
Abstract
All inorganic lead-based perovskites containing bromine-iodine alloys, such as CsPbIBr2, have arisen as one of the most attractive candidates for absorber layers in solar cells. That said, there remains a large gap when it comes to film and crystal quality between the inorganic and hybrid perovskites. In this work, antisolvent engineering is employed as a simple and reproducible method for improving CsPbIBr2 thin films. We found that both the antisolvent used and the conditions under which it was applied have a measurable impact on both the quality and stability of the final product. We arrived at this conclusion by characterising the samples using scanning electron microscopy, X-ray diffraction, UV–visible and photoluminescence measurements, as well as employing a novel system to quantify stability. Our findings, and the application of our novel method for quantifying stability, demonstrate the ability to significantly enhance CsPbIBr2 samples, produced via a static one-step spin coating method, by applying isopropanol 10 s after commencing the spin programme. The antisolvent quenched CsPbIBr2 films demonstrate both improved crystallinity and an extended lifespan.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The Paris Agreement identified the development of solar power as a key to tackling climate change. Perovskites have emerged as a prime candidate for solar cells due to their low cost [1], impressive rise in power conversion efficiency (PCE) [2], and excellent optoelectronic properties [3,4,5,6,7,8]. Perovskites are crystal structures with the composition ABX3, where A and B are cations and X is an anion. The A-site can be occupied by organic molecules, such as MA [9], FA [10], and GA [11], or inorganic ones, such as caesium, giving rise to hybrid- and all-inorganic perovskites, respectively. The B-site cation is the most often occupied by lead, however, due to its toxic nature, tin-based perovskites have also been explored [12]. The anion site is occupied by a halide such as iodine, bromine, or chlorine. Since several different elements or molecules can occupy a given cation [10, 11, 13] or halide site [14, 15], many combinations are known to exist. Perovskites have low stability when compared to silicon, notably in the presence of humidity [9], but other factors have been shown to cause or catalyse degradation [16,17,18,19,20]. While inorganic perovskites have shown superior levels of stability [15, 21,22,23,24], there is still a need to extend their lifespans if they are to compete with silicon-based solar cells.
Of the inorganic mixed halide perovskites, CsPbIBr2 offers the optimum trade-off between stability and bandgap since the bromine content is proportional to both. CsPbIBr2 is especially well suited for use in tandem or multilayer devices due to its relatively wide bandgap of 2.05 eV [25, 26]. One of the limitations of CsPbIBr2-based devices is that their maximum PCE is just over 11% [27], which is around half of the materials Shockley-Quessier limit, and at a more fundamental level, some of the best CsPbIBr2 films reported in the literature have a high number of pinholes [22, 23, 25, 28, 29]. These limitations highlight the existence of a research gap. To date, the solutions explored include: surface passivation [30], Cl− doping [31], antisolvent quenching [30], light soaking [28], intermolecular exchange [23], precursor solution ageing [32], bandgap alignment [31], and doping with Rb+ and Mn2+ [21, 33]. Moreover, CsPbIBr2 films have been deposited via: one-step spin coating [30, 31, 34], a gas-assisted method [35], a spray-assisted method [25], a two-step spin coating method [21] and by dual-source thermal evaporation [15]. While antisolvent quenching has been shown to significantly improve a range of perovskite materials [36,37,38,39], we were surprised to note a lack of literature reporting its use with CsPbIBr2, with Liu et al. reporting its use in a vapour-assisted deposition [40] and only Zhang et al. reporting the use of antisolvents with a one-step spin coating procedure. In their paper, they applied six different antisolvents 20 s after commencing the spin-programme, finding diethyl ether to give the best results. Therefore, it is essential not only to confirm the results of Zhang et al.’s study and to expand upon them but also to establish antisolvent quenching as a trusted and simple method for improving CsPbIBr2.
Here, an antisolvent quenching method is reported which improves both crystal quality and extends the lifespan of CsPbIBr2 thin films. An antisolvent is a solvent, which cannot dissolve the components of a solution, in our case, the perovskite. The application of the antisolvent creates local regions of supersaturation, leading to accelerated heterogeneous nucleation [38]. It is by optimizing the rate of nucleation and crystal growth that a higher quality film is obtained. In general, when antisolvents are used, the concentration is kept within the “seeding zone” which is above the solubility curve but below the metastable limit. Concentrations in these ranges lead to the growth of already existent nuclei, or “seeds”, but do not cause the creation of new ones.
During our review of the literature, we found ourselves asking: could there be more factors that contribute to the quality of antisolvent quenched CsPbIBr2? If so, to what extent do different factors affect the measurable properties of CsPbIBr2? We identified two variables that likely affected the antisolvent quenching method: the antisolvent used and the time at which the antisolvent is applied during the programme (sometimes called dripping time). Following this, we formed the hypothesis that both antisolvent selection and application conditions would have an impact on the final sample. To test this, an experiment was designed where both antisolvent and dripping time were varied, and then the impact that it had on the properties and stability of CsPbIBr2 was measured. We expected, due to Zhang et al.’s paper, that we would find diethyl ether to be the best antisolvent, however, this turned out not to be the case. Moreover, we found the optimization of the dripping time to be a necessary step to maximize the effectiveness of the method.
Methodology
Materials
The precursor chemicals, namely caesium iodide (CsI) and lead bromide (PbBr2), were purchased from Tokyo Chemical Industry. The solvents dimethyl sulfoxide (DMSO 99.9%), isopropanol (IPA), diethyl ether (DEE), toluene (Tl), and chlorobenzene (ClBn) were all purchased from Sigma Aldrich and were used as received.
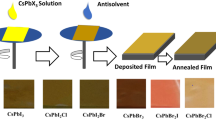
Film synthesis: First, fluorine-doped tin oxide (FTO) glass substrates were cleaned for 15 min in detergent, ethanol, acetone, and isopropanol, before being dried in a stream of compressed air. The substrates were treated by UV-Ozone for 15 min to remove any further impurities and to improve the surface wetness. The substrates were then transported into an inert argon atmosphere, where the samples were prepared and stored until use. A 1 M solution of CsPbIBr2 was prepared by dissolving equimolar ratios of CsI and PbBr2 in DMSO at 85 °C for 90 min. Once the solution was ready, 50 µL of yellow perovskite solution was statically spin-coated at 4000RPM for 50 s. 100 µL of antisolvent was applied after a given time had elapsed from the initiation of the spin programme (referred to as the dripping time). Samples were annealed for 5 min at 50 °C, during the course of which they changed to an orange-red. This initial colour change, signalling the formation of the perovskite structure occurring, happens because relatively little energy is required for caesium to begin intercalating between the PbBr2, due to its small ionic radius of 1.81 Å [41]. Samples are then heated slowly, avoiding thermal shocks, to 250 °C and held at this temperature for 5 min, removing any remaining solvent. During this stage of the annealing, the samples take on a dark-red appearance, confirming the complete formation of the desired perovskite phase. Figure 1 shows the procedure for synthesizing the CsPbIBr2 films.

The CsPbIBr2 solution that is applied to the substrate is yellow to begin with. The solution is spin-coated whilst the antisolvent is applied. The samples are then annealed during which a significant colour change takes place as the solution dries and the perovskite structure is formed
Characterisation techniques: Thin films of CsPbIBr2 were characterised by X-ray diffraction (XRD) using the RIGAKU Ultima IV with Cu kα radiation (λ = 1.5418 Å). Surface morphology was studied using the field emission scanning electron microscope (FESEM) within the Zeiss Auriga Compact with an applied voltage of 1.5 kV. The FESEM was equipped with a focused ion beam (FIB) which was used to make an incision in the surface and measure the film thickness. The Zeiss Auriga Compact was also equipped with the apparatus necessary to carry out electron dispersive spectroscopy (EDS), allowing the atomic proportions of the films to be determined. Optical properties were analysed with an Ocean Optics HR4000 spectrophotometer and photoluminescence (PL) emission was driven by a semiconductor laser with a wavelength of λ = 405 nm.
Experiment structure
A reference sample with no antisolvent was made along with four other samples where Tl, IPA, DEE, and ClBn were used. The dripping time for all samples was 8 s. These samples were characterised by XRD, UV–vis, PL, FESEM and EDS. In the second stage of the experiment, the dripping time of the best sample, IPA, was changed to 5, 10, 15, and 20 s, respectively. The samples were then characterised by XRD, UV–vis, PL, and FESEM. In the final stage of the experiment, stability was quantified in terms of XRD and UV–vis spectra when the samples were 5 weeks old. For this degradation study, all the samples used in the antisolvent selection and dripping time investigation were stored in the laboratory, at 23 °C and with a relative humidity of 20%, for 5 weeks. These conditions were measured daily and only small fluctuations in temperature ± 2 °C were seen to take place. The relative humidity remained constant throughout the experiment except for a few days when it increased to 40% due to heavy precipitation outside. All samples were exposed to the same conditions allowing for a fair comparison between them.
Results
The results section is broken down into four parts: The antisolvent selection, the effect of dripping time, the impact on stability, and then several solar cells are simulated using the parameters of the optimized CsPbIBr2 film.
Antisolvent selection
To determine the effect of antisolvent selection on samples of CsPbIBr2, four different antisolvents were applied under identical conditions, as described in the previous section. These samples were compared with a reference sample that had no antisolvent applied.
Once the XRD spectra of the fresh samples had been collected, a Rietveld refinement was carried out to determine the crystal structure. We found the same parameters as other authors, namely, a cubic structure, pertaining to the PM-3 M space group, with a lattice constant of 5.93423 ± 0.00034 Å [42, 43]. All samples showed two major diffraction peaks, corresponding to the (100) and (200) planes, at 15° and 30°, respectively. Smaller peaks found at 21°, 26° and 37° correspond to the (110), (111) and (211) planes. The calculated Miller indices can be found below in Table 1. These results are based upon an analysis of the reference sample, in which the fit obtained with the refinement was highly precise with the RWP and RP values being 7.1 and 4.72%, respectively. Rietveld refinements were not carried out for all the samples, since their spectra were highly similar, differing only in intensity, meaning that they were unlikely to generate different results.
When comparing the diffraction intensity, the sample in which IPA was used showed an almost two-fold increase when compared to the reference sample, shown in Fig. 2a and b, implying a major increase in the diffraction planes associated with the perovskite structure. The area under the diffraction peaks, which corresponds roughly to the quantity of perovskite material present, gave similar results. The rest of the samples showed a decrease in peak intensity, implying a worsening of the crystal structure as a result of the antisolvent application.

A summary of the results collected for the antisolvent selection. The antisolvents used were IPA, DEE, Tl and ClBn. These results are compared with a reference sample with no antisolvent. a XRD spectra of the samples b peak intensity corresponding to the two main diffraction planes c crystallite size d absorbance spectra e photoluminescence spectra with an integration time of 16 s f time evolution of the PL spectra of the reference sample
The broadening of diffraction peaks is known to be related to the size of the diffraction regions in crystals. Employing the Scherrer equation, it is possible to calculate the crystallite size based on the full-width at half-maximum (FWHM). It should be noted that although the terms “crystallite” and “grain” are often used interchangeably, there are several definitions for the term crystallite. In our case, we define a crystallite as a region in which all the material is orientated in a particular diffractive direction. This is important because it means that a grain can be composed of several crystallites. For this reason, the grains observed using the FESEM may not always coincide with the crystallite sizes calculated from XRD measurements.
In Fig. 2c, it is possible to see that the application of IPA leads to a significant increase in crystallite sizes from around 45 nm in the reference sample to around 55 nm, which could be due to an increased period within the metastable zone and in turn a prolonged period of crystal growth. This increase in crystallite size is important because crystallite and grain boundaries can contribute to charge recombinations. Since an increase in size implies a reduction in the number of these boundaries, this film should yield a higher PCE. Interestingly, while the IPA, the reference, and the ClBn samples had comparable crystallite sizes for both the (100) and (200) diffraction peaks, the other samples showed a divergence, meaning that crystallite sizes in the material were nonuniform.
To characterise the optical properties of the samples, measurements of absorbance and photoluminescence were made. The absorption onset of the samples took place around 600 nm, which agrees with the accepted bandgap value [15, 25, 31, 42]. All samples showed a higher absorbance than the reference sample, with the IPA and DEE samples giving similar results. While ClBn and Tl seem to have increased the absorbance of the samples, they also demonstrate a shallower climb in absorbance, potentially signalling the existence of interband defects. While absorbance measurements are a key tool for understanding the band structure and crystal quality, it should also be noted that the absorption coefficient gives more information since it is also a function of thickness and reflectivity. Any deviations in thickness will create an associated error in the absorbance measurement. This limits our ability to identify the superior samples based solely on its absorbance spectrum. On the other hand, all samples show an increase in absorbance at the accepted wavelength value for the CsPbIBr2 bandgap, signalling that the material has been correctly formed.
As can be seen in Fig. 2f, measurement of the PL spectra revealed that all samples showed a time-dependent spectrum when under illumination, leading to the apparition of a lower energy peak. Moreover, a given spectrum would “reset” if the sample were stored in dark conditions. These observations are consistent with observations made by other authors and with the so-called “Hoke Effect” [44,45,46]. While more detailed explanations can be found in the paper by Hoke et al., the evolution of a PL spectra in a mixed halide perovskite under illumination can be explained by the segregation of different halide phases causing iodine-rich regions to be formed, which in turn are responsible for the lower energy emission.
PL measurements carried out when the sample is initially illuminated, shown in Fig. 2e, give a peak in emission at around 610 nm, which agrees with the established bandgap value of CsPbIBr2 [15, 25, 31, 42]. All samples show a low-energy tail, probably the initial signs of the Hoke Effect. The sample in which Tl was applied gave the highest emission, which is the same result as was found by Zhang et al. The IPA and the reference sample gave very similar PL spectra, suggesting that the antisolvent had a small effect on the band structure.
Figure 3 shows the FESEM images that were taken of the surface. Both the ClBn and Tl samples were nonuniform and had large-scale defects compared with the DEE, IPA and reference samples which showed smooth and uniform surfaces. When magnified 20,000 times, all samples showed pin-hole free regions, demonstrating that film coverage at higher magnifications can be misleading. In general, the DEE, IPA and reference samples were difficult to distinguish; however, the reference sample had slightly fewer defined grains. From the FESEM images alone, it was difficult to distinguish the best sample, however, ClBn and Tl clearly had a negative effect on surface homogeneity.

FESEM images taken of CsPbIBr2, with different antisolvents applied, at magnifications of 2000- and 20,000-times with an accelerating potential of 1.5 keV. Each sample has a large-scale image (above) and a magnified image (below) to see the grain structure
EDS was carried out in the FESEM by increasing the accelerating potential to 15 keV. Table 2 summarises the results. All the EDS spectra gave the correct atomic percentages for the composition CsPbIBr2. The samples showed a slight surplus of iodine and a deficit of bromine; this is beneficial since a higher iodine content will lower the bandgap and lead to an increased Shockley-Quessier limit. Using the FIB inside the FESEM chamber, an incision was made on the surface of the samples and their thickness was measured. The mean value for thickness was 296 nm with a standard deviation of 28 nm.
Dripping time
To ascertain the effect, if any, that the dripping time would have on the finished sample, four samples of CsPbIBr2 were treated with IPA as an antisolvent. IPA was chosen out of the antisolvents as it gave the best results all-round, especially in terms of XRD. The dripping time was increased in steps of 5 s due to the difficulty in carrying out precise time-wise application of antisolvents by hand.
The FESEM revealed that CsPbIBr2 samples treated with IPA over a range of different dripping times, from 5 to 20 s, were difficult to distinguish at magnifications of around 2000 times. However, at very low magnifications of around 24 times, the 5 and 20 s samples showed nonuniformity. Specifically, the 5 s sample showed a patchy structure which could be due to the solution not having enough time to spread out before the antisolvent application. On the other hand, the 20 s sample showed large agglomerations of perovskite material sitting on top of the surface. In Fig. 4, it is possible to see that although the 5 s sample was nonuniform, it had regions with good grain formation at higher magnifications. This shows that often several images are needed to characterise the nature of the surface. The other samples showed increased film coverage, with the 10 s sample having the best-defined grains and the fewest pinholes.

FESEM images of CsPbIBr2 thin films treated with IPA over a range of different dripping times, taken at magnifications of 2000- and 20,000-times with an accelerating potential of 1.5 keV. Each sample has a large-scale image (above) and a magnified image (below) to see the grain structure
In contrast to the FESEM analysis, the XRD results revealed the 10 s sample to be the best one, since it had an intensity far superior to any other. In fact, in Fig. 5b we can see that there seems to be a parabolic relationship between the dripping time and the peak intensity. This parabola takes a maximum, for both the (100) and (200) planes, at around 11 s. Also shown in this graph is the intensity given by the reference sample. The fact that all samples where IPA was used showed greater intensity than the reference samples supports the claim that the method is both reproducible and effective.

A summary of the results collected from varying the dripping time of IPA. a XRD spectra of the freshly prepared samples b intensity of the (100) and (200) peaks compared with the reference sample c absorbance spectra d PL spectra with an integration time of 16 s
In terms of optical properties, the absorbance spectra were all similar, with the reference sample having an absorbance centred between the most and the least absorbent samples. The sample with IPA applied after 8 s showed the highest absorbance followed by the 10 s sample. The samples where antisolvent was applied after 15 and 20 s showed the lowest absorbance. As mentioned in the previous section, the error associated with the sample thickness should be considered when analysing the absorbance measurements since thicker layers will have a higher absorbance. On the other hand, the absorption onset is independent of thickness and agrees with those found in the literature [15, 25, 31, 42].
The PL spectra once again showed signs of the Hoke Effect taking place, as in Fig. 2f, meaning that there was a generic shift to lower energies and the apparition of a new PL peak. The effect could also explain the low energy tail seen in Fig. 5d. The fact that there may be two variables at play, the Hoke Effect and drip time, makes it difficult to confirm the observed small displacements in the peak maxima. However, a clear trend can be seen where samples that had a dripping time higher than 8 s exhibit more than a two-fold increase in emission. This could be a result of improved crystal structure and an associated reduction of non-radiative defects.
Stability study
To quantify the stability of the samples, an XRD analysis was carried out on all the samples used in the previous sections after they were stored for 5 weeks in the laboratory in low-light conditions, at a temperature of 23 °C and relative humidity of 20%.
Since there is no generally accepted procedure for measuring the stability of perovskites, a simple method was devised to quantify it. Since XRD spectra generate some of the most easily quantifiable results, a method was developed which compared values of intensity. That said, values derived from the area could also be used. Assuming significant changes do not occur in the aged spectra compared with their corresponding fresh spectra, such as the apparition of new peaks, it should be possible to correlate a percentage decrease in intensity with stability, since the number of perovskite diffractive planes is proportional to total intensity. This is because any new peaks not found in Table 2 correspond to new phases and demonstrate the partial degradation of the CsPbIBr2 alloy.
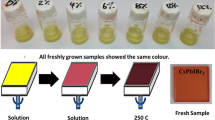
By exploiting the correlation between XRD intensity and stability, we can quantify the stability of a sample by calculating the percentage decrease in its diffraction maxima. The 10 and 15 s samples showed greatly increased stability, up to 65% for the IPA samples in comparison with the 20% that the reference sample gave. Remarkably, the intensity of the aged 10 and 15 s samples is higher than the freshly prepared reference CsPbIBr2 samples. These results suggest that IPA not only improves crystal quality but also stability. Contrastingly, the worst performing sample in terms of percentage decrease was the one where IPA was applied after 5 s. Many new diffraction peaks can also be seen to arise in Fig. 7b. It is likely that the solution did not have enough time to spread out before the antisolvent was applied, leading to inhomogeneous film thickness and crystal nucleation. Moreover, this would explain the patchy structure observed in Fig. 4 and the spiral structure seen in Fig. 6.

Photographs taken of the freshly prepared samples and the 5-week-old samples after they had been stored in the laboratory
Applying this method to measure the stability of the samples treated with different antisolvents, the CsPbIBr2 sample treated with DEE stands out as having extraordinarily high stability in terms of percentage decrease. However, this is misleading, since one can see the apparition of many new peaks in the spectrum in Fig. 7c showing that parts of the structure had degraded into new phases. Similarly, ClBn and Tl would seem to have increased the stability of the samples, however, closer inspection in Fig. 7c reveals that the only sample to have not given rise to new peaks is the reference sample. Seen in this way, not only does the application of these antisolvents seem to adversely affect their properties when fresh, but it also accelerates their degradation into new phases.

Stability measurement based on XRD spectra taken when the samples were freshly prepared and after they were aged in the laboratory. a peak intensity for all the samples mentioned in this work before and after they were aged. The percentage decrease corresponds to the difference between the freshly prepared samples (orange) and the aged samples (green). b XRD spectra of aged samples from the dripping time investigation c XRD spectra of the aged samples from the antisolvent selection
The sample where IPA was applied after 8 s showed a 20% decrease in intensity and the apparition of new peaks. Due to this phase change occurring, which did not take place in the reference sample, it is possible that 8 s is not a long enough amount of time for the solvent to spread out, which is why all the samples carried out at this time show accelerated degradation.
In terms of absorbance (Fig. 8), all samples show a generic shift to lower wavelengths, caused by the transformation of the desired phase, which is metastable at room temperature, into the stable one which has a larger bandgap value. The temperature at which this phase transition occurs in inorganic caesium-based perovskites depends on bromine-iodine content. For CsPbI3 and CsPbBr3, the phase transition occurs at 310 °C [47] and 130 °C [48], respectively, it follows that for CsPbIBr2 it would fall somewhere between the two.

Absorbance spectra of the fresh and 5-week-old CsPbIBr2 samples stored in the laboratory at 23 °C and RH = 20% under low-light conditions a Absorbance spectra of aged samples shown in Fig. 2b absorbance spectra of aged samples shown in Fig. 5. The dotted lines show the bandgap value of the desired phase [25, 31, 34]
Comparing the absorbance spectra of the samples where antisolvent was applied after 8 s, in Fig. 8a, the IPA sample appears to hold up best. If the energy barrier to the phase change has been increased, it could be because of an increase in bond strength resulting from a decrease in defect density, explaining their superior stability. Of all the samples, the worst results were obtained from the samples where ClBn was applied, where a significant decrease in absorbance is seen to take place at wavelengths below the bandgap. This suggests that most of the film had already changed phase.
Figure 8b shows the absorbance spectra of the aged samples that were used in the dripping time investigation. The sample in which IPA was applied after 10 s showed the best stability since it had the highest absorbance out of any of the aged samples. It even showed superior absorbance to the freshly prepared CsPbIBr2 sample, which had no antisolvent treatment. This supports the analysis based on the XRD spectra. The samples where IPA was applied after 15 and 20 s showed remarkably similar absorbance spectra, which were only slightly lower than the freshly prepared sample with no antisolvent applied.
The analysis carried out in this section supports the claim that the application of IPA with a dripping time of 10 s, or higher, will yield significant improvements in stability. This is indicated by the percentage decrease in intensity going from 20%, with the reference sample, to over 60% in Fig. 7a and the superior absorbance spectra in Fig. 8b. The results suggest that to obtain the maximum stability, IPA should be applied with a dripping time of 10 s, however, any period between 10 and 15 s will also enhance the films properties. This is consistent with the parabolic behaviour found in Fig. 5b, which suggests that the ideal application time is 11 s.
CsPbIBr2-based solar cells
Using the measured material properties, namely, thickness and bandgap, a one-dimensional solar cell capacitance simulator (SCAPS) [49] was used to predict the short-circuit current (Jsc), the open circuit voltage (Voc), the filling fraction (FF), and the device PCE. The parameters used in the simulation, which could not be determined experimentally, were taken from the literature, or in some cases, a reasonable estimation was made. All the parameters used in the simulation can be found in the supplementary information in Table 2.
To study the effect that the electron-transport layer (ETL) and the hole transport layer (HTL) had on-device performance, four different cells were simulated. All cells had a layer of FTO as the back contact and used CsPbIBr2 as the absorber layer. In two of the cells, zinc oxide (ZnO) was used as an ETL and in the remaining two titanium dioxide (TiO2) was used. For a given ETL, a simulation was made with and without the commonly used 2,2',7,7'-Tetrakis [N,N-di(4-methoxyphenyl)amino]-9,9'-spirobifluorene (spiro-OMeTAD). The resulting J-V curves and cell properties are shown in Fig. 9 and Table 3, respectively.

J-V curves corresponding to the different CsPbIBr2-based solar cells with different hole- and electron-transport layers
The J-V results revealed that the use of the HTL, spiro-OMeTAD, had a major impact on the performance of the solar cell, shown by the increase in FF from 50% to over 80%. On the other hand, Jsc remained largely unchanged since the bandgap of spiro-OMeTAD, 3.17 eV, is comparable to that of the ETLs, 3.2 and 3.3 eV, and larger than that of CsPbIBr2 2.05 eV. For this reason, a similar number of photons were absorbed with or without the HTL. Interestingly, the Voc increased in the cell where TiO2 was used when the HTL was added, but the opposite took place in the cell where ZnO was used. It is possible that this occurs because of the differences in dopant density and therefore carrier concentration. This finding suggests that when selecting the ETL, one should consider whether an HTL is to be included in the device and how they will perform together. In terms of PCE, the cells in which spiro-OMeTAD was used showed significant increases. The champion cell, where TiO2 was used as an ETL, gave a PCE of 13.7%, which is higher than the record experimental value of 11.53% [42]. To date, much focus has been made on HTL-free CsPbIBr2-based devices [15, 21, 31, 34], and our findings suggest that many of the experimentally tested devices could be significantly enhanced by the addition of an appropriate HTL.
Discussion
Our findings support the hypothesis that both antisolvent selection and application conditions have a measurable impact on the final sample. Our results have helped clarify and quantify the effects that different factors have on the quality of antisolvent quenched CsPbIBr2. We have shown that the antisolvent used and the dripping time both play a major role in the processed samples.
It is reasonable to suppose that antisolvent selection must be important since different solvent-antisolvent combinations will behave differently together due to their varying properties. However, more surprisingly, we discovered that the dripping time, an often-ignored factor, seems to have a large effect on the final sample and therefore should not be overlooked when integrating antisolvent quenching into the film synthesis procedure. It seems important that enough time be given for the solution to spread out, however, when the dripping time is too long, the properties of the film decrease due to the other processes taking place during crystallisation. Although the time needed for the solution to spread out is probably a function of viscosity, surface wetness, and surface tension of the solution, in our case, we believe it to be somewhere between 8 and 10 s.
While our results support the claim that antisolvent quenching can improve the quality of CsPbIBr2 samples, a possible explanation for why antisolvent quenching is not commonplace with CsPbIBr2 is because when other researchers have tried the method, they obtained results like ours for DEE, ClBn and Tl. Namely, that the processed samples decreased in quality (Fig. 2). It might be that DMSO is not extracted as well as dimethylformamide (DMF) by these antisolvents, so antisolvent methodologies are used in hybrid perovskites, such as methylammonium lead iodide, may not extend to inorganic perovskites.
The difficulty in successfully optimizing the antisolvent quenching method may also explain the deviation of our results from the only published method presented by Zhang et al. A possible explanation for our different results is that the antisolvent quenching method is sensitive to small changes in methodology. While on paper our experiments are very similar, there may be subtle differences in the way in which our work was carried out. These differences may include uncontrolled variables in ambient conditions, solution components, or the deposition conditions. In Zhang et al.’s paper, they used the same antisolvents we did, namely ClBn, IPA, Tl and DEE, however, they found DEE to be the best antisolvent, which contradicts our findings significantly. During their experimental work, Zhang et al. employed a dripping time of 20 s, which they did not vary, leaving open the possibility that their method may be further optimized. We optimized this parameter for the best antisolvent, IPA, by varying the dripping time and studying the quality and stability of the synthesized samples as described in the methodology section.
Several studies into antisolvent quenching methods for the sister materials CsPbI2Br and CsPbBr3 [39, 50], found, like us, that IPA was the best antisolvent. Liu et al. reported using ClBn to carry out their antisolvent quenching method [40], however, they used DMF as a solvent, which is unusual in CsPbX3 films containing bromine due to the difficulty it causes in dissolving the bromine-containing precursors. Based on these papers, IPA appears to be an antisolvent that lends itself well to approaches where only DMSO is used as a solvent, however, as soon as DMF is added to the solvent, other antisolvents perform better. Since initial literature reviews of researchers working on CsPbIBr2 may not include sister materials, and the vast majority of CsPbIBr2 films are made with DMSO [31, 34, 35, 42], our findings should be taken into account by other researchers, since they may not be aware that there is an alternative antisolvent quenching method available to the one presented by Zhang et al.
In our experience, DEE was found to be impractical to work with because of its tendency to drop out of the pipette very quickly. It is likely that the low-boiling point of this antisolvent significantly increases the evaporation taking place within the pipette tip, lowering the pressure, resulting in the DEE being ejected from the pipette. We overcame this dripping effect by taking up some air at the end of the pipette, and we also tried an alternative method which involved saturating the interior of the pipette tip with DEE vapour beforehand, and this also seemed to work. The use of these techniques, however, is clearly a limitation if volumetric errors need to be minimized. Furthermore, DEE is not miscible with DMSO [38], which could cause a nonuniform distribution of DMSO and DEE when quenched. This would lead to a nonuniform deposition of perovskite material as well as their different boiling points leading to inhomogeneous drying and crystal nucleation.
Although our results allow us to claim with a high degree of certainty that IPA quenching CsPbIBr2 will result in a more stable film, we acknowledge that the method could be improved by including other diffraction peaks or complementing it with another easily quantifiable piece of data such as bandgap value or absorption coefficient. We have established that FESEM analysis is problematic because it is difficult to quantify and depending on film uniformity, a single image may not be enough to fully capture the nature of the surface.
Our work has established some of the foundations for future studies, where we will extend our investigation of dripping time dependence to the other antisolvents. While considering the dripping time on sample quality and stability, an improved understanding of the effect of the volumetric ratio between the perovskite and antisolvent was identified as necessary to understanding the results. This will be addressed in a forthcoming paper. This is necessary because, although in our methodology we state a volumetric ratio of 2:1 between the antisolvent and the precursor solution, as the spin programme runs the solution is thrown off, therefore changing this volumetric ratio. The lack of information about this variable makes it difficult to explain the observed parabolic behaviour of the intensity in Fig. 5b. While the increase in intensity can be attributed to the settling of the solution, the decrease that happens after around 11 s is likely due to the processes taking place when the crystal precipitates. Our hypothesis is that once the volumetric ratio goes above a certain level, the metastable limit is passed for an extended period, causing rapid nucleation and crystal growth, leading to many defects at the interfaces where these zones meet. To confirm this, more information about the metastable zone width is needed.
Conclusion
Improving the quality of CsPblBr2 samples is vital to enable further its use in solar cells. We believe that the results presented here make an important contribution in understanding how to improve the crystallinity and stability of CsPbIBr2 samples. We have conducted a preliminary analysis to discover which variables and conditions appear to have the greatest impact. In the absence of generally accepted tests for stability, we have developed our own method using XRD spectra. In our method, the percentage decrease in the intensity of diffraction peaks is compared between the samples as they evolve, allowing us to quantify their stability. Our results show that the application of the antisolvent IPA with a dripping time of 10 s, is a simple and reproducible way to substantially improve sample quality.
We report CsPbIBr2 films, whose composition was confirmed via EDS, with a mean thickness of 296 nm (s.d. 28 nm). While carrying out testing to determine the best antisolvent, FESEM analysis revealed that the Tl and ClBn samples showed large-scale defects. Rietveld refinement of the XRD spectra revealed the formation of a cubic structure, pertaining to the PM-3 M space group, with a lattice constant of 5.93423 ± 0.00034 Å. The samples were highly oriented in the (100) direction, with two major diffraction peaks occurring at 15° and 30°, corresponding to the (100) and (200) planes, respectively. From the FWHM, the samples applied with IPA showed an increased crystallite size of 55 nm. This should reduce the number of boundaries at which charge recombination can occur in solar cells, thereby increasing PCE. All samples with antisolvents applied showed an increased absorbance, and PL spectra showed signs of the Hoke Effect taking place.
While investigating the effects of dripping time on IPA quenched samples, FESEM images revealed that dripping times of 5 and 20 s led to large-scale defects. XRD spectra differed only in the intensities of the (100) and (200) peaks. These peak intensities took a parabolic form as a function of dripping time, with a maximum at 11 s. All XRD spectra of IPA quenched CsPbIBr2 showed superior intensities when compared to the reference sample, which had no antisolvent applied. Absorbance spectra of the IPA quenched samples were similar to that of the reference samples, however, PL intensity showed a two-fold increase when the dripping time was 10 s or higher.
To quantify the stability of the samples, we developed and employed a novel system based on XRD spectra and complemented this with an analysis of the absorbance spectra. This was prompted by the lack of established stability tests in the literature, and the need for a quantitative rather than qualitative method for measuring sample stability since qualitative information can be more easily misinterpreted. Our method showed that the samples with IPA applied after 10 and 15 s exhibited an important increase in stability, with only a 65% reduction in the intensity of their XRD spectra after 5 weeks stored in ambient conditions, compared to a 20% reduction in the reference sample. These samples gave superior XRD spectra readings than the freshly prepared reference samples, even after they were aged for 5 weeks in air. The sample where IPA was applied after 20 s also showed a moderate increase in stability. On the other hand, the sample where IPA was applied after 5 s gave poor results across the board, leading us to conclude that a minimum and specified amount of time is necessary for optimum dispersal of the solution. According to XRD data, the samples where other antisolvents were used showed accelerated degradation, along with the appearance of new phases. The absorbance spectra corresponding to the fresh and aged samples showed results that supported all of the findings made with the aforementioned method. Namely, that the sample in which IPA was applied with a dripping time of 10 s showed better results than the freshly prepared reference sample even after it had been stored in air for 5 weeks. This increase in stability also takes place in the samples where the dripping time was higher, but to a lesser degree.
To probe the properties of devices made with the optimized CsPbIBr2 films, four different devices were simulated using the programme SCAPs. The devices were made with either ZnO or TiO2, both with and without spiro-OMeTAD. The addition of spiro-OMeTAD was shown to make an important increase in device performance supporting its use as a simple way to significantly enhance device performance. The champion device was the one, which employed both spiro-OMeTAD and TiO2. This solar cell gave a PCE of 13.7%, which is higher than the best reported single-junction device found in the literature. Moreover, it gave very competitive values for Voc, Jsc and FF which were 1.235 V, 12.894 mAcm−2 and 86.05%, respectively.
We have established that the application of IPA with a dripping time of 10 s is a simple and reproducible method for significantly improving sample quality. We anticipate that our method should greatly enhance CsPbIBr2 sample production and contribute to the development of solar panels as a source of renewable energy. The development of solar panels is of utmost importance in the face of the impending climate crisis and will be vital in achieving the goal of transitioning to net-zero carbon emissions by 2050 as set out in the Paris Agreements.
Data availability
All the data collected in this work relating to FESEM images, XRD readings and optical measurements is available at https://doi.org/10.17632/25cgvvf66j.2
References
Snaith HJ (2013) Perovskites: the emergence of a new era for low-cost, high-efficiency solar cells. J Phys Chem Lett 4:3623–3630
Best Research-Cell Efficiency Chart | Photovoltaic Research | NREL. https://www.nrel.gov/pv/cell-efficiency.html. Accessed 29 December 2020
Wang T, Daiber B, Frost JM et al (2017) Indirect to direct bandgap transition in methylammonium lead halide perovskite. Energy Environ Sci 10:509–515. https://doi.org/10.1039/c6ee03474h
Miyata A, Mitioglu A, Plochocka P et al (2015) Direct measurement of the exciton binding energy and effective masses for charge carriers in organic-inorganic tri-halide perovskites. Nat Phys 11:582–587. https://doi.org/10.1038/nphys3357
D’Innocenzo V, Grancini G, Alcocer MJP et al (2014) Excitons versus free charges in organo-lead tri-halide perovskites. Nat Commun 5. https://doi.org/10.1038/ncomms4586
Herz LM (2017) Charge-carrier mobilities in metal Halide perovskites: fundamental mechanisms and limits. ACS Energy Lett 2:1539–1548
Alcocer MJP, Leijtens T, Herz LM et al (2013) Electron-hole diffusion lengths exceeding trihalide perovskite absorber. Science 342(80):341–344. https://doi.org/10.1126/science.1243982
Xing G, Mathews N, Sun S et al (2013) Long-range balanced electron-and hole-transport lengths in organic-inorganic CH3NH3PbI3. Science 342(80):344–347. https://doi.org/10.1126/science.1243167
Leguy A, Hu Y, Campoy-quiles M et al (2015) The reversible hydration of CHNHPbI in films, single crystals and solar cells the reversible hydration of CH3NH3PbI3 in films, single crystals and solar cells. Chem Mater 27:3397–3407. https://doi.org/10.1021/acs.chemmater.5b00660
Yi C, Luo J, Meloni S et al (2016) Entropic stabilization of mixed A-cation ABX3 metal halide perovskites for high performance perovskite solar cells. Energy Environ Sci 9:656–662. https://doi.org/10.1039/c5ee03255e
Jodlowski AD, Roldán-Carmona C, Grancini G et al (2017) Large guanidinium cation mixed with methylammonium in lead iodide perovskites for 19% efficient solar cells. Nat Energy 2:972–979. https://doi.org/10.1038/s41560-017-0054-3
Stoumpos CC, Malliakas CD, Kanatzidis MG (2013) Semiconducting tin and lead iodide perovskites with organic cations: phase transitions, high mobilities, and near-infrared photoluminescent properties. Inorg Chem 52:9019–9038. https://doi.org/10.1021/ic401215x
Saliba M, Matsui T, Seo JY et al (2016) Cesium-containing triple cation perovskite solar cells: Improved stability, reproducibility and high efficiency. Energy Environ Sci 9:1989–1997. https://doi.org/10.1039/c5ee03874j
Liu C, Li W, Zhang C et al (2018) All-Inorganic CsPbI2Br Perovskite Solar Cells with High Efficiency Exceeding 13%. J Am Chem Soc 140:3825–3828. https://doi.org/10.1021/jacs.7b13229
Ma Q, Huang S, Wen X et al (2016) Hole transport layer free inorganic CsPbIBr 2 perovskite solar cell by dual source thermal evaporation. Adv Energy Mater 6:2–6. https://doi.org/10.1002/aenm.201502202
Han Y, Meyer S, Dkhissi Y et al (2015) Degradation observations of encapsulated planar CH3NH3PbI3 perovskite solar cells at high temperatures and humidity. J Mater Chem A 3:8139–8147. https://doi.org/10.1039/c5ta00358j
Divitini G, Cacovich S, Matteocci F et al (2016) In situ observation of heat-induced degradation of perovskite solar cells. Nat Energy 1. https://doi.org/10.1038/NENERGY.2015.12
Lee SW, Kim S, Bae S et al (2016) UV degradation and recovery of perovskite solar cells. Sci Rep 6:1–10. https://doi.org/10.1038/srep38150
Wang D, Wright M, Elumalai NK, Uddin A (2016) Stability of perovskite solar cells. Sol Energy Mater Sol Cells 147:255–275. https://doi.org/10.1016/j.solmat.2015.12.025
Straus DB, Guo S, Abeykoon AM, Cava RJ (2020) Understanding the instability of the Halide perovskite CsPbI3 through temperature-dependent structural analysis. Adv Mater 32:1–8. https://doi.org/10.1002/adma.202001069
Liang J, Liu Z, Qiu L et al (2018) Enhancing optical, electronic, crystalline, and morphological properties of Cesium Lead Halide by Mn substitution for high-stability all-inorganic Perovskite solar cells with carbon electrodes. Adv Energy Mater 8:1–7. https://doi.org/10.1002/aenm.201800504
Guo Y, Zhao F, Tao J et al (2019) Efficient and hole-transporting-layer-free CsPbI 2 Br planar heterojunction Perovskite solar cells through Rubidium passivation. Chemsuschem 12:983–989. https://doi.org/10.1002/cssc.201802690
Zhu W, Zhang Q, Chen D et al (2018) Intermolecular exchange boosts efficiency of air-stable, carbon-based all-inorganic planar CsPbIBr 2 Perovskite solar cells to over 9%. Adv Energy Mater 8:1–11. https://doi.org/10.1002/aenm.201802080
Mariotti S, Hutter OS, Phillips LJ et al (2018) Stability and performance of CsPbI2Br thin films and solar cell devices. ACS Appl Mater Interfaces 10:3750–3760. https://doi.org/10.1021/acsami.7b14039
Lau CFJ, Deng X, Ma Q et al (2016) CsPbIBr 2 Perovskite solar cell by spray-assisted deposition. ACS Energy Lett 1:573–577. https://doi.org/10.1021/acsenergylett.6b00341
Jošt M, Kegelmann L, Korte L, Albrecht S (2020) Monolithic Perovskite tandem solar cells: a review of the present status and advanced characterization methods toward 30% efficiency. Adv Energy Mater 10. https://doi.org/10.1002/aenm.201904102
Wang H, Li H, Cao S et al (2020) Interface modulator of ultrathin magnesium oxide for low-temperature-processed inorganic CsPbIBr 2 Perovskite solar cells with efficiency over 11%. Sol RRL 4:1–11. https://doi.org/10.1002/solr.202000226
Zhang Q, Zhu W, Chen D et al (2019) Light processing enables efficient carbon-based, all-inorganic planar CsPbIBr 2 solar cells with high photovoltages. ACS Appl Mater Interfaces 11:2997–3005. https://doi.org/10.1021/acsami.8b17839
Pan J, Zhang X, Zheng Y, Xiang W (2021) Morphology control of perovskite film for efficient CsPbIBr 2 based inorganic perovskite solar cells. Sol Energy Mater Sol Cells 221:110878. https://doi.org/10.1016/j.solmat.2020.110878
Zhang B, Bi W, Wu Y et al (2019) High-performance cspbibr2 perovskite solar cells: effectively promoted crystal growth by antisolvent and organic ion strategies. ACS Appl Mater Interfaces 11:33868–33878. https://doi.org/10.1021/acsami.9b09171
Zhu W, Zhang Z, Chai W et al (2019) Band alignment engineering towards high efficiency carbon-based inorganic planar CsPbIBr 2 Perovskite solar cells. Chemsuschem 12:2318–2325. https://doi.org/10.1002/cssc.201900611
Zhu W, Zhang Q, Zhang C et al (2018) Aged precursor solution toward low-temperature fabrication of efficient carbon-based all-inorganic planar CsPbIBr 2 Perovskite solar cells. ACS Appl Energy Mater 1:4991–4997. https://doi.org/10.1021/acsaem.8b00972
Stewart AW, Bouich A, Marí B (2021) Inorganic perovskites improved film and crystal quality of CsPbIBr 2 when doped with rubidium. J Mater Sci Mater Electron 1–9. https://doi.org/10.1007/s10854-021-06941-z
Zhang W, Zhang Z, Jiang Q et al (2020) Charge-transporting-layer-free, all-inorganic CsPbIBr2 Perovskite solar cells via dipoles-adjusted interface. Nanomaterials (Basel) 10:1–10. https://doi.org/10.3390/nano10071324
Li W, Rothmann MU, Liu A et al (2017) Phase segregation enhanced ion movement in efficient inorganic CsPbIBr2 Solar cells. Adv Energy Mater 7:1–8. https://doi.org/10.1002/aenm.201700946
Xiao M, Huang F, Huang W et al (2014) A fast deposition-crystallization procedure for highly efficient lead iodide perovskite thin-film solar cells. Angew Chemie 126:10056–10061. https://doi.org/10.1002/ange.201405334
Shi D, Adinolfi V, Comin R et al (2015) Low trap-state density and long carrier diffusion in organolead trihalide perovskite single crystals. Science (80- ) 347:519–522. https://doi.org/10.1126/science.aaa2725
Paek S, Schouwink P, Athanasopoulou EN et al (2017) From nano- to micrometer scale: the role of antisolvent treatment on high performance Perovskite solar cells. Chem Mater 29:3490–3498. https://doi.org/10.1021/acs.chemmater.6b05353
Chen W, Chen H, Xu G et al (2019) Precise control of crystal growth for highly efficient CsPbI2Br Perovskite solar cells. Joule 3:191–204. https://doi.org/10.1016/j.joule.2018.10.011
Liu X, Li J, Liu Z et al (2020) Vapor-assisted deposition of CsPbIBr 2 films for highly efficient and stable carbon-based planar perovskite solar cells with superior Voc. Electrochim Acta 330:135266. https://doi.org/10.1016/j.electacta.2019.135266
Liu C, Li W, Chen J et al (2017) Ultra-thin MoOx as cathode buffer layer for the improvement of all-inorganic CsPbIBr 2 perovskite solar cells. Nano Energy 41:75–83. https://doi.org/10.1016/j.nanoen.2017.08.048
Guo Y, Yin X, Liu J et al (2019) Inorganic CsPbIBr 2-based perovskite solar cells: fabrication technique modification and efficiency improvement. Sol RRL 3:1–13. https://doi.org/10.1002/solr.201900135
Eperon GE, Paternò GM, Sutton RJ et al (2015) Inorganic caesium lead iodide perovskite solar cells. J Mater Chem A 3:19688–19695. https://doi.org/10.1039/c5ta06398a
Hoke ET, Slotcavage DJ, Dohner ER et al (2015) Reversible photo-induced trap formation in mixed-halide hybrid perovskites for photovoltaics. Chem Sci 6:613–617. https://doi.org/10.1039/c4sc03141e
Bush KA, Frohna K, Prasanna R et al (2018) Compositional engineering for efficient wide band gap perovskites with improved stability to photoinduced phase segregation. ACS Energy Lett 3:428–435. https://doi.org/10.1021/acsenergylett.7b01255
Chen Z, Bobbert PA. Unified theory for light-induced halide segregation in mixed halide perovskites. Nat Commun, pp 1–10. https://doi.org/10.1038/s41467-021-23008-z
Beal RE, Slotcavage DJ, Leijtens T et al (2016) Cesium lead Halide perovskites with improved stability for tandem solar cells. J Phys Chem Lett 7:746–751. https://doi.org/10.1021/acs.jpclett.6b00002
Sutton RJ, Eperon GE, Miranda L et al (2016) Bandgap-tunable cesium lead Halide perovskites with high thermal stability for efficient solar cells. Adv Energy Mater 6:1–6. https://doi.org/10.1002/aenm.201502458
Burgelman M, Nollet P, Degrave S (2000) Modelling polycrystalline semiconductor solar cells. Thin Solid Films 361:527–532. https://doi.org/10.1016/S0040-6090(99)00825-1
Kim BW, Heo JH, Park JK et al (2021) Morphology controlled nanocrystalline CsPbBr 3 thin-film for metal halide perovskite light emitting diodes. J Ind Eng Chem 97:417–425. https://doi.org/10.1016/j.jiec.2021.02.028
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This study was funded by the Generalitat Valenciana and the European Union (ACIF/2020/286) and the Ministerio de Economía y Competitividad (Grant Number PID2019-107137RB-C21).
Author information
Authors and Affiliations
Contributions
AWS: Conceived and designed the experiment, collected the data, performed the analysis, and wrote the paper. AB: Conceived the experiment, carried out the SCAPs simulation and gave critical feedback on the paper. BMS: Gave critical feedback on the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: Pedro Camargo.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stewart, A.W., Bouich, A. & Soucase, B.M. Enhancing the stability and crystallinity of CsPbIBr2 through antisolvent engineering. J Mater Sci 56, 20071–20086 (2021). https://doi.org/10.1007/s10853-021-06552-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-021-06552-3