDHs for the preparation of the carbon materials via CVD method using CH3CN. Moreover, Mn in contrast to Co, Ni and Fe is not able to catalyse the growth of CNT in a common CVD experiments [20]. Co-, Ni- and Fe-containing metal oxides can be more easily reduced to metal than Mn-containing metal oxides. In contrast to Co, Ni and Fe, Mn forms numerous stable carbides with high carbon concentrations. Diffusion of carbon through Mn and formation of its carbides are not as rapid as in the case of Co, Ni and Fe. Therefore, graphite precipitation can be delayed or even inhibited, which prevents the formation of carbon nanotubes [20]. It is well known that Fe catalyses the formation of tubular carbon nanostructures like CNT [20], and since 2007, Fe-containing LDHs have been widely applied for the preparation of CNT via CVD method using CH4 [13, 21, 22].
The morphology of carbon particles is an essential feature as it determines their applications. The existence of CNT in the carbon materials is responsible for the presence of meso- and macropores, which may be less beneficial for the ability of the carbon materials to store charge at electrode/electrolyte interface in electrochemical capacitors. On the other hand, the lack of micropores in the samples made of CNT may be advantageous, when use it as a support for the catalysts/electrocatalysts. The relationship between electrochemical performance of templated carbons and their pore structures is described in a very recent article titled “Recent progress in template-assisted synthesis of porous carbons for supercapacitors” [23].
The aim of this research is to perform systematic studies with cognitive function on finding key factors affecting the quantity and quality of the carbon materials. This research is mostly fundamental and leads to valuable results for future investigations. Application of various techniques/methods provides a complete characterization of the newly prepared materials. This allows to correlate the features of the synthesized carbon materials with synthesis conditions. These data should be useful for the rational design of the carbon materials in terms of their yield and properties.
This work fully describes physicochemical and capacitive properties of the carbon materials produced by CVD with the compounds derived from Mn-containing LDHs. For comparison, a similar characterization is provided for the carbon materials obtained in analogue manner with the use of Mg–Al LDHs.
We demonstrate that the presence of Mn-containing species in a blend of metal oxides derived from LDHs facilitates the formation of carbon deposit on the external surface of the support. They are not only responsible for higher contribution of amorphous carbon but also for lower population of graphitic carbon particles exposing edge plane surfaces. The carbon materials obtained with Mg–Mn–Al LDHs in comparison with those obtained with Mg–Al LDHs are enriched with N, which is likely to be located in graphitic carbon particles. The graphitic domains occurring in the carbon materials obtained with Mg–Mn–Al LDHs are thicker than those in the carbon materials obtained with Mg–Al LDHs. This is associated with the influence of Mn-containing species on the concentration and location of N-containing groups in graphitic array, i.e. decreased population of pyrrolic N (in pentagon site) and increased population of pyridinic and quaternary N (in hexagon site).
The carbon materials synthesized with Mn-containing LDHs are characterized by dominating mesoporous structure and therefore exhibit a potential application, e.g. as catalyst supports in heterogenous catalysis or as metal supports to prepare the electrode materials for low-temperature fuel cells.
Materials and methods
Materials
Magnesium nitrate hexahydrate [Mg(NO3)2 6H2O, min. 99%, pure p.a.], aluminium nitrate nonahydrate [Al(NO3)3 9H2O, min. 98.8%, pure p.a.], sodium hydroxide (NaOH, min. 98.8%, pure p.a.), potassium hydroxide (KOH, min. 85%, pure p.a.), acetonitrile (CH3CN, min. 99.9%, for HPLC) and acetone [(CH3)2CO, min. 99.5%, pure p.a.] were purchased from Avantor Performance Materials Poland S.A. Manganese (II) nitrate tetrahydrate [Mn(NO3)2 4H2O, min. 98%, for analysis] was provided by PanReac AppliChem ITW Reagents. All of the chemicals were used as received. Alumina powder for polishing (particle size of 0.3 μm) was purchased from Buehler. Nafion® perfluorinated ion-exchange resin (20 wt.% in mixture of lower aliphatic alcohols and water, contains 34% water) was purchased from Sigma-Aldrich. Argon (Ar, 99.999%) was purchased from Linde Gas. Water was purified using a Milli-Q purification system (Millipore).
Preparation of the carbon materials
The preparation of the carbon materials started with synthesis of LDHs via method outlined in our previous publication [15]. A set of LDHs was synthesized: Mg3Al LDHs (Mg:Al = 3:1), Mg2.8Mn0.2Al LDHs (Mg:Mn:Al = 2.8:0.2:1), Mg2.6Mn0.4Al LDHs (Mg:Mn:Al = 2.6:0.4:1).
Next, the pyrolysis of CH3CN was carried out at 600 and 700 °C in the presence of metal oxides (magnesia, alumina and manganese oxide species) derived from LDHs. The sample of LDHs (0.5 g) (in an alumina boat) placed in a horizontal tube furnace was heated under flow of Ar (30 mL min−1) to the required temperature (600 or 700 °C). Then, the Ar stream saturated with CH3CN vapour was introduced and the sample was maintained at the target temperature for 180 min, and slowly cooled down under pure Ar stream.
The samples, obtained in above-mentioned CVD experiments, were treated with hydrochloric acid (HCl:H2O 2:1 v/v) for 24 h and ultrasonically agitated for 60 min to remove inorganic residue. Finally, the resulting samples were separated, washed thoroughly with water and dried in a dry box at 50 °C.
In the manuscript the samples before acid treatment are described as the composites, whereas those after acid treatment are described as the carbon materials. The composites prepared at two temperatures, i.e. 600 or 700 °C, with the compounds derived from LDHs of various compositions, i.e. Mg3Al, Mg2.8Mn0.2Al, Mg2.6Mn0.4Al, are further labelled as LDHs-600 or LDHs-700, respectively. The corresponding carbon materials are denoted as C-LDHs-600 or C-LDHs-700, respectively.
Characterization of the physicochemical properties
Powder X-ray diffraction (XRD) analysis was performed using a PANalytical X’Pert PRO powder diffractometer with Cu Kα radiation (40 kV, 30 mA). The XRD patterns were recorded in the range of 5°–139° (2θ) with a step size of 0.05°. The XRD references are provided by International Centre for Diffraction Data (ICDD) database (PDF-4+ 2021).
Fourier transform infrared spectroscopic (FT-IR) measurements were taken by means of Nicolet 380 FT-IR spectrometer. A mixture of the sample (1 mg) and KBr (200 mg) was pressed to form the transparent disc used in spectroscopic measurements. The FT-IR spectra (400–4000 cm−1) were obtained by co-addition of 64 scans recorded with a resolution of 4 cm−1. Spectra processing was performed using Thermo Scientific OMNIC™ software package.
The dynamic contact angle between liquid (water (H2O), diiodomethane (CH2I2)) and the sample was measured using the Drop Shape Analyzer Kruss DSA100M optical contact angle measuring instrument (Hamburg, Germany, GmbH). The sterile syringe needles (NE 44, Kruss GmbH) were used for each new measurement. Kruss DSA100M uses an optical microscope and a digital camera (200 fps) which quickly takes images of the object and then uses a digital image processing algorithm to calculate the droplet’s contact angle by tangents or Laplace–Young approximations. The temperature (22 ± 0.3 °C) and humidity (44 ± 4%) of the test chamber were controlled. For each sample, four successive measurements were taken.
Elemental analysis (EA) was performed using an element analyser (vario EL III) to determine carbon, hydrogen and nitrogen contents.
Raman spectroscopic measurements were taken by means of a Renishaw InVia spectrometer equipped with Ar ion laser (514 nm, power 50 mW), a Leica DMLM confocal microscope, and a CCD detector. The Raman spectra were recorded using 5% of the power, in the spectral range of 200–3500 cm–1 with a resolution of 3 cm–1. Nine scans were accumulated to ensure a sufficient signal-to-noise ratio.
Nitrogen (N2) adsorption/desorption measurement was taken at -196 °C using an Autosorb-1 (Quantachrome) surface area and pore size analyser. Prior to N2 volumetric adsorption/desorption measurement, the samples were preheated and degassed under vacuum at 150 °C for 18 h. Surface area (SDFT) and pore volume (VDFT) were calculated according to density functional theory (DFT) approach. Pore size distribution (PSD) was determined according to quenched solid density functional theory (QSDFT) approach with slit-shaped pore model for pores with diameters ranging between 0.6 and 34.5 nm. Micropore volume (Vmicro) and mesopore volume (Vmeso) were calculated from cumulative pore volume. Vmicro was a part of cumulative volume of pores with diameters up to 2 nm. Vmeso was determined by subtracting Vmicro from VDFT. In addition, PSD was calculated by using the data from the desorption branch of N2 isotherm and the BJH (Barrett–Joyner–Halenda) method. Specific surface area (SBET) was calculated using the BET (Brunauer–Emmett–Teller) method based on adsorption data in the partial pressure of 0.1 < p/p0 < 0.35.
Scanning electron microscopy (SEM) images were recorded using a JEOL JSM-7500F field emission apparatus (with an operating voltage of 15 keV) equipped with INCA PentaFetx3 EDX system. Energy-dispersive spectroscopy (EDS) analysis of SEM micrographs was performed for powder samples deposited on carbon tape and aluminium stub and coated with thin (20 nm) layer of chromium. The estimated thickness of the samples was up to a few micrometres with the EDS resolution commonly 1 µm laterally and about 1–2 µm in depth. The elemental composition by EDS was performed on the surface area of the samples equal (34 μm × 48 μm) 1632 μm2.
Atomic force microscope (AFM) imaging was performed in air using the NT-MDT Solver BIO device with the SMENA SFC050L scanning head. Based on AFM micrographs, the average RMS (root mean square) roughness of the samples shaped in the pallets was estimated by an image analysis software (Nova-1902) and RMS (Gwyddion).
X-ray photoelectron spectroscopic (XPS) measurements were taken using a spectrometer equipped with Al Kα X-ray source (1486 eV, 12 kV, 20 mA) and a hemispherical analyser (R4000, Gammadata Scienta).
Characterization of the electrochemical properties
Electrochemical measurements were taken by means of the potentiostat/galvanostat PGSTAT204 (Metrohm Autolab) using an electrochemical cell in conventional three-electrode configuration. A Pt wire and an Ag/AgCl/KClsat electrode were used as a counter and reference electrodes, respectively. Glassy carbon (GC) disc (5 mm in diameter) was used as a substrate for electrochemical measurements. A GC substrate was polished in the aqueous alumina suspension on a polishing cloth and then cleaned by ultrasonic treatment in Milli-Q water and acetone. The sample of the studied materials (5 mg) was ultrasonically mixed with 1 wt.% Nafion suspension in a mixture of water and aliphatic alcohols (ethanol and n-propanol) (250 μL) for 20 min. Then, the resulting suspension (5 μL) was spread on a GC disc (0.196 cm2) and dried in air at room temperature. The loading of the carbon materials on each electrode (GC) was ca. 0.1 mg (0.51 mg cm−2). The capacitive performance of all the samples was assessed by cyclic voltammetry (CV) measurements. The CV curves were recorded in the aqueous solution of 6 M KOH under Ar atmosphere at potential sweep rates of 5, 10, 20, 50 and 100 mV s−1. The temperature of 6 M KOH (25 ± 1 °C) was maintained by JULABO (ED-5) thermostat. All of the potentials are referred to Ag/AgCl/KClsat.
Results and discussion
This manuscript is an extension of our previous study dealing with the use of various LDHs for the preparation of porous N-doped carbon materials. Up to now, we have applied LDHs containing transition metal cations such as Co2+ and Ni2+ [15, 16, 24,25,26,27] in the following systems, i.e. Mg–Co–Al LDHs, Co–Al LDHs and Ni–Al LDHs, in which the proportion of divalent cation to trivalent cation was equal 2 or 3, and the nitrates/carbonates anions occurred between the layers. Based on our previous observations, the presence of transition metal cations in the composition of LDHs distinctly affects the amount of the deposited carbonaceous product in CVD process carried out at 600–700 °C with CH3CN. For instance, Mg–Al LDHs led to 10–20 wt.% of carbon deposit, while a small incorporation of Co2+ into Mg–Al LDHs matrix (Mg–Co–Al LDHs) led to 14–38 wt.% in CVD processes carried out for 180 min. The replacement of Mg2+ with Co2+ or Ni2+ in LDHs matrix resulted in 30–35 or 33–39 wt.% of carbon deposit for the application of Co–Al LDHs or Ni–Al LDHs, respectively, in analogue CVD experiments carried out for 30 min only. The carbon materials obtained by means of Co–Al LDHs or Ni–Al LDHs consisted of tubular carbon particles, i.e. CNT, in contrast to the carbon materials derived from Mg–Al LDHs, which contained only platy carbon particles. In the case of Mg–Co–Al LDHs the formation of CNT depended on CVD temperature, i.e. CNT were not created at 600 °C, whereas the population of CNT was enormous in the carbon material synthesized at 700 °C.
In this work we apply a set of lab-synthesized Mg–Al LDHs and Mg–Mn–Al LDHs, in which the proportion of divalent cation to trivalent cation is equal 3, to produce N-doped carbon materials. The XRD patterns (Fig. S1) and FT-IR spectra (Fig. S2) confirm that the obtained samples have the layered structure characteristic of LDHs [24] and nitrates/carbonates incorporated between layers. The EDS analysis of SEM images confirms that the determined Mg:Al and Mg:Mn:Al atomic ratios of 2.7:1 and 2.6:0.2:1 or 2.3:0.4:1 for MgAl LDHs and MgMnAl LDHs, respectively, are close to the nominal Mg:Al and Mg:Mn:Al atomic ratios, i.e. 3:1 and 2.8:0.2:1 or 2.6:0.4:1.
LDHs of various compositions, i.e. Mg3Al, Mg2.8Mn0.2Al and Mg2.6Mn0.4Al, served as the sources of metal oxides acting as the support for the formation of carbon deposit by CVD. However, this work mainly provides the results obtained for Mg3Al LDHs and Mg2.6Mn0.4Al LDHs, whereas the data for Mg2.8Mn0.2Al LDHs are presented occasionally to highlight the influence of Mn-containing species on the formation of carbon deposit.
The physicochemical properties of the composites prepared at 600 and 700 °C
The XRD patterns of the composites (Fig. S3a–d) display that the structure of LDHs (illustrated in Fig. S1) is destroyed by heating during CVD experiments. According to the literature, a blend of metal oxides (i.e. MgO, Al2O3, MnO2, Mn2O3, Mn3O4, MgAl spinel and MgMn spinel) is then formed [19, 25, 28,29,30]. Figure S3a–d shows the normalized XRD patterns of the composites and the stick pattern for the data from the ICDD database (PDF 01-085-5619) for MgO. The XRD diffraction peaks attributed to MgO in the composites were compared with those characteristic of referenced MgO in Table S1. Among nine reflections in the range of 2θ = 5°–139° characteristic of MgO, only one reflection at 2θ ~ 106° is not distinctly seen in the XRD patterns of the composites. It cannot be distinguished from the baseline due to its low intensity. The broad reflection at 2θ ~ 35°–37° seems to be a combination of two reflections: one associated with the presence of MgO (at 2θ ~ 36°–37°) and one associated with the existence of MgAl2O4 (at 2θ ~ 35°–36°) (PDF 00-021-1152). Our assumption is based on our previous observations related to the occurrence of MgAl2O4 in the composites prepared via CVD at 800 °C with the use of Mg–Co–Al LDHs [15]. It looks as MgO is a main crystalline phase, while the contribution of MgAl2O4 is insignificant in the composites, whereas other metal oxides such as Al2O3 and manganese oxides seem to be amorphous, because they are not detected by XRD.
Among various metal oxides derived from the studied LDHs, only manganese oxides may be reduced with carbon under applied conditions. It is reported that the carbothermic reduction of manganese oxides (> 450 °C) starts with the transformation of MnO2 into Mn2O3 and is followed by transformation of Mn2O3 into Mn3O4, and finally Mn3O4 is reduced to MnO [31]. Besides, manganese carbide and metallic manganese may be also formed. However, the temperatures required for such deep reduction are relatively high [31]. So it is likely that thermal decomposition of Mg–Mn–Al LDHs at 600 and 700 °C under flow of CH3CN–Ar results in partial reduction yielding a mixture of various manganese oxides (MnxOy). Our hypothesis is additionally supported by other published data: (i) the reduction of MnO by carbon under atmospheric pressure is only feasible over 1410 °C [32], (ii) the deep reduction of manganese oxides is restricted by covering and wrapping manganese oxide particles by carbon species, thereby impeding the reduction of manganese oxides as observed for MnO2 heated under flow of CH4–N2 [33], (iii) CoMn spinel (obtained via calcination of Co–Mn LDHs at 450, 600 and 750 °C) used as the catalyst for pyrolysis of toluene can be reduced to metallic cobalt (Co) and manganese oxide (MnO); the spent catalysts are covered with CNT grown by means of Co [34].
It is worth to underline that some amount of carbon deposit in the composites could be consumed by the reduction of manganese oxides during CVD experiments. So, the real amount of carbon deposit is higher than that estimated by means of EA.
A partial reduction of manganese oxides by carbon could be considered as a part of activation process. Providing that manganese oxides act as activation reagents they should facilitate the production of micropores on the outer surface of the carbon materials by etching carbon atom as other metal oxides, i.e. ZnO, can do [35]. However, taking into account that the contribution of activation reagent (manganese oxides) is insignificant, the extent of activation on the properties of carbon deposit may not be observable. So, too small concentration of Mn-containing species may not affect the overall properties of carbon deposit in a typical way like in the case of other activation reagents being responsible for the creation of microporosity. In fact, we do not observe an increase in microporosity for the carbon materials produced with Mn-containing compounds derived from Mg–Mn–Al LDHs compared with those produced with the compounds derived from Mg–Al LDHs, but we notice an increase in mesoporosity as revealed by N2 sorption measurements, which is discussed further. There is no evidence of activation of the carbon deposit during CVD experiments by the presence of the compounds derived from studied LDHs, in particular, manganese oxides.
According to the literature [36, 37], MgO and Al2O3—the products of thermal decomposition of Mg3Al LDHs and Mg2.6Mn0.4Al LDHs, can act as hard templates for the preparation of porous carbons. EDS analysis of SEM images (Fig. S4a, b) reveals that all metal oxide species derived from Mg2.6Mn0.4Al LDHs are well-intermixed (uniform distribution of Mg, Mn, Al and O) and they do not show any phase separation.
According to the literature [30], the porosity of the hard template may be responsible for the yield of carbon deposit and its porous structure. We determine SBET and total pore volume (Vtot) of the blend of metal oxides (prepared separately in similar experiments like CVD using pure Ar), which are 156–237 m2 g−1 and ca. 0.3 cm3 g−1, respectively. Shi et al. [38] reported comparable data for thermally treated Mg3Al LDHs at 700 °C for 120 min, i.e. SBET = 124 and Vtot = 0.45 cm3 g−1.
SBET of thermally treated Mg3Al LDHs at 600 and 700 °C is 179 and 206 m2 g−1, respectively, while SBET of thermally treated Mg2.6Mn0.4Al LDHs at 600 and 700 °C is 237 and 156 m2 g−1, respectively. The content of carbon deposit in the composites estimated by means of EA is equal 8.9, 13.7, 19.4 and 23.1 wt.% in C-Mg3Al-600, C-Mg2.6Mn0.4Al-600, C-Mg3Al-700 and C-Mg2.6Mn0.4Al-700, respectively (Table 1). There is no simple relation between SBET of the support and the yield of carbon deposit. For instance, the greatest quantity of carbon deposit (23.1 wt.%) is formed in the presence of the support having the smallest specific surface area, i.e. SBET ~ 156 m2 g−1. On the other hand, the amount of the carbon deposit is only moderate (13.7 wt.%) in the presence of the support having the largest specific surface area, i.e. SBET ~ 237 m2 g−1. It reveals that porosity of the studied metal oxides is not a main parameter affecting the yield of carbon deposit. In general, the amount of carbon deposit increases with increasing synthesis temperature. The application of Mn-containing LDHs is another variable promoting the carbon deposition process.
During CVD process a blend of metal oxide particles become covered with carbon deposit. However, there is no XRD reflections characteristic of carbon deposit in the XRD patterns of the composites (Fig. S3a–d) since the amount of carbon deposit is small. It seems that carbon deposit is well-distributed in the samples and it does not form a condensed material.
The content of carbon deposit (determined by EA) formed with the use of both Mn-containing LDHs is 13.7–14.4 wt.%, which is ca. 1.5–1.6 times higher than that (8.9 wt.%) obtained by using Mg3Al LDHs at 600 °C. Similar trend is observed for the samples prepared at 700 °C (Table 1).
We employ additional methods to characterize the surface properties of a blend of metal oxides in order to figure out the reasons of the differences in the deposition of carbonaceous product on the surface of metal oxides derived from Mg–Al LDHs and Mg–Mn–Al LDHs. Knowing that the product of thermal decomposition of Mg–Mn–Al LDHs is a multi-component system, with a relatively low contribution of Mn-containing species (Fig. S4a, b), we may not observe drastic differences in the measured values.
We measure the contact angle of water (H2O) and diiodomethane (CH2J2) droplets on the surface of the products of thermal decomposition of Mg3Al LDHs and Mg2.6Mn0.4Al LDHs obtained by heating at 600 and 700 °C for 180 min under flow of Ar. The results of the measurements and experimental errors are presented in Table S2. We use the average contact angle data to estimate the free energy of the samples, in particular, to study the polar and dispersive components of the surface energy. We apply the Owens–Wendt method [39], which assumes that interactions between molecules of two substances, present in their surface layer, are equal to the geometric average of intermolecular interactions within each substance. Rudawska and co-workers presented an exact and detailed introduction to the Owens–Wendt methods in [40]. It can be generally concluded that dispersive energy describes the Lifshitz–van der Waals interactions, while the polar energy: polar or acid-based interactions. The results of our analysis are presented in Table S2. As can be seen, the compounds derived from LDHs have an affinity for both covalent and polar substances, as evidenced by the balance of polar and dispersive energies in the studied samples, regardless of the temperature and their compositions (manganese oxide addition or lack thereof). The addition of manganese oxides causes a slight reduction in the hydrophilicity of the sample (increasing the contact angles of water from 11.1° to 15.5°), with simultaneous variations of the contact angles of diiodomethane. However, the level of all these differences does not cause any critical impact on the hydrophilic and hydrophobic properties of the sample surface and in polar and dispersive surface energies. The surface properties of the studied compounds seem to be comparable so the obtained data do not allow us to explain the observed differences in the deposition of carbonaceous products. It is worth mentioning that during CVD experiments, carbon coating can change the wetting behaviour of the supports and generate the hydrophobic state characteristic of the carbon materials.
We estimate the surface roughness of the supports. The comparison of these values for a series of studied samples allows us to get qualitative information on the fraction of nucleation sites, which affects the nucleation, clustering and growth mechanism of carbon structures. The values of RMS (root mean square) roughness estimated by analysis of AFM images (40 × 40 µm2) (Fig. 1) of thermally treated Mg3Al LDHs at 600 and 700 °C are 180 ± 10 and 190 ± 10 nm, respectively, while the surface roughness of thermally treated Mg2.6Mn0.4Al LDHs at 600 and 700 °C is 180 ± 10 and 200 ± 10 nm, respectively. The values of RMS roughness are comparable. The presence of manganese oxides in a blend of metal oxides derived from Mg–Mn–Al LDHs does not affect the roughness determined by means of AFM. Their population on the surface of the samples is minor (Fig. S4a, b), therefore, it does not affect the average RMS roughness as it does not have an impact on SBET. So, we do not have a proof that the roughness of a blend of metal oxides derived from either Mg–Al LDHs or Mg–Mn–Al LDHs is influenced by its composition. Nevertheless, we have evidences that the selection of metal oxides is important and it has an impact on the quantity of carbon deposit formed during CVD at 600 and 700 °C.

AFM topography of thermally treated Mg3Al LDHs at a 600 °C, b 700 °C, and thermally treated Mg2.6Mn0.4Al LDHs at c 600 °C, d 700 °C
A lack of XRD reflections (Fig. S3a–d) characteristic of manganese oxides suggests that they are either amorphous or consist of small crystallites, thereby providing more defects than crystalline particles of MgO. So the grains of the support derived from Mg2.6Mn0.4Al LDHs due to the presence of manganese oxides may have enhanced number of edges and steps on the surface, which may be involved in resting of the diffused particles formed in CVD experiments. According to the literature, pyrolysis of CH3CN below 1000 °C leads to HCN, CH4 and N-containing polymeric residue formed via polymerization of the initial products [41]. Defective manganese oxides provide large number of “nucleation sites”, which participate in the growth of carbon deposit. As a consequence more carbonaceous product is deposited on the grains of the support derived from Mg2.6Mn0.4Al LDHs than in the case of Mg3Al LDHs, which is revealed by the amount of carbon deposit shown in Table 1.
We divide the weight percentage of carbon deposit, i.e. 8.9 and 19.4 wt.%, in the composites prepared at 600 and 700 °C, respectively, by the sum of atomic percentages of metals (3.7 at.%), i.e. Mg and Al, in Mg3Al LDHs. The calculated values are equal 2.4 and 5.2, respectively. The factor is twofold higher for 700 °C than for 600 °C, which is consistent with the fact that at higher temperature CH3CN decomposition is faster and consequently the concentration of decomposition products interacting with the surface of the supports is higher. Assuming that “carbon deposit productivity” of Mg- and Al-containing sites is the same in the supports derived from Mg2.6Mn0.4Al LDHs, we attempt to estimate roughly “carbon deposit productivity” provided by Mn-containing sites (0.4 at.%). To do so we take into account the following data: 13.7 and 23.1 wt.%—the content of the carbon deposit in the composites obtained at 600 and 700 °C; 3.3 at.%—the sum of atomic percentages of Mg and Al in Mg2.6Mn0.4Al LDHs. The calculated values are equal 14.4 and 14.5 for the composites prepared at 600 and 700 °C, respectively. The obtained values are much higher than 2.4 and 5.2 calculated as “carbon deposit productivity” of Mg + Al species. Although this estimation method is only considered as a speculation, it clearly shows very effective participation of Mn species in carbon deposit formation, much effective than that of Mg + Al ones.
Based on above-mentioned calculations, 58% of carbon deposit is formed on Mg + Al species, while as much as 42% of carbon deposit is collected on much smaller amount of Mn species at 600 °C. At 700 °C, the majority of carbon deposit (75%) is formed on Mg + Al species, whereas the minority (25%) of carbon deposit is collected on Mn species. We do not have any other evidence to explain such observation in detail. Additional experiments are needed to verify this interesting effect, which may involve the influence of temperature on the crystallization of metal oxides and/or the reduction of MnxOy.
The physicochemical properties of the carbon materials prepared at 600 and 700 °C
The XRD patterns (Fig. 2) of the carbon materials display two broad reflections at 2θ ~ 24 and ~ 43°, and a shoulder at 2θ ~ 80° corresponding to the (002) diffraction plane of the graphitic layer-by-layer structure, the (101) and (112) diffraction planes for graphite, respectively. As clearly illustrated in Fig. S5a–d, the reflections are shifted towards lower angle compared with those in referenced graphite (PDF 04-014-0362) (Table S3). It indicates that the interlayer distance d in graphitic carbon is expanded (d002 ~ 0.358, 0.371, 0.375, 0.358 nm for the studied samples > d002 ~ 0.338 nm for graphite), which implies that graphitic carbon existing in the carbon materials is disordered. Besides low temperature of carbonization, i.e. 600 or 700 °C, the incorporation of N into carbon framework may induce disruption to the structure of graphite being responsible for buckling of the basal planes.

The XRD patterns of the carbon materials prepared at 600 and 700 °C via CVD with the compounds derived from Mg3Al LDHs and Mg2.6Mn0.4Al LDHs
Partial graphitization of the carbonaceous component can be also deduced from the C/H atomic ratio. In a single hydrogen-terminated graphene ribbon (one ring wide and infinite in length), the C/H atomic ratio equal 2 would be expected. The C/H atomic ratio increases with growing contribution of graphitic carbon. The data in Table 2 show that the C/H atomic ratio is slightly below 2 for the samples prepared at 600 °C or slightly above 2 for the samples prepared at 700 °C. It implies that the carbon materials prepared at 700 °C have slightly improved structural ordering compared with those prepared at 600 °C. EA determines the total concentration of elements, i.e. C and H, independently whether they come from graphitic or amorphous carbons. Knowing that the carbon materials produced by CVD always contain some amorphous carbon, elemental analysis should not be considered as the most reliable source of information on graphitization degree.
EA (Table 2) reveals that the carbon materials contain inorganic residue and the proportion of carbonaceous to non-carbonaceous components is roughly 7 to 3 and 8 to 2, in the samples prepared at 600 and 700 °C, respectively. It shows higher content of carbonaceous component in the carbon materials formed at 700 °C than at 600 °C.
According to XRD analysis (Fig. 2), inorganic residue consists of metal oxides and their derivatives, which are likely to be encased in carbon deposit. The reflection at 2θ ~ 18° in the XRD patterns of C-Mg3Al-600 and C-Mg3Al-700 may correspond to the (010) diffraction plane of Al(OH)3 (PDF 00-024-0006). Al(OH)3 is produced during acid treatment via hydrolysis of Al2O3 and HCl leading to the formation of AlCl3, which converts into aluminium hydroxide. The shoulders at 2θ ~ 37, ~ 45, ~ 59 and ~ 65° in the XRD pattern of C-Mg2.6Mn0.4Al-700 may correspond to the (311), (400), (511) and (440) diffraction planes of MgAl2O4 (PDF 00-021-1152).
XPS confirms that carbon dominates on the surface of C-Mg3Al-700 and C-Mg2.6Mn0.4Al-700 as its concentration reaches the values of 84.3 and 78.1 wt.% (Fig. 3a, Table 3), respectively.

The XPS survey spectra a of the carbon materials prepared at 700 °C via CVD with the compounds derived from Mg3Al LDHs or Mg2.6Mn0.4Al LDHs and the XPS spectra showing b C 1s, c N 1s, d O 1s and e Cl 2p signals
The C 1s core level spectra of C-Mg3Al-700 and C-Mg2.6Mn0.4Al-700 are displayed in Fig. 3b. Both C 1s signals consist of five peak components. The main peak component at binding energy of 284–285 eV can be assigned to the sp2–hybridized graphitic carbon (C=C) [27]. Its contribution to the total area of the C 1s peak is 49.2 and 62.0% for C-Mg3Al-700 and C-Mg2.6Mn0.4Al-700 indicating that approximately 50–60% of the surface carbon detected by XPS is represented by the sp2-hybridized graphitic units.
XPS also confirms the presence of N-containing groups incorporated into the carbon framework. The N 1s core level spectra of C-Mg3Al-700 and C-Mg2.6Mn0.4Al-700 are displayed in Fig. 3c. The N 1s signals consist of four peak components ascribed to pyridinic N (at 398–399 eV), pyrrolic N (at ~ 401 eV), quaternary N (at 403–404 eV) and oxidized N (at 406–407 eV) [42].
According to the literature [43], the pyrrolic N and pyridinic N always have nitrogen located on the graphitic edge, while quaternary N can be both ‘‘edge-N’’ and ‘‘bulk-like-N’’. The edge-N quaternary assignment is bonded to three carbons: one interior carbon and two edge carbons. Judging by the analysis of the XPS spectra, in particular, N 1s signal, N-doping mainly takes place on edges because the contribution of N-containing functional groups including pyrrolic N and pyridinic N is prominent (84.5 and 79.3%). In addition, quaternary N can also exist near the edge. It is consistent with the information prevailing in the literature that functional groups are preferably created at edge plane orientations of graphitic lattice.
Based on the calculations involving full geometry optimizations [44], incorporating nitrogen in a pentagon instead of in a hexagon in pentagon-containing hexagonally arranged carbon atoms results in lower total energy of the system. Thus, nitrogen atoms prefer nonplanar surrounding as in the curved structure around a pentagon. Our results are in agreement with such experimental data, among various N-containing functional groups, a majority of nitrogen (60.5 and 53.1%) is identified as pyrrolic N. Our previous experimental results reveal that pyrrolic N is also dominating in the carbon materials prepared at 600, 700 and 800 °C via CVD with the compounds derived from Mg–Co–Al LDHs [26].
The bulk N contents in C-Mg3Al-700 and C-Mg2.6Mn0.4Al-700 determined by EA (6.7 and 8.3 wt.%, respectively) are very similar to the surface N contents (7.3 and 9.0 wt.%, respectively) calculated from XPS analysis [45]. The values of N/C molar ratio obtained by both techniques are also alike (0.079 vs. 0.075 and 0.104 vs. 0.098, respectively). It implies that N is rather uniformly distributed throughout the N-doped carbon framework. In addition, it may indicate that nitrogen is rather located in graphitic carbon particles than in amorphous ones.
Besides N-containing groups also O-containing groups are present on the surface of synthesized carbon materials. The carbon materials are capable of chemisorbing oxygen with the formation of C–O and C=O surface oxides just by exposing to air at laboratory temperature. As a result of chemisorption of oxygen on the surface of the carbon materials O-containing function groups are formed.
The O 1s core level spectra for C-Mg3Al-700 and C-Mg2.6Mn0.4Al-700 are shown in Fig. 3d. The O 1s signals consist of five peak components such as O atoms connected to metal atoms (at ~ 531 eV), O atoms in –OH groups (at ~ 532 eV), O atoms in C=O and C–O–C groups (at ~ 534 eV), O atoms in carboxyl (COOH) and carbonate (CO32−) groups and in adsorbed water (H2O) (at ~ 535 eV) and O atoms in the oxygen molecules (O2) or bonded to aromatic carbon compounds (at ~ 538 eV) [27]. The major part of oxygen (77.5–79.2%) is associated with oxygen atoms incorporated in the carbon framework, whereas the minor part of oxygen (20.8–22.5%) originates from inorganic compounds, i.e. Al(OH)2 and MgAl2O4, covered with a relatively thin carbonaceous layers.
Taking into account that the surface oxygen concentration for both samples is comparable and equal 6.5 and 6.9 wt.% (Table 3) and that the contributions of O-containing functional groups (represented by peak components of O 2s signal at ~ 532 eV, ~ 533 eV and ~ 535 eV) (Fig. 3d) are almost constant and equal 73.9 and 73.8%, respectively, we assume that for the difference in the contributions of two peaks components of C 1s signal at ~ 286 eV (assigned to C–O, C–N) and at ~ 288 eV (assigned to C=O, C=N), is responsible nitrogen, for which the surface concentration increases from 7.3 up to 9.0 wt.%. Thus, increasing content of graphitic carbon (expressed by the contribution of C=C, 49.2 vs. 62.0% for C-Mg3Al-700 and C-Mg2.6Mn0.4Al-700, respectively) is accompanied by decreasing population of C–N (29.8 vs. 15.3%) but increasing population of C=N (7.0 vs. 10.8%), which is in agreement with reduced number of pyrrolic N (60.5 vs. 53.1%) and increased number of pyridinic N (24.0 vs. 26.2%) and quaternary N (9.7 vs. 14.0%). Reduced population of pyrrolic N (53.1%) indicates the reduced number of 5-membered rings (pentagon defects), so the graphene array resembles more that in graphitic carbon. Therefore, we observe increase in the contribution of C=C, despite the fact that the population of nitrogen in hexagon positions increases from 33.7 up to 40.2% for C-Mg3Al-700 and C-Mg2.6Mn0.4Al-700, respectively.
XPS reveals the traces of aluminium or magnesium and aluminium on the surface of C-Mg3Al-700 or C-Mg2.6Mn0.4Al-700 (Fig. 3a, Table 3), respectively. Al is detected in C-Mg3Al-700 as the samples contains Al(OH)3 (evidenced by XRD), while Mg and Al are detected in C-Mg2.6Mn0.4Al-700 as the sample contains Mg–Al spinel (evidenced by XRD).
The carbon deposit obtained by CVD contains structural defects in graphitic lattice, such as pentagon or edge defects, which can act as adsorption sites for chlorine anions during acid treatment of the composites. As a consequence, chlorine is bonded to carbon. In addition, XPS identifies another chlorine surrounding, i.e. chlorine-metal (e.g. AlCl3). Figure 3e demonstrates the XPS spectra showing Cl 2p signal for C-Mg3Al-700-30 and C-Mg2.6Mn0.4Al-700, which consists of two components, i.e. Cl–C and Cl-metal.
It is well known in the literature that the carbon materials obtained by means of CVD method contain defects in the graphene layers, for example 5-membered rings. During carbonization process via pyrolysis of CH3CN, nitrogen atoms are incorporated into the carbon framework [26, 27]. Replacing carbon atoms with nitrogen atoms leads to the distortion in the graphene layers and as a consequence, the interlayer distance (d002) increases above 0.338 nm as mentioned earlier. As a result, the carbon materials synthesized by means of CVD method contain structural defects within graphene layers, also in their arrangement and ordering. In addition, carbon deposit obtained in low-temperature pyrolysis (like in our case: 600 and 700 °C) also contains some amorphous carbon, which does not show any structural ordering. Overall, the surface of the carbon materials is heterogenous as they are a mixture of various forms of carbonaceous components. Therefore, the information on carbonaceous component of the carbon materials provided by XPS is limited to ordered/disordered graphitic carbon. The studied carbon materials contain also amorphous carbon, which is XPS-silent species. Thus Raman spectroscopy is employed to verify the existence of ordered/disordered graphitic but also to estimate the contribution of amorphous carbon.
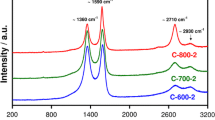
Two strong bands centred at about 1360 and 1590 cm−1 observed in the spectra of the carbon materials (Fig. 4) are commonly considered to be a combination of four Lorentzian-shaped bands (G, D1, D2 and D4) at about 1580, 1360, 1600 and 1190 cm−1, respectively, and a Gaussian-shaped band (D3) at 1500 cm−1. These bands can be attributed to an ideal graphitic lattice (G), graphene layer edges (D1), graphene layer surface (D2), amorphous carbon (D3) and other types of disordered graphitic lattice (D4) [46]. The area ratios, i.e. ID1/IG + ID1 + ID2; ID2/IG; ID3/IG; ID1/IG of various components (D1, D2, D3, D4 and G) of the Raman spectra (Fig. S6a–d), are presented in Table 4.

The Raman spectra of the carbon materials denoted as C-Mg3Al-600, C-Mg2.6Mn0.6Al-600, C-Mg3Al-700, C-Mg2.6Mn0.6Al-700
Raman spectroscopy supports XPS data, i.e. higher content of graphitic carbon for C-Mg2.6Mn0.4Al-700 than for C-Mg3Al-700, as graphitic domains are thicker for C-Mg2.6Mn0.4Al-700 than for C-Mg3Al-700 (ID2/IG ~ 0.87 vs. 1.09).
Raman spectroscopy reveals that the production of amorphous carbon (ID3/IG ~ 0.93 vs. 1.59 and 1.16 vs. 1.69) and the population of graphitic carbon particles exposing edge plane surfaces (ID1/IG ~ 4.37 vs. 4.93 and 4.23 vs. 4.68) increase with increasing CVD temperature. A mixture of metal oxides derived from Mg2.6Mn0.4Al LDHs in comparison with that derived from Mg3Al LDHs facilities the formation of amorphous carbon. C-Mg2.6Mn0.4Al-600 contains more amorphous carbon than C-Mg3Al-600 (ID3/IG ~ 1.16 vs. 0.93), but the number of structural defects is only slightly higher for C-Mg2.6Mn0.4Al-600 than for C-Mg3Al-600 (ID1/(IG + ID1 + ID2) ~ 0.70 vs. 0.69). Similar tendency is observed for the corresponding samples prepared at 700 °C (ID3/IG ~ 1.69 vs. 1.59, (ID1/(IG + ID1 + ID2) ~ 0.72 vs. 0.70).
Raman and EA data are in agreement with the information prevailing in the literature [47] that increased N-doping extent (7.4 vs. 4.2 wt.% and 8.3 vs. 6.7 wt.%) can be accompanied by increased contribution of disordered carbon (ID1/(IG + ID1 + ID2) ~ 0.70 vs. 0.69 and 0.72 vs. 0.70). It is observed that N content (4.2–8.3 wt.% determined by EA) in the carbon materials depends on the choice of CVD temperature and/or the type of LDHs. Higher temperature of carbonization and/or Mn-containing LDHs are the factors which facilitate incorporation of N (4.2, 6.7, 7.4 and 8.3 wt.%) into the carbon framework leading to increased number of defects (ID1/(IG + ID1 + ID2) ~ 0.69, 0.70, 0.70 and 0.72). More precise analysis of the XPS spectra reveals that the population of nitrogen in pentagon position decreases, whereas that in hexagon position increases. As a result, the thickness of graphitic domain increases (ID2/IG ~ 0.87 vs. 1.09 for Mg2.6Mn0.4Al-700 vs. C-Mg3Al-700, respectively).
Although a mixture of metal oxides derived from Mg2.6Mn0.4Al LDHs in comparison with that derived from Mg3Al LDHs facilities the formation of carbon deposit, the population of graphitic carbon particles exposing edge plane surface (ID1/IG ~ 4.23 vs. 4.37 and 4.68 vs. 4.93) decreases. The possible explanation for such tendency may be that the presence of defective/amorphous manganese oxides prefers the production of amorphous carbon to graphitic carbon.
N2 adsorption/desorption isotherms (Fig. S7a) of the carbon materials synthesized at 600 °C can be classified as type IV, characteristic of mesoporous materials [48]. The hysteresis loop with type H4 is clearly noticed for C-Mg2.6Mn0.4Al-600, which indicates that the carbon material consists of aggregates of platy particles possessing slit-shaped mesopores. The hysteresis loop is very narrow in the case of C-Mg3Al-600, which may be a consequence of small contribution of mesopores. The shape of N2 sorption isotherm of C-Mg3Al-600 suggests that during removal of the supports the carbon material can shrink due to low content of carbon deposit, i.e. 8.9 wt.% in the composite. Analogue situation can be observed for the carbon material (Fig. S8) prepared with the use of Mg3Al LDHs at 700 °C via CVD process lasting 30 min, which was extracted from the composite of low content of carbon deposit, i.e. 7 wt.%. Some pores can collapse after removal of the support especially when carbon structure is poorly interconnected. Similar behaviour was described in our previous publication for the carbon material prepared with the use of Mg2Al LDHs at 600 °C via CVD process lasting 180 min, which was extracted from the composite of low content of carbon deposit, i.e. 11 wt.% [28]. Shi et al. [38] also reported that when the ration of the support (metal oxides derived from Mg3Al LDHs via calcination at 700 °C for 120 min) to sucrose was lower than 1:1 the mesopores in the carbonaceous material collapsed and connected with each other to form holes during washing process because the sucrose amount was too little.
The pore size distribution (PSD) profiles (Fig. 5) shows that micro- and mesopores are present in both carbon materials obtained at 600 °C, but they differ in their contributions. The micropores strongly dominates in C-Mg3Al-600 reflecting in high value of Vmicro/Vmeso = 2.99 (Table 5). On the other hand, this ratio is only equal 0.89 in C-Mg2.6Mn0.4Al-600, thus showing comparable contributions of micro- and mesopores. The total pore volume is higher for C-Mg2.6Mn0.4Al-600 than for C-Mg3Al-600 by 1.7 times, which demonstrates that Mn-containing LDHs facilitates the formation of pores compared to Mg3Al LDHs.

Micro/mesopore size distribution calculated by means of QSDFT method using N2 adsorption/desorption data for the carbon materials prepared at 600 °C and 700 °C, via CVD with the compounds derived from Mg3Al LDHs and Mg2.6Mn0.4Al LDHs
N2 adsorption/desorption isotherms (Fig. S7b) of the carbon materials synthesized at 700 °C can be classified as type IV with type H2 (b) hysteresis loop [48], which indicates that they consist of aggregates of platy particles possessing slit-shaped mesopores with diverse pore diameters (e.g. narrow entrance and large pore body; bottleneck pore). The pore structure of mesopores existing in the samples prepared at 700 °C seems to be more complex than those in the samples prepared at 600 °C.
PSD curves (Fig. 5) illustrate that both samples obtained at 700 °C have higher total pore volume than those synthesized at 600 °C. The values of Vmicro/Vmeso are 0.14–0.21 only, showing a distinct predomination of mesoporous structures in both carbons obtained at 700 °C. It seems that the mesopores are especially grown at 700 °C, while micropores decreases compared to the samples synthesized at 600 °C.
Figure S9a, b demonstrates that the chemical composition of LDHs has no significant influence on the diameter of micropores present in the carbon materials, whereas it has profound impact on the formation of mesopores. The use of Mg2.6Mn0.4Al LDHs enlarges their mesopores (Table 5). The diameter of mesopores also depends on the choice of synthesis temperature.
The pore volumes for C-Mg3Al-700 and C-Mg2.6Mn0.4Al-700 are higher than those for the analogue carbon materials obtained at 600 °C (Table 5). The possible explanation for such observation may be as follows. At 700 °C CH3CN decomposition is quicker and as a result the concentration of decomposition products interacting with the surface of the supports is higher. Thus, increased population of “building blocks” affects not only the content of carbon deposit but also its properties. The carbon deposit formed at 700 °C contains more pores and seems to be more loose than the carbon deposit formed at 600 °C. Besides the concentration of carbonaceous species in CH3CN–Ar flow at 600 and 700 °C, the properties of the support, such as textural and hydrophobic, also may affect the amount of deposited carbon and its characteristics. It is worth to empathize that carbon deposit is collected not only on pure support particles but also on support particles coated with carbonaceous layers, which offer their own structural defects and hydrophobic state. Such circumstances are especially pronounced when the content of carbon deposit is relatively high, particularly in the composites obtained at 700 °C. Our experimental results support the previous results reported by other authors that the porosity of the obtained carbon materials depends on the proportion of carbon deposit to the support [49].
To sum up, the type of metal oxides derived from LDHs and CVD temperature affect the process of carbon deposition as well as the properties of the carbon materials such as chemical composition (N-doping), structural ordering and porous structure. There is no evidence of creating replicas. The formation of carbon deposit happens mostly on the external surface of the support grains. The choice of CVD temperature affects the porosity of the studied carbon materials, which are micro- and mesoporous. In particular, 600 °C facilitates the formation of the carbon materials exhibiting relatively high contribution of micropores, while 700 °C leads to the formation of the carbon materials, which are predominantly mesoporous. The carbon materials prepared at 700 °C with the compounds derived from Mg–Al LDHs and Mg–Mn–Al LDHs have bottleneck pores and characterized by wider mesopores in comparison with the corresponding samples prepared at 600 °C, which contains narrower mesopores with slit-like shape. The differences in the shape and diameter of mesopores may be responsible for the electrochemical performance of carbon materials for charge storage.
SEM micrographs of the carbon materials obtained at 600 and 700 °C using Mn-containing LDHs (Fig. S10a, b, e, f, Fig. S11a–d) as well as the analogous carbon samples derived from Mg3Al LDHs (Fig. S10c, d) have similar morphologies. They consist of irregular aggregates of plate-like particles.
Functional groups are incorporated into the graphitic matrix via edge or basal plane orientations. The edge orientation is more preferable for the functional groups to become incorporated into the graphitic lattice than the basal orientation [43]. The studied carbon materials consist of aggregates of platy particles showing both edge and basal orientations on the surface. However, they exhibit relatively high edge plane exposure providing the appropriate locations for oxygen to be covered by O-containing functional groups as evidenced by XPS.
The capacitive properties of the carbon materials prepared at 600 °C
The CV curves (Fig. S12a–d) illustrate the current response associated with charging/discharging the electrical double layer at the electrode/electrolyte interface. Since the studied samples consist mostly of carbon deposit (70.1–82.5 wt.%, Table 2), their ability to store charge is provided by electrically conductive and porous carbonaceous constituent. The electrical double layer arises from adsorption of positive ions from the electrolyte on the negatively charged electrode under the effect of the electric field imposed by the negative polarization at the potentials between − 0.3 and − 0.9 V. According to the literature, potassium ions (K+) in a hydrated form enter the pores of the electrode. The diameter of the solvated K+ in aqueous solution is estimated to be 0.26 nm [50]. The diameter of electrolyte ions determines the accessibility of the pores in the electrode materials. Chmiola et al. [51] described the ion penetration in the pores with diameters less than 1 nm and revealed the influence of the pore diameter on the capacitance. In the case of our samples, the smallest pores (the diameter of 0.6 nm, Fig. 5) are larger than twice of the diameter of the solvated K+ therefore the capacitance is unaffected by the micropore diameter. The ion sieving effect described in the literature [52] for ions too large to be accommodated into the pore system is not observed in our case.
The shape of the CV curves recorded for C-Mg3Al and C-Mg2.6Mn0.4Al deviates from rectangular mirror image revealing that ohmic resistance is involved in the non-faradaic process [53]. The observed deviation is larger for the samples prepared at 600 °C than that for those prepared at 700 °C, probably because the former are less porous than the latter (Table 5). In particular, the samples obtained at 600 °C contain lower volume of mesopores than the counterparts obtained at 700 °C (Table 5).
The capacitive behaviour of the carbon materials prepared at 600 °C is a consequence of distinctly higher ratio of micropores to mesopores, i.e. Vmicro/Vmeso = 2.99 and 0.89, for C-Mg3Al-600 and C-Mg2.6Mn0.4Al-600, respectively. The capacitances calculated using the current measured at the potential of − 0.6 V (for the potential sweep rate of 100 mV s−1) for these two carbon materials are lower than those for the corresponding samples prepared at 700 °C (Table 6). The same trend is also observed while comparing their corresponding specific capacitances (Table 7). It seems that the electrolyte can more easily penetrate the pore systems even at high potential sweep rate when the volume of mesopores is distinctly higher than that of micropores as it is observed for C-Mg3Al-700 and C-Mg2.6Mn0.4Al-700, for which Vmicro/Vmeso is equal 0.21 and 0.14, respectively. It is consistent with the opinion prevailing in other works, i.e. that mesopores mostly participate in the transport of electrolyte [54].
Figure 6a illustrates that electrochemical interface between electrode/electrolyte increases with decreasing potential sweep rate for both samples prepared at 600 °C. Such correlation was previously reported in the literature for micro-mesoporous carbon materials examined in 6 M KOH [55]. C-Mg3Al-600 and C-Mg2.6Mn0.4Al-600 reach their maximum capacitances, i.e. 166 and 197 F g−1, respectively, at 10 mV s−1. Knowing that CV measurements carried out under slow potential sweep rate facilitates the adsorption of the ions having diameters comparable with the pore diameters [56], we may suppose that only slightly better capacitive performance of C-Mg2.6Mn0.4Al-600 than that of C-Mg3Al-600 may be associated with only slightly higher volume of micropores (Table 5) in the former, which according to the literature [54, 57] can offer the adsorption sites for electrolyte ions. Our CV measurements are in agreement with the results published elsewhere [58] that the microporous structure hinders the access of electrolyte ions to the overall porosity.

Capacitive properties of the carbon materials prepared at 600 and 700 °C via CVD with the compounds derived from Mg3Al LDHs and Mg2.6Mn0.4Al LDHs, evaluated by CV measurements performed in the aqueous solution of Ar-bubbled 6 M KOH applying various potential sweep rates: capacitance calculated from the CV data using the current at the potential of − 0.6 V (a) and specific capacitance calculated by dividing the capacitance by the surface area (SDFT) (b). The electrode material loading was 0.1 mg (0.51 mg cm−2)
C-Mg2.6Mn0.4Al-600 has significantly higher volume of mesopores than C-Mg3Al-600 (Table 5), which may serve as corridors for electrolyte transport. Figure S9b reveals that C-Mg2.6Mn0.4Al-600 have larger mesopores compared to C-Mg3Al-600, which additionally may facilitate electrolyte mobility in porous electrode material. Despite differences in mesoporosity, the difference in the capacitive behaviour of C-Mg3Al-600 and C-Mg2.6Mn0.4Al-600 (166 vs. 197 F g−1 at 10 mV s−1) is not significant. The capacitive performances are similar, especially their specific capacitances, i.e. 25 and 22 μF cm−2, even though their textural properties, i.e. the contribution of mesopores, are not alike. It shows that not all pores, which participate in N2 sorption measurements, are accessible for the electrolyte ions in CV measurements. It also suggests that even accessible pores may not efficiently contribute to the electrochemical interface. In particular, not all adsorption sites offered by micropores are involved in the formation of electrical double layer. In addition, mesopores not only facilitate electrolyte transport but also they offer adsorption sites located on their walls.
Based on the observations mentioned above, both carbon materials synthesized at 600 °C using Mn-free and Mn-containing LDHs exhibit similar capacitive properties because the volume of micropores (0.338 vs. 0.364 cm3 g−1) and their surface area (587 vs. 648 m2 g−1) are comparable. The role of mesopores (0.113 vs. 0.407 cm3 g−1) seems to be minor due to the distinct contribution of micropores (Vmicro/Vmeso = 2.99 and 0.89). Reaching the maximum value of the capacitances for both carbon materials prepared at 600 °C is hindered. It suggests that micropores are directly involved in the charge accumulation process.
The capacitive properties of the carbon materials prepared at 700 °C
In contrast to the samples prepared at 600 °C, the capacitances measured at 100 mV s−1 for C-Mg3Al-700 and C-Mg2.6Mn0.4Al-700 are the maximum values as the capacitance decreases with decreasing potential sweep rate (Table 6). This capacitive behaviour is a consequence of distinctly higher proportion of mesopores to micropores, i.e. Vmicro/Vmeso = 0.21 and 0.14, respectively.
The pore size distribution profiles (Fig. S9b) show that both samples prepared at 700 °C have a broad distribution of mesopore width. It looks that the formation of larger mesopores is rather the matter of synthesis temperature than the presence of Mn in the LDHs matrix. However, the maximum of the curve obtained for C-Mg2.6Mn0.4Al-700 is slightly shifted towards higher values of pore width. The PSD curve (Fig. S13) for C-Mg3Al-700 is asymmetric with one maximum at 39 Å, while in the case of C-Mg2.6Mn0.4Al-700 PSD is clearly bimodal with the maxima at 39 and 49 Å.
Migration of ions is easier in the carbon materials prepared at 700 °C in comparison with the ones prepared at 600 °C because the access to the micropores is facilitated by high volume of mesopores (Table 5). We suppose that the population of K+ electroadsorbed at 100 mV s−1 is higher for the samples prepared at 700 °C than for the samples prepared at 600 °C, probably because the former offer larger number of accessible adsorption sites. The amount of K+ attracted by negatively polarized surface of the carbon materials at 10 mV s−1 is lower for the samples prepared at 700 °C than for the samples prepared at 600 °C, probably because the pores in the samples prepared at 700 °C, which are explored during CV measurements carried out at slow potential sweep rate, are too wide for the effective interaction between K+ and the pore walls, as previously reported for micro-mesoporous carbon materials examined in organic medium [59]. Therefore, a large fraction of the pore volume is underused.
Similarly like in the carbon materials prepared at 600 °C, the use of Mn-containing LDHs instead of Mn-free LDHs does not affect the capacitive properties of the carbon materials prepared at 700 °C, even though the volume of micropores and their surface area are distinctly different. It is against our expectations especially that the results for the carbon materials prepared at 600 °C suggest that micropores are directly involved in charge storage. Perhaps the different volumes of micropores may not affect the process of charge storage as the ratios of micropore to mesopore volumes are relatively low and there are other adsorption sites associated with mesopores.
Figure 6b demonstrates that the ability to accumulate the charge increases with decreasing potential sweep rate for the carbon materials prepared at 600 °C, whereas the performance of the corresponding samples prepared at 700 °C is opposite. We do not know the real reason of such behaviour. Therefore, we cannot explain precisely its mechanism. We suggest that such abnormal tendency based on CV experiments may be associated with the presence of mesopores of specific geometry—bottleneck pores, identified by means of N2 sorption measurements in both carbon materials prepared at 700 °C.
In the case of the carbon materials prepared at 600 °C having narrow slit-shaped mesopores, the capability for charge storage increases with decreasing potential sweep rate, because electrolyte ions are able not only to penetrate more deeply carbon particles but also they can more efficiently use the surface of the carbon materials for charge accumulation as they can reach the adsorption sites, for example located in micropores.
In the case of the carbon materials prepared at 700 °C having bottleneck mesopores, the capability for charge storage decreases with decreasing potential sweep rate, which is in contrast to the samples prepared at 600 °C. Electrolyte ions can penetrate more deeply carbon particles but they are not able to use efficiently the surface of carbon materials for charge storage because they do not approach the adsorption sites, which could participate in the formation of electrical double layer at electrode/electrolyte interface.
According to the paper [60] on the influence of pore shape on charge redistribution, during charging of a pore containing a bottleneck, essentially only the pore area above the bottleneck participates in the charging process, with the pore area below the bottleneck accumulating only a small amount of charge during charging of the electrode, and this significantly decreases the overall amount of charge within a pore during charging.
Our experimental results are consistent with the above-mentioned observation published in the literature. It is worth to highlight that both the diameter of pore mouth and diameter of pore body are in the mesoporosity range (2 nm < pore diameter < 50 nm). We suppose that electrolyte ions first penetrate the narrow entrance of the pore (pore mouth) and then the wide pore body. So the increasing amount of electrolyte ions entering the pore during charging, especially at slow potential sweep rate, does not lead to effective interaction between electrolyte ions with the surface of mesopores. As a consequence the capacitance of the samples decreases with decreasing potential sweep rate despite the fact that more electrolyte ions can enter the interior of the electrolyte materials. It seems that electrolyte ions entering the interior of the carbon particles do not approach adsorption sites because deeper parts of the electrode material are still built of mesopores (pores with large diameters).
In our previous paper [25], we characterized capacitive properties of the carbon material produced by CVD with the compounds derived from Mg2Al LDHs by means of CV and galvanostatic charging/discharging (GCD). The values of capacitances determined by CV (applying potential sweep rates: 5, 10, 20, 50 and 100 mV s−1) were in the range of 125–134 F g−1, whereas those evaluated by GCD (using currents: 10, 20, 50 and 100 µA) were slightly higher, i.e. 156–208 F g−1, than those basing on CV data. The differences were more pronounced when low currents such as 10 or 20 µA were applied. Galvanostatic charging/discharging, in particular using low currents, lasts longer than the CV measurements; therefore, it may both generate and/or detect additional small currents (residual faradaic current) insignificant in CV. In addition, the periodic course of current in the galvanostatic experiments may be also approximated by a sinusoid. However, when low current is used in the galvanostatic experiments the equivalent frequency is very low, much lower than corresponding frequency for the CV experiments. As the capacitance increases with the decreasing frequency the capacitance measured in GCD experiments must be higher. Learning from our previous experiments we can expect that the capacitances of the carbon materials studied in this work determined by GCD would be higher than those already determined by CV.
We estimate the surface area occupied by K+ during CV measurement. The calculations are based on the maximum values of capacitances, i.e. measured either at 10 or 100 mV s−1 for the carbon materials prepared at 600 or 700 °C, respectively. The calculations take into account the surface area of micro- and mesopores (SDFT). In order to do this we convert the experimental values (charge) into the number of K+ and then into their surface area. We adopt the surface area per K+ (5.3 × 10–20 m2) to be the surface area of a circle of radius, which can be derived from the van der Waals volume. The surface area of K+ involved in the formation of electrical double layer in the pores is lower than the surface area of the pores (SDFT) and it is 8.4, 7.4, 6.5 and 7.1% of the nominal value (Table 8). It shows that only small part of the surface area (determined by N2 sorption measurements) acts as the electrochemical interface.
The difference between the surface area available for N2 and the surface area occupied by K+ during CV measurement seems to be a consequence of (a) the surface of the carbon material may not be completely covered with K+-containing solution, (b) not all K+ being present in the pores may participate in the electrochemical interface as the contact of electrolyte with electrode surface is necessary but insufficient for electrostatic interaction, (c) due to electrostatic repulsion of K+ in the solution of high KOH concentration (6 M) the accumulation of K+ near the surface of the carbon materials may be weak and/or the concentration of K+ within the pore body may be lower than outside the pores. (The location of K+ near the pore entrance may prevent other K+ entering the pores.)
The above-mentioned calculations demonstrate that the microporous samples prepared at 600 °C have a similar potential for the utilization of K+ for charge storage as the mesoporous samples prepared at 700 °C.
Feng et al. [61] computationally demonstrated that depending on micropore width, K+ form either a well-hydrated single layer in the centre of negatively charged slit-shaped pores or separate layers near each slit wall. It suggests that the calculated number of K+ accumulated after immersing in 6 M KOH solution is probably underestimated because we do not take into account the region of single cation layer between the pore walls. In addition, we take into account all the mesopores, also with larger diameters, which may be useless for charge storage as they do not interact sufficiently with hydrated K+.
According to the literature [9, 62], N-doping of the carbon materials may improve their abilities for charge storage. The presence of N in the carbon framework is responsible for the creation of the structural defects, which can serve as adsorption centres. Besides structural defects, there are other defects induced by incorporation of N in the carbon framework, i.e. charge delocalization between N and C, as N is more electronegative than C. In addition, functional N-containing groups may be responsible for the interaction with metal ions, i.e. K+. Theoretical investigations show that pyrrolic N and pyridinic N have a positive influence, whereas the role of quaternary N is ambiguous [63,64,65,66].
Both carbon materials derived from Mn-containing LDHs, i.e. C-Mg2.6Mn0.4Al-600 and C-Mg2.6Mn0.4Al-700, have higher N/C molar ratios (0.108 and 0.104, respectively) than their counterparts derived from Mn-free LDHs, i.e. C-Mg3Al-600 and C-Mg3Al-700, (N/C = 0.056 and 0.079, respectively), but their maximum specific capacitances are either slightly lower for the samples prepared at 600 °C or slightly higher for the samples prepared at 700 °C compared to their counterparts containing less nitrogen (Table 7).
Although we do not observe correlation between the capacitive properties of the studied samples and the concentration of N in their structure like other authors [65], the influence of N-doping on electrochemical performance cannot be excluded as we prove that C-Mg3Al-700 and C-Mg2.6Mn0.4Al-700 contain N-containing functional groups (Fig. 3c) being claimed to give an advantage for the accumulation of charge also via an improved wettability of the carbon surface by the electrolyte solution.
The comparison of the capacitive performance of the studied materials with the literature data for other porous carbon materials is shown in Table S4. It is difficult to make a clear comparison because other authors used various carbon/support precursors and they applied different synthesis conditions, which had impact on structure, morphology, porosity and surface properties of those carbon materials and consequently on their ability to store the charge. The specific capacitance of the samples derived from CH3CN and (Mg–Al and Mg–Mn–Al) LDHs studied in this work is comparable to that (10–20 μF cm−2) for other carbon materials described in the literature.
Conclusions
Our investigations reveal that a mixture of metal oxides derived from Mg–Mn–Al LDHs compared to that derived from Mg–Al LDHs leads to higher carbon deposit yield. In particular, more amorphous carbon is produced. The possible explanation for such tendency may be the presence of additional structural defects in thermally treated Mg–Mn–Al LDHs due to the existence of defective/amorphous manganese oxides, which provide “nucleation sites” participating in the growth of carbon deposit.
Judging by the analysis of the SEM images, the carbon materials obtained with addition of Mn into Mg–Al LDHs still consist of plate-like particles like the corresponding samples obtained with the compounds derived from Mg–Al LDHs. However, Raman spectroscopy informs that Mn-containing LDHs leads to higher amounts of amorphous carbon and lower contribution of graphitic carbon particles exposing edge plane surfaces, which suggests that the population of platy carbon materials may decrease.
The added value of using Mn-containing LDHs is that the addition of Mn into Mg–Al LDHs matrix results in increased N-doping of carbon particles. The highest concentration of N (N/C = 0.108) is observed for the carbon material prepared at 600 °C with the compounds derived from Mg–Mn–Al LDHs, while the lowest concentration of N (N/C = 0.056) occurs in the carbon material prepared at 600 °C with the compounds derived from Mg–Al LDHs. XPS and Raman spectroscopies prove that the concentration of nitrogen in the carbon materials obtained with addition of Mn into Mg–Al LDHs increases in graphitic carbon. N/C determined by XPS and EA are similar, which implies that nitrogen is rather distributed throughout graphitic carbon particles than in amorphous forms.
The carbon materials prepared via CVD with the compounds derived from LDHs differ not only in the total pore volume and the contribution of micropores and mesopores but also in geometry of mesopores. In particular, the samples obtained at 600 °C contain slit-shaped mesopores, while those obtained at 700 °C contain bottleneck mesopores.
The integration of the results obtained by means of N2 sorption and CV applying various potential sweep rates demonstrates how the porosity of carbon materials affect their ability to store the charge. The maximum values of measured capacitances, i.e. 166, 197, 180 and 148 F g−1, for the carbon materials produced by CVD at 600 and 700 °C with the compounds derived from Mg–Al and Mg–Mn–Al LDHs, respectively, are comparable. The only differences observed for the studied samples are associated with the formation of electrical double layer at electrode/electrolyte interface, which is easier for the carbon materials prepared at 700 °C than for the materials prepared at 600 °C. In the case of the former, the charge is stored in the shallow parts of the carbon particles via the adsorption sites available for the electrolyte ions before passing through bottleneck mesopores. In the case of the latter, the charge is mainly stored in the deep parts of the carbon particles via the adsorption sites, which are mostly accessible for the electrolyte ions after their penetration of narrow slit-shaped mesopores.
In addition, the estimations based on N2 sorption and CV measurements indicate that only ca. 6–8% of specific surface area of N-doped carbon material is involved in charge storage using 6 M KOH.
References
Bandosz TJ, Putyera K, Jagiełło J, Schwarz JA (1993) Application of inverse gas chromatography to the study of the surface properties of modified layered minerals. Micropor Mater 1:73–79
Putyera K, Bandosz TJ, Jagiełło J, Schwarz JA (1994) Sorption properties of carbon composite materials formed from layered clay minerals. Clay Clay Miner 42:1–6
Putyera K, Bandosz TJ, Jagiełło J, Schwarz JA (1995) Hydrotalcite-like structures as molecular containers for preparation of microporous carbons. Appl Clay Sci 10:177–186
Putyera K, Bandosz TJ, Jagiełło J, Schwarz JA (1995) Micropore structure development in poly(sodium-4-styrenosulfonate) derived carbon. Carbon 33:1047–1052
Putyera K, Bandosz TJ, Jagiełło J, Schwarz JA (1996) Effect of template constraints on adsorption properties of synthetic carbons prepared within the gallery of layered double hydroxides. Carbon 34:1559–1567
Mostafa Moujahid E, Besse J-P, Leroux F (2003) Poly(styrene sulfonate) layered double hydroxide nanocomposites. Stability and subsequent structural transformation with changes in temperature. J Mater Chem 13:258–264
Leroux F, Taviot-Guého C (2005) Fine tuning between organic and inorganic host structure: new trends in layered double hydroxide hybrid assemblies. J Mater Chem 15:3628–3642
Leroux F, Raymundo-Piñero E, Nedelec J-M, Béguin F (2006) Textural and electrochemical properties of carbon replica obtained from styryl organo-modified layered double hydroxide. J Mater Chem 16:2074–2081
Stimpfling T, Leroux F (2010) Supercapacitor-type behavior of carbon composite and replica obtained from hybrid layered double hydroxide active container. Chem Mater 22:974–987
Stimpfling T, Leroux F (2010) Carbon composites and replicas from intercalated layered double hydroxides. Appl Clay Sci 50:367–375
Li Y, Chen J, Chang L (1997) Catalytic growth of carbon fibers from methane on a nickel-alumina composite catalyst prepared from Feitknecht compound precursor. Appl Catal A: Gen 163:45–57
Shaijumon MM, Bejoy N, Ramaprabhu S (2005) Catalytic growth of carbon nanotubes over Ni/Cr hydrotalcite-type anionic clay and their hydrogen storage properties. Appl Surf Sci 242:192–198
Zhao Y, Jiao Q, Li C, Liang J (2007) Catalytic synthesis of carbon nanostructures using layered double hydroxides as catalyst precursors. Carbon 42:2159–2163
Lv D (2022) Layered double hydroxides functionalized by carbonaceous materials: from preparation to energy and environmental applications. Environ Sci Pollut Res
Pacuła A, Ikeda K, Masuda T, Uosaki K (2012) Examination of the electroactive composites containing cobalt nanoclusters and nitrogen-doped nanostructured carbon as electrocatalysts for oxygen reduction reaction. J Power Sources 220:20–30
Pacuła A, Socha RP, Zimowska M, Ruggiero-Mikołajczyk M, Mucha D, Nowak P (2013) Application of as-synthesized Co-Al layered double hydroxides for the preparation of the electroactive composites containing N-doped carbon nanotubes. Appl Clay Sci 72:163–174
Wang H-F, Tang C, Zhu X, Zhang Q (2016) A ‘point-line-point’ hybrid electrocatalyst for bi-functional catalysis of oxygen evolution and reduction reactions. J Mater Chem A 4:3379–3385
Shi J-L, Peng H-J, Zhu L, Zhu W, Zhang Q (2015) Template growth of porous graphene microspheres on layered double oxide catalysts and their applications in lithium-sulfur batteries. Carbon 92:96–105
Shi J-L, Tian G-L, Zhang Q, Zhao M-Q, Wei F (2015) Customized casting of unstacked graphene with high surface area (> 1300 m2g–1) and its application in oxygen reduction reaction. Carbon 93:702–712
Deck CP, Vecchio K (2006) Prediction of carbon nanotube growth success by the analysis of carbon-catalyst binary phase diagrams. Carbon 44:267–275
Xiang X, Zhang L, Hima HI, Li F, Evans DG (2009) Co-based catalysts from Co/Fe/Al layered double hydroxides for preparation of carbon nanotubes. Appl Clay Sci 42:405–409
Cao Y, Zhao Y, Jiao Q (2010) Fe-based catalysts from Mg/Fe layered double hydroxides for preparation of N-doped carbon nanotubes. Mater Chem Phys 122:612–616
Wang C, Yan B, Zheng J, Feng L, Chen Z, Zhang Q, Liao T, Chen J, Jiang S, Du C, He S (2022) Recent progress in template-assisted synthesis of porous carbons for supercapacitors. Adv Powder Mater 1:100018
Pacuła A, Nowak P, Socha RP, Ruggiero-Mikołajczyk M, Mucha D, Bielańska E (2013) Preparation and characterization of the electroactive composites containing nickel nanoparticles and carbon nanotubes. Electrochim Acta 90:563–572
Pacuła A, Nowak P, Makowski W, Socha RP (2015) The influence of layered double hydroxide composition on the morphology, porosity and capacitive properties of nitrogen-doped carbon materials prepared via chemical vapour decomposition. Mesoporous Mesoporous Mater 201:1–9
Pacuła A, Uosaki K, Socha RP, Bielańska E, Pietrzyk P, Zimowska M (2016) Nitrogen-doped carbon materials derived from acetonitrile and Mg-Co-Al layered double hydroxides as electrocatalysts for oxygen reduction reaction. Electrochim Acta 212:47–58
Pacuła A, Socha RP, Pietrzyk P, Zimowska M, Ruggiero-Mikołaczyk M, Mucha D, Kosydar R, Mordarski G (2018) Physicochemical and electrochemical properties of the carbon materials containing nitrogen and cobalt derived from acetonitrile and Co-Al layered double hydroxides. J Mater Sci 53(16):11292–11314. https://doi.org/10.1007/s10853-018-2427-9
Pacuła A, Mokaya R (2007) Layered double hydroxides as templates for nanocasting porous N-doped graphitic carbons via chemical vapour deposition. Mesoporous Mesoporous Mater 106:147–154
Fernández JM, Barriga C, Ulibarri M-A, Labajos FM, Rives V (1994) Preparation and thermal stability of manganese-containing hydrotalcite, [Mg0.75MnII0.04MnIII0.21(OH)2](CO3)0.11·nH2O. J Mater Chem 4:1117–1121
Velu S, Shah N, Jyothi TM, Sivasanker S (1999) Effect of manganese substitution on the physicochemical properties and catalytic toluene oxidation activities of Mg–Al layered double hydroxides. Mesoporous Mesoporous Mater 33:61–75
Şeşen FE (2017) Practical reduction of manganese oxide. J Chem Tech App 1(1):1–2
Welham NJ (2002) Activation of the carbothermic reduction of manganese ore. Int J Miner Process 67:187–198
Liu B, Zhang Y, Su Z, Peng Z, Li G, Jiang T (2017) Thermodynamic analysis of reduction of MnO2 by methane-hydrogen gas mixture. JOM 69:1669–1675
Wang C, Xu X, Ma X, Chen R, Liu B, Du Y, Zeng Z, Li L (2020) Insight into an invisible active site presented in the low temperature pyrolysis of toluene by CoMn spinel catalyst. J Catal 391:111–122
Wang Y, Qu Q, Gao S, Tang G, Liu K, He S, Huang S (2019) Biomass derived carbon as binder-free electrode materials for supercapacitors. Carbon 155:706–726
Inagaki M, Kobayashi S, Kojin F, Tanaka N, Morishita T, Tryba B (2004) Pore structure of carbons coated on ceramic particles. Carbon 42:3153–3158
Morishita T, Soneda Y, Tsumura T, Inagaki M (2006) Preparation of porous carbons from thermoplastic precursors and their performance for electric double layer capacitors. Carbon 44:2360–2367
Shi L, Chen Y, Chen G, Wang Y, Chen Z, Song H (2017) Fabrication of hierarchical porous carbon microspheres using porous layered double oxide templates for high-performance lithium ion batteries. Carbon 123:186–192
Owens DK, Wendt RC (1969) Estimation of the surface free energy of polymers. J Appl Polym Sci 13:1741–1747
Rudawska A, Jacniacka E (2009) Analysis for determining surface free energy uncertainty by the Owen-Wendt method. Int J Adhes Adhes 29:451–457
Britt PF (2002) Pyrolysis and combustion of acetonitrile (CH3CN). Technical report ORNL/TM-2002/113
Feng L, Yan B, Zheng J, Chen J, Wei R, Jiang S, Yang W, Zhang Q, He S (2022) Soyabean protein-derived N, O co-doped porous carbon sheets for supercapacitor applications. New J Chem 46:10844–10853
Biddinger EJ, Ozkan US (2010) Role of graphitic edge plane exposure in carbon nanostructures for oxygen reduction reaction. J Phys Chem C 114:15306–15314
Sjöström H, Stafström S, Boman M, Sundgren J-E (1995) Superhard and elastic carbon nitride thin films having fullerenelike microstructure. Phys Rev Lett 75:1336–1339
Yang Z, Xia Y, Mokaya R (2005) Hollow shells of high surface area graphitic N-doped carbon composites nanocast using zeolite templates. Mesoporous Mesoporous Mater 86:69–80
Sadezky A, Muckenhuber H, Grothe H, Niessner R, Poschl U (2005) Raman microspectroscopy of soot and related carbonaceous materials: spectral analysis and structural information. Carbon 43:1731–1742
Charreteur F, Jaouen F, Ruggeri S, Dodelet J-P (2008) Fe/N/C non-precious catalysts for PEM fuel cells: influence of the structural parameters of pristine commercial carbon blacks on their activity for oxygen reduction. Electrochim Acta 53:2925–2938
Thommes M, Kaneko K, Neimark AV, Olivier JP, Rodriguez-Reinoso F, Rouquerol J, Sing KSW (2015) Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl Chem 87:1051–1069
Morishita T, Tsumura T, Toyoda M, Przepiórski J, Morawski AW, Konno H, Inagaki M (2010) A review of the control of pore structure in MgO-templated nanoporous carbons. Carbon 48:2690–2707
Torchała K, Kierzek K, Machnikowski J (2012) Capacitance behavior of KOH activated mesocarbon microbeads in different aqueous electrolytes. Electrochim Acta 86:260–267
Chmiola J, Yushin G, Gogotsi Y, Portet C, Simon P, Taberna PL (2006) Anomalous increase in carbon capacitance at pore sizes less than 1 nanometer. Science 313:1760–1763
Eliad L, Pollak E, Levy N, Salitra G, Soffer A, Aurbach D (2006) Assessing optimal pore-to-ion size relations in the design of porous poly(vinylidene chloride) carbons for EDL capacitors. Appl Phys A 82:607–613
Kado Y, Soneda Y (2019) Preparation of porous carbons by templating method using Mg hydroxide for supercapacitors. Mesoporous Mesoporous Mater 287:101–106
Hsieh W, Allen Horng T-L, Huang H-C, Teng H (2015) Facile simulation of carbon with wide pore size distribution for electric double-layer capacitance based on Helmholtz models. J Mater Chem A 3:16535–16543
Liu H, Zhang Y, Ke Q, Ho KH, Hu Y, Wang J (2013) Tuning the porous texture and specific surface area of nanoporous carbons for supercapacitor electrodes by adjusting the hydrothermal synthesis temperature. J Mater Chem A 1:12962–12970
Salitra G, Soffer A, Eliad L, Cohen Y, Aurbach D (2000) Carbon electrodes for double-layer capacitors I. Relations between ion and pore dimensions. J Electrochem Soc 147:2486–2493
Redondo E, Carretero-González J, Goikolea E, Ségalini J, Mysyk R (2015) Effect of pore texture on performance of activated carbon supercapacitor electrodes derived from olive pits. Electrochim Acta 160:178–184
Liu K-C, Anderson MA (1996) Porous nickel oxide/nickel films for electrochemical capacitors. J Electrochem Soc 143:124–130
Ania CO, Pernak J, Stefaniak F, Raymundo-Piñero E, Béguin F (2009) Polarization-induced distortion of ions in the pores of carbon electrodes for electrochemical capacitors. Carbon 47:3158–3166
Black JM, Andreas HA (2010) Pore shape affects spontaneous charge redistribution in small pores. J Phys Chem C 114:12030–12038
Feng G, Qiao R, Huang J, Sumpter BG, Meunier V (2010) Ion distribution in electrified micropores and its role in the anomalous enhancement of capacitance. ACS Nano 4:2382–2390
Wu K, Liu Q (2016) Nitrogen-doped mesoporous carbons for high performance supercapacitors. Appl Surf Sci 379:132–139
Chen C, Wang Z, Zhang B, Miao L, Cai J, Peng L, Huang Y, Jiang J, Huang Y, Zhang L, Xie J (2017) Nitrogen-rich hard carbon as a highly durable anode for high-power potassium-ion batteries. Energy Storage Mater 8:161–168
Xu Y, Zhang C, Zhou M, Fu Q, Zhao C, Wu M, Lei Y (2018) Highly nitrogen doped carbon nanofibers with superior rate capability and cyclability for potassium ion batteries. Nat Commun 9:1–11
Zhang W, Bu S, Yuan Q, Xu Q, Hu M (2019) Controllable nitrogen-doping of nanoporous carbons enabled by coordination frameworks. J Mater Chem A 7:647–656
Nazir G, Rehman A, Park S-J (2020) Sustainable N-doped hierarchical porous carbons as efficient CO2 adsorbents and high-performance supercapacitor electrodes. J CO2 Util 42:1013
Acknowledgements
This research received funding from National Science Centre in Poland within decision number DEC-2013/11/B/ST5/01417. This work was also supported by National Centre for Research and Development (NCBiR, Poland) agreement number EIG CONCERT-JAPAN/1/2019. Paweł Nowak, Paweł Weroński and Grzegorz Mordarski (Jerzy Haber Institute of Catalysis and Surface Chemistry, Polish Academy of Sciences) are gratefully acknowledged for fruitful discussions. This research paper is dedicated to the memory of Professor Alicja Drelinkiewicz (1951-2022), our dear co-worker and great teacher/mentor, who died while this paper was being peer-reviewed.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: Christopher Blanford.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pacuła, A., Drelinkiewicz, A., Ruggiero-Mikołajczyk, M. et al. N-doped carbon materials produced by CVD with the compounds derived from LDHs. J Mater Sci 57, 18298–18322 (2022). https://doi.org/10.1007/s10853-022-07760-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-022-07760-1