
Abstract
A methodology to enrich epoxy coatings of an effective self-healing feature on wet surfaces was developed as a further step on for practical corrosion protection issues. To this aim, a polyetheramine was chemically engineered by grafting catechol units and then successfully encapsulated in microcapsules (MCs) to be finally embedded into an epoxy resin deposited on steel panels. Fourier transform infrared spectroscopy (FTIR), thin-layer chromatography, and 1D and 2D nuclear magnetic resonance spectroscopy confirmed the successful polyetheramine modification by dopamine units. Different dosages of catechol-modified polyetheramine were encapsulated within poly(styrene-co-acrylonitrile) shell via electrospray method to study the influence of dopamine grafting on the healing performance. Scanning electron microscopy (SEM) analysis revealed the formation of the spherical MCs, while FTIR and TGA analyses confirmed the successful encapsulation. The highly responsive self-healing coatings were then prepared by embedding amine- and isocyanate-containing MCs (1:1 weight ratio; 3 wt% overall) as a dual-capsule system exploiting the polyurea formation as a fast healing reaction. In operando electrochemical impedance spectroscopy (EIS) tests were employed to study the underwater self-healing performance. According to the EIS results, monotonically increasing variation with time of the charge transfer resistance was correlated with a fast and effective underwater self-healing performance for the sample using 40 wt% of a catechol-modified healing agent. Such results, combined with others including SEM investigation on the underwater healed samples, point to an improved adhesion of the growing dopamine-bearing polymer to both underlying metal and epoxy edges of the scratch.
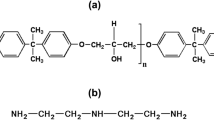
Graphic abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nowadays, several self-healing techniques have been developed as promising approaches for increasing the service life of the organic coatings via controlling the crack propagation in their matrix [1,2,3,4]. Although many of the reported self-healing materials have shown acceptable performance, most of them were evaluated at standard environmental conditions (e.g., temperature and humidity) [5,6,7]. Considering the direct relationship between the steel corrosion phenomena and the presence of moisture and water in the environment, the ability for fast and efficient underwater self-healing is indispensable for effective self-healing anticorrosion coatings [8,9,10,11]. The sensitivity of polymerization reactions to the presence of external factors, such as water, and the low adhesion of the newly formed polymer layer onto the wet surfaces are the most critical challenges toward the development of underwater self-healing systems [2, 9].
Several methods have been proposed to address the adhesion loss in the presence of interfacial water. Cho et al. studied the underwater healing performance through the incorporation of a poly(dimethylsiloxane)-based healing system in a vinyl ester matrix that revealed stable in the presence of moisture and water. They used methylacryloxypropyl triethoxysilane as the adhesion promoter, leading to enhance in the underwater self-healing performance. However, lower stiffness and fracture toughness of PDMS with respect to the matrix material showed to be two important limitations in reaching a suitable overall efficiency [9].
Huang et al. encapsulated hexamethylene diisocyanate through interfacial polymerization of methylene diphenyl diisocyanate and 1,4-butanediol and subsequently studied its healing efficiency in an epoxy coating at submerged corrosion conditions [12, 13]. The healing process, triggered by the reaction between the released isocyanate and environmental present water molecules, increased the corrosion resistance of the damaged coatings. Zhang et al. synthesized an intrinsically self-healing copolymer composed of a hexafluorobutyl methacrylate monomer, as a hydrophobic monomer, and an isocyanate-containing acrylate monomer, as the water-reactive agent, responsible for the healing reaction in case of damage to the integrity of the coating [14]. Independently by the specific engineering strategy, the slow kinetics of the isocyanate/water reaction represents a real challenge for isocyanate-based approaches, especially in low humidity/temperature environments where the healing reaction would not proceed efficiently [10, 15].
Mussel-inspired materials have shown great performance in underwater self-healing applications due to their unique chemistry driven by catechol groups [16,17,18]. The superior adhesion of mussels to wet surfaces is provided by the salt-triggered production of natural adhesive proteins that contain side catechol groups in their structure [19,20,21]. In catechol-containing compounds, two vicinal hydroxyl groups on the aromatic ring, acting as a bidentate ligand and tend to make stronger hydrogen bonds with oxide surfaces, are responsible to address the typical poor adhesion of materials on the wet surfaces. This behavior can be justified by Bell bond lifetime theory that predicts the hydrogen bonding lifespan of the ortho-hydroxyl groups on the aromatic ring (catechol) acting as a bidentate site being 108 times longer than the hydrogen bond lifetime of water molecules [22]. Ahn et al. reported underwater self-healing of an elastomer based on catechol-modified polyacrylate and polymethacrylate [23]. Although a fast and efficient healing response was developed, the adopted system needed a low pH environment and molecular mobility driven by a brief compression force for the activation. Li et al. synthesized poly(dopamine acrylamide-co–n-butyl acrylate) with more stable catechol units and pH-independent performance. The corresponding polymer film showed good healing properties in seawater due to the coordination reactions induced by Ca2+/Mg2+ cations present in the medium, while its healing efficiency was not appropriate in the air or pure water [24].
Although the catechol-based chemistry has been effectively used in several intrinsic self-healing polymers and coatings, their anti-corrosion properties have not been well studied yet due to the limitations engaged with applying intrinsic systems to common industrial polymer matrices [25,26,27]. The catechol chemistry has been only very recently exploited to extrinsic self-healing coatings through modifying the surfaces of healing agent containers due to their pH sensitivity and water compatibility [28, 29].
To do a further step on, in the present paper, a polyurea-based extrinsic healing system recently proposed by our group [30] was enriched of an effective underwater self-healing feature through the direct chemical engineering of the healing agent with catechol units. The rationale of the proposed work born from two solid observations properly merged. On the one hand, the polyurea formation (i.e., the repairing process), resulting from the reaction between the released amines and isocyanates, is of great potential for underwater self-healing applications due to both its insensitivity to water and its high reaction rate [31]. On the other hand, the novelty to apply the mussel-inspired biomimetic engineering directly on one of the healing components (and not on its containers [28, 29]) is introduced to address the underwater adhesion between the growing healing polymer and the metal surface, generally hampered by the presence of interfacial water. In doing so, polyetheramine, acting as a component of a dual-capsule-based system, was grafted with dopamine units as a source of the vicinal diol units that strengthen the underwater healing feature of the polyurea film grown by the condensation reaction with diisocyanate reactant. To prove the efficacy of the methodology, the performance of the self-healing process was evaluated for underwater scratched epoxy coatings through electrochemical impedance spectroscopy (EIS), a noninvasive technique that allowed the in operando monitoring of the self-repairing event occurring at the metal interface as a result of the formation of polyurea within the crack.
Materials and methods
Materials
Dopamine hydrochloride, methanol, dichloromethane, and sodium hydroxide were purchased from Sigma-Aldrich and used as received. Diglycidyl ether of 1,6-hexanediol (1,6-HDGE; commercial name: ED 180) was provided by Inchem Ltd and Jeffamine™D230 [poly(propylene glycol) bis(2-aminopropyl ether)], was purchased from Huntsman. These chemicals were used to prepare the dopamine-modified amine healing agent. CORONATE™1391 (isocyanate prepolymer based on 4,4′-diphenylmethane diisocyanate) was purchased from TOSOH Corporation and used as the second healing agent of the dual capsule system. N,N′-dimethylformamide (DMF) and poly(styrene-co-acrylonitrile) (SAN) (185 kDa, 30 wt% acrylonitrile content) were purchased from Sigma-Aldrich and used for the encapsulation process [30]. To prepare the epoxy coatings, diglycidyl ether of bisphenol-A epoxy resin (EPON 828) and a polyaminoamide-based hardener (Merginamid A280) were purchased from Hexion and Hobum oleochemicals, respectively [32].
Synthesis of dopamine grafted prepolymer
A two-step procedure based on epoxide ring-opening reaction was used to synthesis the catechol-modified amine as a highly responsive healing agent (Fig. 1). In the first step of the synthesis, an excess of 1,6-HDGE (1.82 g, 7.8 mmol) followed by dopamine hydrochloride (0.5 g, 2.6 mmol) were dissolved in 100 mL of methanol in a 250-mL two-neck round-bottom flask and bubbled with nitrogen for 60 min. The molar ratio between 1,6-HDGE and dopamine hydrochloride was set as 3:1 to control the degree of polymerization as well as to favor the reaction of the amine groups of the dopamine. NaOH (0.1 g, 2.6 mmol) was then dissolved in 50 mL of methanol under nitrogen atmosphere. The alkaline solution was added dropwise into the reaction mixture to progressively activate the dopamine –NH2 groups through neutralizing the hydrochloric acid molecules. The reaction mixture was stirred under a nitrogen atmosphere for 4 h at room temperature until its color changed from clear to yellow.

Synthesis of the dopamine-modified polyetheramine prepolymer 1
The second step of the reaction was performed to prepare a stable dopamine-containing prepolymer with amine functional end groups (1). In this regard, 1.79 g (7.8 mmol) of Jeffamine™D230 (a linear polyether with amine functional end groups) was added dropwise to the reaction mixture, under the nitrogen atmosphere. The nominal molar ratio between the added polyetheramine and the starting dopamine hydrochloride was again set at 3:1, to control the reaction and consuming as much available epoxide groups as possible. The mixture was then stirred at room temperature under a nitrogen atmosphere for another 24 h to ensure the completion of the reaction. Finally, methanol was removed under vacuum, and the crude product was dissolved in the minimum amount of dichloromethane to precipitate and filtrate the NaCl (resulting from the neutralization reaction between HCl and NaOH). Dichloromethane was finally removed by vacuum to obtain the amine-containing prepolymer 1. Yield: ca. 70% (from 1H NMR). FTIR (KBr): \(\overline{\nu }\) (cm−1) = 3565–3115 (aromatic O–H, aliphatic N–H), 2996–2787 (C–H), 1100 (aliphatic ether). Additional characterizations of the resulting product are provided in Result and Discussion section and in Supporting Information file.
Preparation of the microcapsules
The electrospray method was used to encapsulate the healing agents owning to its facility and cost-efficiency with respect to Pickering emulsion and interfacial polymerization techniques. In particular, poly(styrene-co-acrylonitrile) was employed as the shell material, while isocyanate-based (CORONATE™1391) and amine-based prepolymers were separately encapsulated as the two core components. To study the effect of the dopamine grafting on the performance of modified prepolymer 1, three different amine-based core materials were prepared as described in Table1. The shell solution (SAN in DMF, 4%w/v) was prepared as described in our previous work [30]. The isocyanate-based core–shell solution was prepared by adding CORONATE™1391 (i.e., core) directly to the shell solution at 0.5 wt% core/shell ratio. Similarly, three different amine-based core–shell solutions were prepared by separate incorporation of CTRL (as the control amine), D10, and D40 core materials to the shell polymer solutions (Table 1). Likewise, isocyanate solution, a 0.5 wt% core/shell ratio was invariably kept for all amine-based polymer solutions. All the four prepared core–shell containing polymer solutions were then sprayed separately by a SP102 (Fnm co. Ltd) electrospray setup through a G23 stainless steel needle. Grounded aluminum foil sheets were used as the collector for the sprayed MCs. The injection flow rate, the applied DC potential, and the distance between the needle tip and the collector were fixed at 0.3 mL h−1, 24 kV, and 15 cm, respectively.
Preparation of the self-healing coatings
EPON 828 epoxy resin was mixed with ED 180 reactive diluent (3:1 weight ratio) to reduce its viscosity. Subsequently, a 1:1 weight mixture of amine and isocyanate MCs was added to the epoxy resin followed by mechanical mixing at 200 rpm for 5 min. To cure the epoxy resin, the polyaminoamide-based hardener (Merginamid A280) was added to the resin mixture at a stoichiometric weight ratio. The total MC content in the matrix was kept at 3 wt% for all the samples, being the optimal amount in terms of the corrosion protection ability [30]. The mixtures were then immediately degassed by a vacuum pump and coated on the previously sanded and cleaned steel panels (Q-PANEL) using a wet film applicator. The thickness was adjusted at 150 µm for all of the applied coatings. Table 2 lists the detailed composition of the coatings prepared to investigate the effect of dopamine grafting on the underwater healing process. After 7 days of curing in laboratory conditions, the prepared coatings were scratched by a scalpel cutter (equipped with a #11 blade) doing a 1 × 1 cm length cross in the film down to the substrate and immediately immersed in a 3.5 wt% NaCl aqueous solution for further corrosion testing. The same blade and pressure were used for all tests, in order to assure the as high as possible reproducibility in the cut width.
Characterization
Characterization of the dopamine grafted prepolymer
The grafting reaction of dopamine on HDGE (the first step of the synthesis, Fig. 1), was monitored by analyzing the intermediate reaction mixture by Fourier transform infrared spectroscopy (PerkinElmer Spectrum 100 FTIR spectrometer). To perform the analysis, two samples were taken by syringe from the reaction mixture before (as control) and after the addition of NaOH solution. Then, one drop of each sample was dipped on KBr pellets. Immediately after solvent evaporation, the analysis was carried out recording the spectrum in the range of 4000–400 cm−1. A similar investigation was performed also onto the final reaction product (dopamine-grafted prepolymer 1) after filtration and solvent evaporation.
Thin-layer chromatography (TLC) and nuclear magnetic resonance (NMR) spectroscopy were used to study the final reaction product 1. TLC experiments were conducted on the diluted solutions of each single reagents (as control) and of the crude reaction product before and after filtration, in both normal and reverse phase. In the normal phase, a 9:1 mixture of dichloromethane/methanol was used as eluent coupled with silica gel 60 F254 plates (Merck Millipore), while a 1:1 mixture of methanol/water and silica gel 60 RP-18 F254 plates (Merck Millipore) were utilized for the reverse phase. In both eluents, 1 vol% of triethanolamine (Merck Millipore) was added for neutralizing the HCl resulting from dopamine hydrochloride. A Camag UV cabinet (CAMAG, Switzerland) was used to observe TLC plates under 254 nm and 366 nm UV lights. To observe the components that were not detectable under UV light, TLC plates were immersed in sulfuric acid/methanol solution for a couple of seconds and then dried by heating at 150 °C.
1H, 13C, and 2D NMR (1H, 13C-HSQC; 1H, 13C-HMBC; DOSY) spectra were recorded in DMSO-d6 on a Bruker Avance III 400 MHz (Billerica, MA, USA) instrument for both pristine dopamine hydrochloride and the reaction product 1 to confirm the success of the reaction. Chemical shifts (δ) for 1H and 13C spectra are expressed in ppm relative to the standard Me4Si. Heteronuclear 1H-13C long-range HMBC was acquired with inverse detection using the following parameters (hmbcgplpndqf, Bruker library): relaxation delay (d1) = 1.5 s, 256 increments in t1 (128 scans for each spectrum), JC–H delay = 145 Hz and long-range JC–H delay = 8 Hz. DOSY experiments were carried out with the pulse program ledbpgp2s (Bruker library) setting: 32 gradient increments in t2, 16 scans for each spectrum, d1 = 2 s, length of the magnetic field pulse gradient (δ) = 2.5 ms and diffusion time (Δ) = 200 ms.
Characterization of the microcapsules
Scanning electron microscopy (JEOL JSM-5500 LV, JEOL) was employed to study the morphology and size distribution of the prepared MCs. Image J software was used for the size distribution analysis (50 measurements). The core content and thermal stability of the MCs were studied by thermogravimetric analysis (TGA/DSC 2 STAR-Mettler-Toledo) under N2 atmosphere and with 10 °C min−1 heating rate. Fourier transform infrared spectroscopy (PerkinElmer Spectrum 100 FTIR spectrometer) was used to assess the chemical stability of the MCs, excluding possible reactions within the core material and between core and shell one.
Electrochemical characterization of the coatings
EIS tests were carried out to evaluate the self-healing ability of the prepared coatings. To simulate the loss of physical integrity of the coating and study the consequent self-healing process via polyurea formation reaction in underwater conditions, the coatings were scratched, as above described, and immediately immersed in 3.5 wt% NaCl solution as a synthetic seawater model. The evolution in time of the electrochemical behavior of each coating was monitored recording in operando EIS spectra at 0.5, 4, 24, 48, and 72 h after being scratched. EIS measurements were conducted by an Ivium Compactstat (Netherlands) at open circuit potential (OCP). A home-made three-electrode cell was used for the experiments, equipped with an aqueous saturated calomel electrode (SCE), a platinum rod, and the coated steel panel serving as a reference, counter and the working electrode, respectively. For all the samples, the geometric area in contact with the electrolyte was ca. 1 cm2, defined by the diameter of the open bottom of a tapered polyethylene tube sealed to the panel (See Figure S1). The potentiostatic EIS spectra were obtained scanning frequencies from 104 to 10–2 Hz, using a 10-mV amplitude single-sine signal as excitation voltage.
Scanning electron microscopy (SEM) was employed as an objective method to visually assess the effect of the self-healing process and to study the morphology of the resulting repairing polymer as a function of the content of dopamine.
Salt spray test
To provide a further proof of the effectiveness of the here proposed healing strategy, the salt spray test (ASTM B117) was used as an accelerated corrosion test to assess the protection ability of the coatings under different environment conditions with respect to those of the EIS analyses. The coated Q-panels (5 × 15 cm), were prepared and scratched diagonally following the above described procedures, before being inserted into the salt spray chamber for 72 h (5 wt% NaCl solution, 35 ± 2 °C).
Results and Discussion
Characterization of the dopamine-grafted polyetheramine
FTIR and TGA analyses
Figure 2a depicts the FTIR spectra of the reaction mixture at the first modification stage, before and after NaOH addition (to deprotonate the amine group of the dopamine hydrochloride), as well as the FTIR spectrum of the final modified prepolymer 1. As it is marked with pink color, the disappearance of the bands at 3056 cm−1 and 915 cm−1, after the addition of NaOH, reveals the opening of the epoxy rings of 1,6-HDGE. Concurrently, a notable increase in the intensity of the band at 1638 cm−1 is observed, which proves the formation of aliphatic –OH groups as a result of the opening of the ethylene oxides [33]. According to the FTIR results, the intermediate HDGE-grafted dopamine was successfully produced as the first stage toward the synthesis of the dopamine-modified prepolymer 1 (Fig. 1). The excess of non-reacted 1,6-HDGE is maintained into the reaction mixture, directly used in the subsequent reaction step.

FTIR spectra for the Stage 1 intermediate before and after activation of dopamine via the NaOH addition as well as the final dopamine-grafted amine prepolymer 1 (a); TGA/DTGA analyses of modified prepolymer 1 and CTRL (Jeffamine™D230) (b)
FTIR analysis was performed also on the crude reaction mixture (dopamine-grafted prepolymer 1). The FTIR spectrum shows a wide and strong absorbance peak at 3565–3115 cm−1 attributed to O–H stretching vibrations of the catechol groups (invariably present in both the reagent mixture and the first step intermediate), as well as N–H stretching vibrations of primary amine end groups [34]. Accordingly, the appearance of signals around 900–800 cm−1 are attributable to N–H oop of the primary amines. The other absorbance in the range 2996–2787 cm−1 corresponds to C–H stretching vibrations of the alkyl portions. The aliphatic ether part of the structure is responsible for the strong band at 1100 cm−1 (from both HDGE and Jeffamine™D230 components) [35].
TGA and the related DTGA curves for the modified polyetheramine 1 in comparison with that for neat polyetheramine D230 are presented in Fig. 2b. According to the results, three stages of decomposition (spreading from 60 °C to 400 °C) can be detected for compound 1. The first stage, which is starting from 60 °C and is ending at 110 °C, may be attributed to evaporation of the unreacted ingredients (i.e., 1,6 HDGE) or solvents (i.e., methanol and DCM). The second stage, ending at around 260 °C in good agreement with the single process recorded for the pristine control sample, is attributed to the decomposition of free Jeffamine™D230 in the reaction product [30]. The total weight loss for these two stages was around 25%. The last and more significant thermal decomposition stage occurs in the range 260–400 °C and is attributed to the desired reaction product 1 due to the fact that it occurred at a higher temperature with respect to Jeffamine™D230. The total weight loss for this stage was 75% that can be considered an estimation of the yield of the dopamine grafting reaction. This value is in qualitatively good agreement with the stoichiometry of the reaction.
NMR analyses
According to the 1H NMR spectrum of dopamine hydrochloride (Figure S2) the aromatic ring protons show a characteristic fingerprint constituted by a doublet at 6.69 ppm (J = 8 Hz) of the He proton, a doublet of doublets at 6.48 ppm (J = 8 Hz and 4 Hz) of the adjacent, coupled Hf proton and, finally, a doublet at 6.64 ppm of the Hb proton (J = 4 Hz). Peaks at higher fields correspond to the protons of the aliphatic portion of the molecule, Hb′ (2.91 ppm) and Ha′ (2.71 ppm) [36]. The absence of any aromatic protons in the structure of the polyetheramine made the signals of phenyl ring protons of the dopamine useful to prove the presence of the catechol derivative in the prepolymer. Indeed, in the 1H NMR spectrum of 1 (Fig. 3a), some peaks are detected in a window typical of the aromatic protons, with chemical shifts (ranging from 6.66 to 6.37 ppm) compatible with the fingerprint signals of the catechol unit. The more complex structure of the multiplets recorded for 1 could be easily ascribed to the polydispersity of the polyetheramine itself and the fluxional equilibrium of the polymer backbones. The additional signals in the high field region (4.0–3.0 ppm and 1.5–0.7 ppm) are attributable to the aliphatic protons (–CH3, –CH2, and –CH) of the polyetheramine backbone. In particular, peaks centered at 1.47 ppm and 1.27 ppm were attributed to Cβ and Cγ, respectively, of the 1,6-hexanediol units linked to the amine group of dopamine through a 2-hydroxypropyl bridge (Fig. 3a and Figure S3).

1D and 2D NMR characterizations of polyetheramine prepolymer 1 (red curves). From top to bottom: 1H NMR (a), 1H, 13C-HMBC (b), DOSY (c). Dopamine hydrochloride spectra are also reported (green curves) for the sake of comparison
Further confirmation of the presence of dopamine units in the polyetheramine 1 was provided by 13C NMR spectroscopy, recorded with the Attached Proton Test (APT) protocol to further improve the available peak resolution. The proton-decoupled spectrum of 1 (Figure S4) clearly indicates the signals in the region of aromatic carbons, which are not attributable to the pristine polyetheramine but they well match the peaks detected for pure dopamine. Analog conclusions can be derived from heteronuclear single quantum coherence (HSQC) NMR spectrum, which is reported in Supplementary Information (Figure S4).
Heteronuclear Multiple Bond Correlation (HMBC) could provide a first proof of the grafting of dopamine by recording long-range correlations between the dopamine unit (i.e., catechol ring or ethyl sidechain) and the aliphatic backbone of the polyetheramine (Fig. 3b and Figure S5). Most of the net cross-peaks are attributable to long-range interactions already present in the HMBC spectrum of the free dopamine hydrochloride (Figure S6), thereby, circumscribed within the dopamine molecular scaffold (like between the two ortho-positions and the two methylenes of the sidechain). The only exception is given by the -CH2 carbon signal at 32 ppm that shows additional cross-peaks with the multiplets at ca. 3.3 and 1.3 ppm, attributable to protons of the polyether backbone (Fig. 3b).
The final confirmation of the effective grafting of dopamine to product 1 was provided by DOSY NMR experiments. This 2D spectroscopic technique allows to resolve the signals of different molecular species according to their diffusion capability. Each proton (corresponding to a signal in the 1H NMR, recorded in the direct dimension and reported on the horizontal axis in Fig. 3c) is sorted along the indirect dimension (vertical axis) depending on the diffusion coefficient of the molecule to which the proton belongs. As a consequence, protons belonging to molecules with sufficiently different dimensions (i.e., different diffusion capability) will result not aligned on the vertical axis. The more interesting proton signals are, again, those related to the aromatic ring of dopamine, ranging around 6.5 ppm. According to the DOSY spectrum of the reaction product 1 (Fig. 3c), the diffusion coefficient of the aromatic hydrogens (around 7–8 × 10–7 cm2 s−1) is in the same range of the other resonant protons. The good alignment along the indirect dimension of the spectrum of the aliphatic protons (belonging to polyetheramine backbone) and the aromatic ones (belonging to catechol unit) confirms that dopamine units are effectively grafted on the polymeric chain, acting as pendants that move solidly with the rest of the polymer. To validate the hypothesis, DOSY analysis was also performed on free dopamine hydrochloride in the same experimental conditions (Fig. 3c), to ensure that its diffusion coefficient is sufficiently different from that estimated for the related protons in the reaction product sample. In order to avoid any reaction between the free dopamine and the air oxygen as well as dopamine self-polymerization reaction, DMSO-d6 solvent was thoroughly pre-degassed with nitrogen into the NMR tube and the acquisition started immediately after the sample dissolution. According to the results presented in Fig. 3c, the diffusion coefficient of the free dopamine was around 2.3 × 10–6 cm2 s−1, 3-times higher than that of the reaction product 1. Therefore, it can be concluded that the dopamine detected in the reaction product is bonded to a larger molecule that determines a significant increase of the bulkiness with respect to the pure dopamine molecule. The DOSY results confirmed the qualitative conclusions derived from TLC analyses (Figure S7).
Characterization of the amine-containing microcapsules
Morphology and size distribution
For the dual capsule self-healing system, core–shell microcapsules (MCs) embedding separately isocyanate-based and amine-based prepolymers within a poly(styrene-co-acrylonitrile) shell were prepared by electrospray method. Figure 4 shows the SEM micrographs and the corresponding size distribution diagrams of the amine microcapsules containing 10 wt% (MC-D10) and 40 wt% (MC-D40) of prepolymer 1, respectively. According to both images, the prepared MCs are spherical in shape, with a smoother outer shell wall for MC-D40 sample with respect to that of MC-D10. The size distribution diagrams showed that the diameters for both samples were less than 2 μm, with an average particle size of 0.83 ± 0.59 µm and 0.84 ± 0.60 µm for MC-D10 and MC-D40, respectively. This relatively wide range of the capsule size (two orders of magnitude, from a few tens of nanometers to one micron), is beneficial because it will provide better self-healing performance [37, 38]. Comparable shape, diameter, and size distribution were already reported in our previous work for MCs containing isocyanate prepolymer and pure Jeffamine™D230 [30].

SEM micrographs (marker = 1 µm) and size distribution diagrams of the amine-containing microcapsules MC-D10 (a) and MC-D40 (b)
Chemical and thermal characteristics
FTIR analysis was conducted to study the chemical composition of the modified amine MCs and to identify any possible chemical reactions that occurred between the core and shell material. The FTIR spectra of the neat shell material (SAN), neat catechol-modified amine 1, and the crushed core–shell MC-D40 are presented in Fig. 5. FTIR spectrum for the neat SAN (Fig. 5a) shows the main characteristic peaks at 3085–3029, 2924, and 2238 cm−1 attributed to styrene ring C–H stretching, aliphatic C–H stretching, and nitrile group C≡N stretching vibrations, respectively [30]. Regarding the crushed core–shell MC-D40 spectrum (Fig. 5c), the main characteristic peaks of both core (marked with green shadow) and shell (pink one) materials can be easily recognized, diagnosing the successful encapsulation in absence of any significant chemical reaction between the two components. For the sake of clarity, the already discussed spectrum of dopamine-grafted prepolymer 1 is also presented as a representative of core material (Fig. 5b).

FTIR spectra of neat SAN (a), neat prepolymer 1 (b), and crushed MC-D40 (c)
Thermogravimetric analysis was conducted on the neat shell material, the incorporated core materials, and the corresponding assembled MCs to study their thermal stability and calculate the encapsulation yield.
Figure 6a shows the TGA/DTGA curves of the shell polymer (SAN) and the polyetheramine-based core materials (D10 and D40). According to the results, the neat shell material was stable up to 390 °C and its thermal decomposition was majorly recorded around 430 °C. On the other hand, three decomposition stages were recorded for the core materials. The first two stages at lower temperatures (110 °C and 230 °C) correspond to the Jeffamine™D230 while the third thermal process (at 370 °C) is attributed to the modified polyetheramine 1 (see also Fig. 2b) [30]. Interestingly, the relative intensity (i.e., weight variation) of the last two processes well correlates with the relative amount of the two components in each of the core solutions.

Thermogravimetric analysis of: neat SAN (shell), D10 and D40 amine mixtures (core) (a); and related core–shell MC-D10 and MC-D40 (b)
Similar thermogravimetric analyses were carried out on the prepared core/shell MCs (Fig. 6b). According to the results, all the described four thermal-induced processes ascribed to the core and shell components were invariably detected in the MC thermograms, pointing to the concomitant presence of the corresponding components in the MCs. Once again, the relative intensity of each single process perfectly scales with the composition of the microcapsules: SAN contribution to the total weight loss is almost the same, while the contribution of the two components of the core changes accordingly to their relative amount (e.g., the intensity of the decomposition stage of 1 increases going from MC-D10 to MC-D40). For sake of clarity, the temperature ranges and the weight loss percentages for each of the recorded stage is listed in Table S1.
Using TGA results, the percentage yield of the encapsulation process, %α, can be calculated as follows [30, 39]:
where %Wcore-tga is the weight percentage of core material calculated using the TGA results and %Wcore-feed is the core material weight percentage used as feed in the encapsulation process. The amounts of %Wcore-tga, %Wcore-feed, and %α calculated from Eq. 1 are listed in Table S2. The encapsulation process yield for the MC-D10 and MC-D40 was estimated to be 79% and 82%, respectively. The slight, net increase in the encapsulation yield for both mixed-amine MCs with respect to the ca. 71% for the MC-CTRL (i.e., unmodified amine-based ones) [30] could be tentatively due to the change in the amphiphilic nature of the core material as a result of the addition of the catechol-bearing polyetheramine 1 to the main component (i.e., JeffamineTMD230).
The thermogravimetric features of the isocyanate-containing MCs (constituting the second actor of the dual capsule self-healing system) revealed two decomposition stages mainly at 328 °C and 418 °C attributed to the core and shell materials respectively [30].
Assessment of the coatings' underwater self-healing performance through in operando EIS technique
Self-healing coatings for corrosion protection were prepared by embedding into an epoxy resin matrix the isocyanate- and amine-based MCs, with a 1:1 weight ratio as presented in Table 2. The total amount of MCs was adjusted at 3 wt%, based on the formulation that offered the best healing results in the air [30]. To test the underwater self-healing performance, after having assembled the electrochemical cell on the coated steel panel and filled it with the electrolyte (see Materials and Methods section), the coating was cross scratched in such a way as to expose the underlying steel.
EIS was selected as a noninvasive probing technique able to indirectly monitor the growth of the polyurea within the split in the polymer coating (as a result of the spill of the two reactive core materials) and its adhesion features to both the underlying metal surface and the lateral polymeric edges [40]. The monitor action was performed without affecting the self-evolution of the system. The resulting in operando working condition of EIS was assured by a specific instrument setting: the completely self-adjustment of the bias potential (i.e., at the OCP) and the small amplitude of the voltage perturbation (i.e., 10 mV) applied to the electrochemical system to record the output signal. The OCP was the potential spontaneously assumed by each system (i.e., coating + underlying steel) at each specific period of time, while the small amplitude of the sinusoidal exciting signal was small enough not to bring the system away from the equilibrium condition.
Underwater self-healing behavior of the control coating
The extrinsic self-healing strategy adopted for the coatings forced to perform a preliminary test aimed to check that the embedded MCs do not affect the barrier-like protection ability of the intact coatings. This was done comparing the EIS response of the intact self-healing coating (coat-CTRL) with a coating prepared in the same way but without embedding MCs (i.e., a not self-healing system; coat-NO_MCs). Both coatings showed the typical behavior of a system with one time constant (Fig. 7a), which means the systems are modeled by an equivalent circuit made of a unique RC-like parallel connection (inset of Fig. 7a). No electrolyte resistance is detected in both cases, accounting for a very efficient physical/electric isolation of the metal from the aqueous medium by the polymer coatings [41]. Fitting the experimental spectra with the simple circuit made of a resistor in parallel with a constant phase element, CPE (accounting for the non-ideality of the system), the values listed in Table S3 were obtained. In particular, the dielectric feature of the coatings is almost invariant (with an estimated coating capacitance of ca. 0.2 nF cm−2), according to the fact that the composition of the bulk matrix, defining the relative permittivity of the dielectric, is essentially the same in both coatings. On the other hand, the physical barrier feature of the coatings decreases after addition of 3 wt% of MCs (with pore resistance, Rpo, varying from ca. 400 to 30 MΩ cm2 for coat-NO_MCs and coat-CTRL, respectively), but still preserving very high corrosion protection of the underlying metal (being 4 orders of magnitude higher than impedance recorded for naked steel, Figure S8). The observations point out that the MC embedding process causes only a limited increase of the porosity and/or of the defects of the coating that, in turn, produce more conduction pathways into the polymer matrix and a higher water uptake [32, 42].

Bode modulus (filled symbols) and phase (empty symbols) graphs for: undamaged coat-CTRL and the coat-NO_MCs coating (a); coat-CTRL scratched in air and in underwater conditions (b). Lines are the corresponding fitted spectra. Electrolyte: 3.5 wt% NaCl solution
Producing a macroscopic defect (e.g., a crossed scratch) in the self-healing coat-CTRL significantly modified the impedance spectrum of the sample because of the direct exposure of the naked metal surface to the highly corrosive 3.5 wt% NaCl solution (Fig. 7b). The new interface (i.e., metal|solution one) is involved in the redox reactions that trigger the corrosion process of the metal, with a characteristic time constant spanning from seconds to few tens of seconds (corresponding to characteristic frequencies from ca. 1 to 0.1 Hz, respectively). The underwater self-healing performance of the control coat-CTRL was evaluated with respect to an analogous sample that was scratched and kept in air for seven days, to allow the self-repairing process, before being immersed into the test solution. For a better comparison, the EIS spectrum for the air-healed sample was recorded immediately after the immersion in the testing solution, while for wet-healed samples 24 h were waited to allow the healing reaction (see also the discussion in “Time dependence study of self-healing and adhesion features through in operando EIS” section). The EIS spectra of the wet versus air-healed samples are qualitatively comparable in both the shape of the curve and the magnitude of the impedance (Fig. 7b). The results demonstrated a good water insensitivity of the dual-capsule system employed; in particular, water seems not to significantly affect neither the release of the core contents of the microspheres nor the amine-isocyanate reactions. As a result, after 24 h in an aqueous solution, a protective film was produced within the crack, with a protection ability toward water penetration comparable to that of the film grown in the air.
Time dependence study of self-healing and adhesion features through in operando EIS
Besides the effectiveness of the self-repairing performance of the dual-capsule system, the stability and the adhesion of the grown polyurea film to the underlying metal and/or the epoxy edges is another critical key parameter to be considered especially for underwater applications. The modeling of the EIS response of polymer-coated metals, as well as the specific subset made by scratched self-healing coatings, is not a simple matter, according to the many different equivalent circuits proposed in the open scientific literature [13, 43, 44]. It can be attributed to the high number of possible coexisting interfaces, which relative relevance changes also with the exposition time [43]. By the outermost, the following interfaces could be found: electrolyte|coating, electrolyte|healing layer, electrolyte|metal oxide, electrolyte|metal. The higher number of interfaces, the higher number of time constants of the equivalent circuit (corresponding to the number of peaks in a Bode phase diagram), and the higher electric circuit elements that should be fitted together. In this study, a two-time constant equivalent circuit was used for modeling EIS results according to the two breakpoint frequencies identifiable in many of the recorded spectra [41, 43, 45, 46] (Figure S9). In ascending order of the time constant, the fitted parameters are (Fig. 7b): Rcoat and CPEcoat, Rct and CPEdl corresponding to the coating resistance to the passage of electrolyte, the capacitance of the coating, the charge transfer resistance across the metal interface (i.e., corrosion phenomenon) and the capacitance of the double layer at metal|solution interface, respectively. Rs corresponds to the sum of the ohmic resistances of the system, typically the electrolyte resistance. The employment of a constant phase element (CPE) was preferred to a capacitor, as mentioned above, to better take into account the non-ideality of the metal/coating system (e.g., current leakage, non-uniform porosity, surface inhomogeneity). Due to the restricted value of the frequency scan's upper vertex, the solution resistance cannot be properly evaluated, and so it will no longer be considered. The best-fitting values of the circuit elements are listed in Table S4.
Control coating
The underwater self-healing process and the stability of the resulting repaired coat-CTRL were indirectly monitored by following the variations of the EIS spectra recorded at different times of the healing process after the scratch (Fig. 8, Figure S9). The charge transfer resistance (Rct), inversely proportional to the corrosion rate [47], was selected as the suitable probe, being related to the barrier effect of the coating. According to literature, variation to Rct values is proportional to the extent of the metal surface in contact with the solution (i.e., corroding area): positive variation (i.e., increase of Rct) is related to coverage phenomena (i.e., thickening or enlargement of the protective coating), while negative variation is related to delamination phenomenon (i.e., detachment of the coating). The relationship can be summarized as follows:
where Rct and Rct0 stand, respectively, for the charge transfer resistance of the coated and uncoated metal, and Ad the delaminated area (equal to the corroding area), with both Rct and Ad varying with the exposure time [48]. For coat-CTRL, the charge transfer resistance showed an inverted V-shape trend, with a maximum after 24 h from the underwater scratching (Fig. 9). This trend evidences a competition between two antithetic processes. One leads to a reduction (the healing process) and the other causes an acceleration (polyurea detachment) of the corrosion rate. The needle of the balance moves toward the delamination process for an exposition time longer than 1 day, proving a limited adhesion of the fresh polyurea film to the wet surfaces of metal and/or original polymer matrix. The higher slope of the growing branch with respect to the decreasing one suggests that the healing process was faster than the detachment process for the times lower than 24 h. In contrast, the situation is reversed for a longer period, even if the rate of the delamination is partially mitigated by the continuous formation of polyurea resulting from the reaction of amines and isocyanates leaked into the widening crack. Comparing the EIS spectra of the defective coating after 24 h (Fig. 7b) with that of the intact coating (Fig. 7a) reveals that the underwater healing efficiency is relatively low, tentatively hampered by the competing detachment process caused by an insufficient adhesion.

Nyquist plots of carbon steel plates coated with coat-CTRL (a), coat-D10 (b), and coat-D40 (c) in 3.5 wt% NaCl solution. Points are experimental values, while lines are fitted data. Spectra were recorded at the self-adjusting OCP at different times after the underwater scratch

Dependence of charge transfer resistance (Rct) of the scratched coatings by the exposition time in a 3.5 wt% NaCl solution (a). Comparison of Nyquist plots after 72 h of immersion, where points are experimental values while lines are fitted data (b)
Dopamine-bearing coatings
The adhesion of the repairing polymer significantly affects the self-healing efficiency, especially for underwater conditions where the interfacial water, that is the layer of water molecules adsorbed onto the metal surface (after the formation of a defect in the coating), has deleterious effects on the tackiness of the repairing polymer. To address the observed adhesion issue of the coat-CTRL, the anchoring ability of catechol units [19, 23] are adopted through the modification of the prepolymer constituting the core of the amine MCs (coat-D10 and coat-D40 samples; Table 2).
The evolution of the self-repairing systems was monitored again by recording EIS spectra at open circuit potential at different times after the creation of a defect in the polymer coating (Fig. 8 and Figure S9). The variation over time of the computed Rct parameter is reported in Fig. 9a. Coat-D10 exhibited improved behavior in comparison with the control system, reaching the best corrosion protection performance after 1 day of the exposition. Two significant differences can be found out: (i) the slope of the increasing branch of the curve is steeper than the coat-CTRL; (ii) the decreasing branch exhibits a comparable steepness with respect to coat-CTRL, while Rct is invariably higher. All these clues point to a modest increment of the adhesion of the resewing polymer, allowing the healing process to better compete with the delamination one. The situation is substantially boosted for the coat-D40. The corrosion protection offered by polyurea monotonically increases (Fig. 9a), reaching 4-times higher charge transfer resistance with respect to that of the coat-D10 loaded with one-fourth of the modified prepolymer 1, pointing to a linear correlation between the amount of catechol-bearing amine and the healing rate. Clearly, the last hypothesis demands a future better assessment. The monotonic increase of Rct value with time for coat-D40 is attributable to a very efficient adhesion of modified polyurea to the wet surfaces of the crack that makes detachment only a marginal event. A final, but interesting, observation comes from the comparison of the slopes of the increasing branch of Rct versus time plots for the three coatings (Fig. 9a). As derived by Eq. 2, the slope defines the coverage rate of the exposed metal surface due to the self-healing reaction. In other words, the slope relates to the actual polymerization reaction rate, if no delamination process occurs. Considering that for all the three coatings the amount of healing components is the same, in both absolute (3 wt%) and relative amount (1:1 amine-isocyanate weight ratio), an identical intrinsic rate for the polymerization reaction is expected. Considering the constant width of the scratches, a similar rate of healing (slope) is expected for the three coatings. The different steepness experimentally observed allows us to rank the adhesion strength of the growing polymer: higher the slope, stronger is the adhesion. Therefore, all experimental data prove that the rate of the repairing and, as a consequence, the corrosion protection ability of the coating improves by increasing the content of dopamine-grafted amine (Fig. 9) that in turns efficiently hampers the delamination process due to a stronger adhesion provided by a higher amount of effective anchoring sites (i.e., catechol units).
Besides EIS, the healing process was also assessed through an independent electrochemical technique, that is open circuit potential (OCP) monitoring (Figure S10). Similar to the EIS study, OCP recording definitely does not affect the natural evolution of the process under investigation, being a passive monitoring of the evolution of the free corrosion potential of the system. Thus, it represents a useful tool to monitor self-healing performance [46, 49]. At the initial stage (30 min after the scratch), all the scratched coatings had almost the same OCP value, more negative than that of an intact coating (ca. −0.68 V and + 0.20 V vs SCE, respectively), compatible with the exposition of a portion of bare steel to the aqueous environment. By increasing the exposure time, OCP shifts toward more positive values, a fact attributed to the release of the healing agents and their subsequent underwater reaction. In more detail, a monotonically increase of the OCP was recorded over the whole immersion time for coat-D40, while a slightly deviation toward more negative values was observed for both coat-CTRL and coat-D10 samples after 24 h. The two types of trend of the free potential can be the result of a different morphology of the growing layer: uniform/compact for coat-D40, and poorly continuous for coat-D10 and the coat-CTRL (see also SEM investigation). As a result, the more compact healing layer hampers (or prevents) further permeation of the medium in the scratched down to the underlying steel. These observations are quite well in agreement with the previous reported EIS results, confirming the positive effect of the catechol groups on the healing performance.
Visual evaluation of the self-healing process
SEM investigation was carried out to study the healing process [50, 51]. Figure 10 shows the micrographs of the scratched samples after immersing in 3.5 wt% NaCl solution. Figure 10a reveals that the artificial scratch is not healed for the epoxy coating without MCs, while the scratch on the coat-CTRL is partially filled by the released healing agents that form an irregular and porous matrix (Fig. 10b). As a consequence, the grown healing polymer could not make a compact barrier film possibly due to its low tendency to stick on the wet surfaces (both of the cut edges of the coating and of the growing polymer itself). The uniformity and compactness of the healed area increase for the samples containing dopamine-bearing healing agents (Fig. 10c and d). This improvement might be due to their higher water compatibility and better reaction with the wet surfaces derived by grafted catechol groups that allowed better resistance against the invasion of corrosive medium. According to the SEM micrographs, the artificial scratch-made to the coat-D40 sample (Fig. 10d) is properly healed with a dense and continuous barrier film confirming the hypotheses deduced from both Rct and OCP trends.

SEM micrographs of the scratched coatings after 72 h of immersion in a 3.5 wt% NaCl solution: coat-NO_MCs (a), coat-CTRL (b), coat-D10 (c), and coat-D40 (d)
Salt spray test
To further test the healing performance of the here proposed engineered coatings, the specimens were scratched and then immediately moved into a salt spray chamber. Figure 11 shows the scratched coatings after 72 h of exposure to salt spray. According to the results, the corroded area is visible along with the scratch for the coat-CTRL (Fig. 11a) due to the NaCl solution penetration, while the corrosion is significantly decreased in the dopamine-bearing samples. By visual inspection, the amount of the reddish corrosion products, accumulated into the scratch, decreases by increasing the content of dopamine-grafted healing agent into the protective coating (Fig. 11b and c). Again, this phenomenological result points to an effective chemically-driven adhesion process between the dopa-modified healing agent and the wet substrate/matrix surfaces. The results of the visual inspection of steel panels after salt spray test well correlate with the outcomes of the electrochemical investigations.

Salt spray results (72 h) for Q-panel of steel covered with coat-CTRL (a), coat-D10 (b), and coat-D40 (c)
Possible mechanism of the healing process with the dopamine-bearing agent
Figure 12 shows a cartoon of the plausible, but tentative, reactions occurring in dopamine-containing coating samples, during the underwater healing process, which can explain their improved adhesion. As can be seen, there are three main reactions that can occur upon the capsules rupture.

The plausible reactivity of the functional groups of the healing agents during underwater self-repairing for dopamine-bearing coatings
The first one (blue shadow, in Fig. 12) is the condensation reaction between the amine and isocyanate functionalities giving raise to polyurea formation. This is a fast and water insensitive crosslinking reaction that occurs either between the two released healing agents or the NCO-bearing core material and the amine groups of the coating matrix (dark blue shadow).
The second possible reaction (green shadow) is the polyurethane formation reaction. This side reaction is not fast but is effective when the released isocyanates do not combine with any amine groups. Therefore, these isocyanate groups react with water or hydroxyl groups of the matrix and/or of the metal oxide layer.
The third process (red shadow) is the most relevant one for assuring a superior adhesion to the underwater growing polymers. Contrary to the above-described processes, this last one does not lead to any fully covalent bond but induces an intermolecular bonding interaction between an electron donor and a hydrogen-bonded to an electronegative atom [22]. A fast and strong hydrogen bonding formation is expected between the hydroxyl units of two catechol groups, as well as between the catechol functionality and the hydroxylated metal and/or matrix surface. Except for the homo-interactions (light red shadow), the two hetero ones ensure effective adhesion of the self-healing film and, hence, a superior underwater corrosion protective performance. This result is obtained due to the tendency of the catechol unit to make stronger hydrogen bonds with oxide surfaces with respect to water molecules [22, 36], the last invariably adsorbed onto the surfaces of immersed samples causing the poor adhesion commonly observed on wet surfaces.
Conclusions
The present work has presented a proof-of-concept methodology to enrich epoxy coatings of an effective underwater self-healing feature through the chemical engineering of the healing agent with catechol units acting as anchoring sites. The modification reaction was conducted in two stages. Dopamine (i.e., catechol group donor) was firstly grafted to 1,6 HDGE glycidyl ether that was then condensed with a commercial-grade polyetheramine (Jeffamine™D230). The resulting prepolymer with primary amine functional end groups and catechol-containing side groups (1) was then encapsulated (at different dilution ratio) within SAN shells and used in a dual capsule-based self-healing system exploiting polyurea as sewing material. The amine MCs were finally embedded, together with isocyanate-based ones, in epoxy coatings for further healing and corrosion protection assessment. In operando EIS tests revealed for coat-D40 the fastest and most effective healing performance in underwater conditions. Analyzing the different trends in time of charge transfer resistance (from EIS) and open circuit potential for the coated specimens, the best performance of coat-D40 was attributed to the improved adhesion features offered by dopamine units that, undermining interfacial water from the wet surfaces, act as strong anchoring sites. This hypothesis was further supported by the denser and more compact morphology shown by the healed polymer according to a SEM investigation.
As a result, dopamine grafting has been proven a valuable approach to increase the self-repairing ability of damaged epoxy coatings even in presence of water. Therefore, a highly responsive healing agent was proposed for the condition that many coated metal objects can encounter during their real service life, or because constantly dipped into an aqueous environment (e.g., submerged infrastructures) or subjected to wet conditions (e.g., car bodyworks under rain).
References
Toohey KS, Sottos NR, Lewis JA, Moore JS, White SR (2007) Self-healing materials with microvascular networks. Nat Mater 6(8):581–585. https://doi.org/10.1038/nmat1934
Blaiszik BJ, Kramer SL, Olugebefola SC, Moore JS, Sottos NR, White SR (2010) Self-healing polymers and composites. Annu Rev Mater Res 40:179–211. https://doi.org/10.1146/annurev-matsci-070909-104532
Wang Z, Scheres L, Xia H, Zuilhof H (2020) Developments and challenges in self-healing antifouling materials. Adv Funct Mater 30:1908098. https://doi.org/10.1002/adfm.201908098
Esmaeely Neisiany R, Enayati MS, Sajkiewicz P, Pahlevanneshan Z, Ramakrishna S (2020) Insight into the current directions in functionalized nanocomposite hydrogels. Front Mater 7:25. https://doi.org/10.3389/fmats.2020.00025
Billiet S, Hillewaere XK, Teixeira RF, Du Prez FE (2013) Chemistry of crosslinking processes for self-healing polymers. Macromol Rapid Commun 34(4):290–309. https://doi.org/10.1002/marc.201200689
Wang Z, Lu X, Sun S, Yu C, Xia H (2019) Preparation, characterization and properties of intrinsic self-healing elastomers. J Mater Chem B 7(32):4876–4926. https://doi.org/10.1039/C9TB00831D
Ghomi ER, Khorasani SN, Kichi MK, Dinari M, Ataei S, Enayati MH, Koochaki MS, Neisiany RE (2020) Synthesis and characterization of TiO2/acrylic acid-co-2-acrylamido-2-methyl propane sulfonic acid nanogel composite and investigation its self-healing performance in the epoxy coatings. Colloid Polym Sci 298(2):213–223. https://doi.org/10.1007/s00396-019-04597-0
Kim D-M, Cho Y-J, Choi J-Y, Kim B-J, Jin S-W, Chung C-M (2017) Low-temperature self-healing of a microcapsule-type protective coating. Materials 10(9):1079. https://doi.org/10.3390/ma10091079
Cho SH, Andersson HM, White SR, Sottos NR, Braun PV (2006) Polydimethylsiloxane-based self-healing materials. Adv Mater 18(8):997–1000. https://doi.org/10.1002/adma.200501814
Yang J, Keller MW, Moore JS, White SR, Sottos NR (2008) Microencapsulation of isocyanates for self-healing polymers. Macromolecules 41(24):9650–9655. https://doi.org/10.1021/ma801718v
Wang Q, Pan X, Lin C, Ma X, Cao S, Ni Y (2020) Ultrafast gelling using sulfonated lignin – Fe3+ chelates to produce dynamic crosslinked hydrogel/coating with charming stretchable, conductive, self-healing, and ultraviolet-blocking properties. Chem Eng J 396:125341. https://doi.org/10.1016/j.cej.2020.125341
Huang M, Yang J (2011) Facile microencapsulation of HDI for self-healing anticorrosion coatings. J Mater Chem 21(30):11123–11130. https://doi.org/10.1039/C1JM10794A
Huang M, Yang J (2014) Salt spray and EIS studies on HDI microcapsule-based self-healing anticorrosive coatings. Prog Org Coat 77(1):168–175. https://doi.org/10.1016/j.porgcoat.2013.09.002
Zhang Z, Hu Y, Liu Z, Guo T (2012) Synthesis and evaluation of a moisture-promoted healing copolymer. Polymer 53(14):2979–2990. https://doi.org/10.1016/j.polymer.2012.04.048
Guo M, Li W, Han N, Wang J, Su J, Li J, Zhang X (2018) Novel dual-component microencapsulated hydrophobic amine and microencapsulated isocyanate used for self-healing anti-corrosion coating. Polymers 10(3):319. https://doi.org/10.3390/polym10030319
Holten-Andersen N, Harrington MJ, Birkedal H, Lee BP, Messersmith PB, Lee KYC, Waite JH (2011) pH-induced metal-ligand cross-links inspired by mussel yield self-healing polymer networks with near-covalent elastic moduli. In: Proceedings of the National Academy of Sciences, vol 7, pp 2651–2655
Krogsgaard M, Behrens MA, Pedersen JS, Birkedal H (2013) Self-healing mussel-inspired multi-pH-responsive hydrogels. Biomacromol 14(2):297–301. https://doi.org/10.1021/bm301844u
Habibiyan A, Ramezanzadeh B, Mahdavian M, Bahlakeh G, Kasaeian M (2019) Rational assembly of mussel-inspired polydopamine (PDA)-Zn (II) complex nanospheres on graphene oxide framework tailored for robust self-healing anti-corrosion coatings application. Chem Eng J 391:123630. https://doi.org/10.1016/j.cej.2019.123630
Ahn BK (2017) Perspectives on mussel-inspired wet adhesion. J Am Chem Soc 139(30):10166–10171. https://doi.org/10.1021/jacs.6b13149
Petrone L, Kumar A, Sutanto CN, Patil NJ, Kannan S, Palaniappan A, Amini S, Zappone B, Verma C, Miserez A (2015) Mussel adhesion is dictated by time-regulated secretion and molecular conformation of mussel adhesive proteins. Nat Commun 6:8737. https://doi.org/10.1038/ncomms9737
North MA, Del Grosso CA, Wilker JJ (2017) High strength underwater bonding with polymer mimics of mussel adhesive proteins. ACS Appl Mater Inter 9(8):7866–7872. https://doi.org/10.1021/acsami.7b00270
Yu J, Kan Y, Rapp M, Danner E, Wei W, Das S, Miller DR, Chen Y, Waite JH, Israelachvili JN (2013) Adaptive hydrophobic and hydrophilic interactions of mussel foot proteins with organic thin films. Proc Natl Acad Sci 110(39):15680–15685. https://doi.org/10.1073/pnas.1315015110
Ahn BK, Lee DW, Israelachvili JN, Waite JH (2014) Surface-initiated self-healing of polymers in aqueous media. Nat Mater 13(9):867–872. https://doi.org/10.1038/nmat4037
Li J, Ejima H, Yoshie N (2016) Seawater-assisted self-healing of catechol polymers via hydrogen bonding and coordination interactions. ACS Appl Mater Inter 8(29):19047–19053. https://doi.org/10.1021/acsami.6b04075
Amendola V, Meneghetti M (2009) Self-healing at the nanoscale. Nanoscale 1(1):163–164. https://doi.org/10.1039/B9NR00146H
Wang L, Shi Y, Chen S, Wang W, Tian M, Ning N, Zhang L (2017) Highly efficient mussel-like inspired modification of aramid fibers by UV-accelerated catechol/polyamine deposition followed chemical grafting for high-performance polymer composites. Chem Eng J 314:583–593. https://doi.org/10.1016/j.cej.2016.12.015
Li Z, Shan Y, Wang X, Li H, Yang K, Cui Y (2020) Self-healing flexible sensor based on metal-ligand coordination. Chem Eng J 394:124932. https://doi.org/10.1016/j.cej.2020.124932
Qian B, Zheng Z, Michailids M, Fleck N, Bilton M, Song Y, Li G, Shchukin D (2019) Mussel-inspired self-healing coatings based on polydopamine-coated nanocontainers for corrosion protection. ACS Appl Mater Inter 11(10):10283–10291. https://doi.org/10.1021/acsami.8b21197
Chen C, Xiao G, He Y, Zhong F, Li H, Wu Y, Chen J (2020) Bio-inspired superior barrier self-healing coating: Self-assemble of graphene oxide and polydopamine-coated halloysite nanotubes for enhancing corrosion resistance of waterborne epoxy coating. Prog Org Coat 139:105402. https://doi.org/10.1016/j.porgcoat.2019.105402
Koochaki MS, Khorasani SN, Neisiany RE, Ashrafi A, Magni M, Trasatti SP (2019) Facile strategy toward the development of a self-healing coating by electrospray method. Mater Res Express 6(11):116444. https://doi.org/10.1088/2053-1591/ab4d1b
Chiriac CI, Tanasǎ F (2000) Polyureas. In: Chadwick SS (ed) Ullmann's Encyclopedia of Industrial Chemistry. Reference Services Review
Ataei S, Khorasani SN, Torkaman R, Neisiany RE, Koochaki MS (2018) Self-healing performance of an epoxy coating containing microencapsulated alkyd resin based on coconut oil. Prog Org Coat 120:160–166. https://doi.org/10.1016/j.porgcoat.2018.03.024
Nikolic G, Zlatkovic S, Cakic M, Cakic S, Lacnjevac C, Rajic Z (2010) Fast fourier transform IR characterization of epoxy GY systems crosslinked with aliphatic and cycloaliphatic EH polyamine adducts. Sensors 10(1):684–696. https://doi.org/10.3390/s100100684
Jiang J, Zhu L, Zhu L, Zhu B, Xu Y (2011) Surface characteristics of a self-polymerized dopamine coating deposited on hydrophobic polymer films. Langmuir 27(23):14180–14187. https://doi.org/10.1021/la202877k
Chen B, Li J, Liu T, Dai Z, Zhao H (2018) Facile preparation of epoxy based elastomers with tunable T gs and mechanical properties. RSC Adv 8(24):13474–13481. https://doi.org/10.1039/C8RA00894A
Xu LQ, Pranantyo D, Neoh K-G, Kang E-T, Teo SL-M, Fu GD (2016) Synthesis of catechol and zwitterion-bifunctionalized poly(ethylene glycol) for the construction of antifouling surfaces. Polym Chem 7(2):493–501. https://doi.org/10.1039/C5PY01234A
Sun J, Wang Y, Li N, Tian L (2019) Tribological and anticorrosion behavior of self-healing coating containing nanocapsules. Tribol Int 136:332–341. https://doi.org/10.1016/j.triboint.2019.03.062
Kouhi M, Mohebbi A, Mirzaei M, Peikari M (2013) Optimization of smart self-healing coatings based on micro/nanocapsules in heavy metals emission inhibition. Prog Org Coat 76(7):1006–1015. https://doi.org/10.1016/j.porgcoat.2013.02.014
Yao L, Yuan YC, Rong MZ, Zhang MQ (2011) Self-healing linear polymers based on RAFT polymerization. Polymer 52(14):3137–3145. https://doi.org/10.1016/j.polymer.2011.05.024
Mohammadkhani R, Ramezanzadeh M, Akbarzadeh S, Bahlakeh G, Ramezanzadeh B (2020) Graphene oxide nanoplatforms reduction by green plant-sourced organic compounds for construction of an active anti-corrosion coating; experimental/electronic-scale DFT-D modeling studies. Chem Eng J 397:125433. https://doi.org/10.1016/j.cej.2020.125433
Mansfeld F (1990) Electrochemical impedance spectroscopy (EIS) as a new tool for investigating methods of corrosion protection. Electrochim Acta 35(10):1533–1544. https://doi.org/10.1016/0013-4686(90)80007-B
Gan SN, Shahabudin N (2019) Applications of Microcapsules in Self-Healing Polymeric Materials. In: Microencapsulation-Processes, Technologies and Industrial Applications. IntechOpen
Njoku DI, Cui M, Xiao H, Shang B, Li Y (2017) Understanding the anticorrosive protective mechanisms of modified epoxy coatings with improved barrier, active and self-healing functionalities: EIS and spectroscopic techniques. Sci Rep 7(1):1–15. https://doi.org/10.1038/s41598-017-15845-0
Shchukin DG, Zheludkevich M, Yasakau K, Lamaka S, Ferreira MG, Möhwald H (2006) Layer-by-layer assembled nanocontainers for self-healing corrosion protection. Adv Mater 18(13):1672–1678. https://doi.org/10.1002/adma.200502053
Wang W, Xu L, Li X, Lin Z, Yang Y, An E (2014) Self-healing mechanisms of water triggered smart coating in seawater. J Mater Chem A 2(6):1914–1921. https://doi.org/10.1039/C3TA13389C
Hasanzadeh M, Shahidi M, Kazemipour M (2015) Application of EIS and EN techniques to investigate the self-healing ability of coatings based on microcapsules filled with linseed oil and CeO2 nanoparticles. Prog Org Coat 80:106–119. https://doi.org/10.1016/j.porgcoat.2014.12.002
Bard AJ, Faulkner LR, Leddy J, Zoski CG (1980) Electrochemical methods: fundamentals and applications. Wiley, New York
Mansfeld F, Tsai C (1991) Determination of coating deterioration with EIS: I basic relation. Corrosion 47(12):958–963. https://doi.org/10.5006/1.3585209
Zhou S, Mao J (2020) Evaluation of anticorrosive and self-healing performances of TiO2-added cerium conversion coatings developed on 211Z aluminium alloy. Mater Res Express 7(2):026556
Li H, Cui Y, Li Z, Zhu Y, Wang H (2018) Fabrication of microcapsules containing dual-functional tung oil and properties suitable for self-healing and self-lubricating coatings. Prog Org Coat 115:164–171
Khorasani SN, Neisiany RE (2020) Chapter 5 - Characterization of self-healing polymeric materials. In: Thomas S, Surendran A (eds) Self-healing polymer-based systems. Elsevier, Amsterdam, pp 123–140. https://doi.org/10.1016/B978-0-12-818450-9.00005-2
Acknowledgements
Iranian Ministry of Science, Research, and Technology (MSRT) is sincerely acknowledged for the monetary support of M. S. K. during his research visit to Università degli Studi di Milano (UniMi) and accomplishment of this research. SmartMatLab Centre and Dr. Serena Cappelli (Department of Chemistry, UniMi) are appreciated for the thermal-gravimetric analyses. The authors thank the UNITECH NoLimits and Dr. Nadia Santo for the SEM investigation. Prof. Giuseppe Cappelletti (Department of Chemistry, UniMi) is also appreciated for the access to the universal film applicator. M. M. deeply thanks Pasquale Illiano (Department of Chemistry, UniMi) for allowing machine time for the NMR characterization.
Funding
Open access funding provided by Università degli Studi di Milano within the CRUI-CARE Agreement. This work was supported by the Iran National Science Foundation [Grant No. 97008660].
Author information
Authors and Affiliations
Contributions
MSK performed the experiments (syntheses and characterizations), interpreted the results, and wrote the paper draft; MM designed and supervised the EIS and NMR study, interpreted related results, and performed the review of the draft; REN and AA interpreted the results and edited the final draft; SNK and SPT edited the final draft and supervised the project.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Handling Editor: Maude Jimenez.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koochaki, M.S., Khorasani, S.N., Neisiany, R.E. et al. A highly responsive healing agent for the autonomous repair of anti-corrosion coatings on wet surfaces. In operando assessment of the self-healing process. J Mater Sci 56, 1794–1813 (2021). https://doi.org/10.1007/s10853-020-05332-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-020-05332-9