
Abstract
During the investigations, functionalization of graphene oxide synthesized using modified Hummers’ method and its reduced form was performed. Mixtures of graphene oxide and reduced graphene oxide with pimelic acid and calcium hydroxide were prepared for functionalization. During the reaction, the molecules of pimelic acid were attached to the surface of graphene sheets. By forming links between the carboxylic groups of pimelic acid and graphene oxide, the durable connection was achieved. The functionalized graphene oxide and the reduced graphene oxide were used as additives in isotactic polypropylene crystallization. The influence of additives on crystallisation in non-isothermal conditions was examined using polarized optical microscopy and differential scanning calorimetry. The effect of the additives on the polypropylene structure was analysed using wide-angle X-ray scattering. For both functionalized compounds, the nucleating ability towards β-form of polypropylene was detected. Both additives showed the increase in the nucleation rate and promotion of growth of the β-form crystals. Nucleation efficiency similar to other nucleating agents used in the crystallization of the β-form of polypropylene was revealed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Isotactic polypropylene is one of the commonly and widely used high-performance polymers. Due to a low price, good processability, low density, high chemical and biological resistance and various additional advantages, the polypropylene is widely applied for the production of fibres, textiles, foils, mouldings and other products utilized in various areas of human activity.
As a typical thermoplastic polymer, polypropylene crystallizes forming several polymorphic modifications. The best-known forms include monoclinic α, trigonal β, orthorhombic γ and smectic mesophase [1, 2]. The α-form, with the unit cell parameters: a = 0.666 nm, b = 2.078 nm, c = 0.650 nm and β = 99.62° [3], is the most thermodynamically stable. It usually forms in ordinary processing conditions and occurs commonly in the polypropylene articles. The β-form is less stable and less frequently found in the final products. The presence of the β-phase within the crystalline portion of the material is beneficial to its macroscopic toughness. Products with high β-form content display the greatest impact strength, the largest elongation at break, and the highest heat deformation temperature [4]. For this reason, for many years the β form has been drawing researchers’ attention.
The β-form, with the unit cell parameters: a = b = 1.101 nm, c = 0.65 nm, α = β = 90° and γ = 60° [5], is formed under special crystallization conditions [6, 7]. The methods of preparation of the β-form described in the literature include: crystallization after quenching to a certain temperature range [8], crystallization in a specific temperature gradient [9], crystallization of melt subjected to shear or elongation [10, 11] or crystallization in the presence of selective β-nucleating agents (β-NA) [4]. The last method, based on the application of selective β-NA, is the most convenient and reliable. The technique was repeatedly used in laboratory and processing conditions to prepare samples or products rich in the β-modification.
The investigation revealed that various substances have the ability to nucleate the β-form. In earlier publications, the nucleating ability towards the β-form was reported for: aromatic aminosulphur compounds with coplanar phenyl rings [12], pimelic acid and calcium stearate [13,14,15], calcium salts of suberic and pimelic acids [16], N′,N″-dicyclohexylnaphthalene-2,6-dicarboxamide [17, 18], calcium salts of dicarboxylic acids [19], aryl amide derivative [20] and organic pigments [21,22,23]. During the studies special attention was given to the quinacridone pigment considered as one of the most effective β-NA. By adding the quinacridone pigment injection mouldings [24], films [25, 26] and fibres with high β-form content were obtained [27,28,29].
In recent investigations, a novel effective β-NA based on zinc salt of bicycle hept-5-ene-2-dodecanamide-3-carboxylate (NBDA30) [30] and zinc phthalate generated in situ by adding phthalic anhydride and zinc oxide during polypropylene extrusion were developed [31].
Moreover, attention of researches has been drawn in particular to a new group of nucleating agents based on graphene and graphene oxide (GO) derivatives. During the examinations of crystallization behaviour of polypropylene modified with exfoliated graphite, the reduction of the barrier to nucleation was observed [32, 33]. Similarly, the nucleating ability of the GO nanosheets was revealed [34].
The efficiency of nucleating agents in the polymer crystallization depends on their dispersal in the polymer matrix. It is difficult to achieve a homogeneous and fine dispersion of nucleating agents based on graphene or GO in hydrophobic polypropylene. Graphene built from sheets of carbon atoms arranged in a honeycomb structure shows a great tendency to agglomerate. The GO composed of graphitic domains and oxidation regions with carbonyl and carboxyl groups at the edges and epoxy and hydroxyl groups located on the basal planes is strongly hydrophilic and hardly dispersed in non-polar polypropylene. Low dispersibility of both graphene and GO considerably limits their nucleation efficiency.
To improve the ability to form a fine dispersion in the polypropylene matrix, the functionalization of GO was performed. Through grafting, the hydrophobic groups on the GO surface products compatible with hydrophobic polypropylene were obtained. The GO modified with linear alkyl chain [35], dodecyl amine [36] and phosphorus acid [37] exhibited better dispersion. Simultaneously, with better dispersibility the nucleating ability of the functionalized GO was improved.
During the examinations of the PP/GO composites, it was stated that functionalized GO nucleated first all the α-form of polypropylene. To obtain the PP/GO composites rich in the β-form, the tests with the addition of selective β-NA were performed. The investigations on the polypropylene doped with bi-component β-NA composed from pimelic acid/calcium stearate revealed the competition between α-nucleation effect of GO and β-nucleation effect of pimelic acid [38]. It was stated that the nucleation efficiency of GO is higher than the efficiency of conventional β-NA and that it is difficult to obtain the high content of the β-form. Enhancement of the β-form content in β-nucleated PP/GO composites was possible through the crystallization of the pre-ordered structure [39, 40].
In addition to the studies involving the classic β-NA added to PP/GO composites, the literature includes two papers discussing the preparation of selective β-NA based on the functionalized GO. In the first examinations, the β-NA was obtained in two-stage modification by supporting N,N′-dicyclohexyl-1,5-diamino-2,6-naphthalenedicarboxamide onto the surface of previously octadecylamine-functionalized GO [41]. The β-NA obtained in the two-stage process enabled the formation of samples with the high β-form content characterised by enhanced toughness and tensile strength. In other examinations, the effective β-NA in the single-stage process was prepared. In this case, the GO was functionalized with pimelic acid in the presence of calcium hydroxide [42]. The β-NA formed in this process possessed high thermal stability and effectively promoted the formation of the β-form even at low concentration.
In our investigations on the functionalization of graphene derivatives, we applied the technique similar to the second procedure. Graphene oxide (GO) and reduced graphene oxide (rGO) were obtained, and then both products were subject to functionalization with pimelic acid (PA) in the presence of calcium hydroxide. The structure and morphology of both functionalized compounds were examined. Then the crystallization behaviour of isotactic polypropylene doped with the functionalized GO and rGO in non-isothermal conditions was analysed.
Experimental
Materials
Graphite powder < 20 µm was purchased from Sigma-Aldrich. Other reagents, including sulphuric acid (98% H2SO4), permanganate potassium (KMnO4), hydrogen peroxide (30% H2O2), acetone, calcium hydroxide (Ca(OH)2), were supplied by Chempur® and used directly without further purification. Pimelic acid (PA) was supplied by Sigma-Aldrich. Commercial isotactic polypropylene Moplen HP462R (MFI = 25 g/10 min) was supplied by LyondellBasell.
Preparation of graphene oxide (GO) and reduced graphene oxide (rGO)
Graphene oxide (GO) was obtained by means of the modified Hummers method [43]. Twenty grams of graphite powder was added to 460 cm3 of H2SO4, and the suspension obtained was stirred for 30 min in an ice bath. Subsequently, 60 g of KMnO4 was being slowly added to the solution, in such a way that the temperature did not exceed 20 °C. The contents of the beaker were warmed to 35 °C and stirred for 2 h. This was followed by adding 920 cm3 of distilled water. To remove the remaining KMnO4, 800 cm3 of warm distilled water (60 °C) and 500 cm3 of a 3% aqueous solution of H2O2 were added. The obtained sample was centrifuged and rinsed with distilled water several times to achieve pH of 7. GO was then rinsed with acetone in order to obtain the brown GO precipitate.
In the second stage, GO was thermally reduced. Low-temperature thermal reduction was carried out under a nitrogen atmosphere in the temperature below 300 °C at the heating rate of 30 °C/min. The final product was a fluffy powder and was used directly in the next step.
Functionalization of GO and rGO with pimelic acid
The functionalization of GO with pimelic acid was carried out by mixing the GO and PA mixture (1:1, wt/wt) in the presence of Ca(OH)2 (1:0.5 wt/wt) until a homogeneous powder was obtained. The mixture was then heated in an aluminium vessel at 120 °C for 1.5 h.
For rGO functionalization, rGO and PA were preliminarily mixed (1:2 wt/wt), and then Ca(OH)2 was added (1:0.5 wt/wt). The mixture was homogenized to obtain a unitary powder. Like GO, the homogenous powder was being heated in an aluminium vessel at 120 °C for 1.5 h.
Preparation of polypropylene nanocomposites
The GO/PA and rGO/PA were mixed with the polypropylene resin in the two-stage process. In the first stage, the masterbatches containing 5 wt% of functionalized compounds were prepared. During the second stage, the masterbatch was compounded with the appropriate amount of the polypropylene resin to obtain a mixture with a concentration of 0.5 wt%. The mixing was carried out in the molten state in the temperature of 230 °C. The twin-screw extruder EHP-2x16S supplied by Zamak Mercator (Poland) was used for compounding.
Methods
SEM/EDS
Morphology and elemental composition of GO, rGO and products of their functionalization were examined by scanning electron microscopy (SEM). During the investigations a Phenom ProX scanning electron microscope coupled with the PhenomWorld EDS (energy-dispersive X-ray spectroscopy) detector was used. Micrographs were recorded in the image mode. The EDS measurements were performed in the map mode.
FTIR
Functionalized and not functionalized GO and rGO were characterized using Fourier transform infrared spectroscopy (FTIR). For the examinations, the FTIR spectrometer Nicolet 6700 (Thermo Electron Corporation, USA) equipped with photoacoustic device MTEC model 300 was applied. The spectra were registered at the resolution of 4 cm−1 in the range from 4000 to 400 cm−1 with 128 scans per spectrum. Data collection and post-processing were performed using the OMNIC software (v. 8.0, Thermo Electron Corp.).
DSC
The investigations of melting and crystallization behaviour of polypropylene doped with GO/PA and rGO/PA were carried out with differential scanning calorimetry (DSC). The samples were melted at a heating rate of 10 K/min. Melted samples were kept in 210 °C for 5 min and then cooled to 50 °C at a rate of 5 K/min. Endothermal curves were recorded during the heating and cooling.
The investigations involved using an analytical system (TA Instruments, USA) with a calorimeter (MDSC 2920) equipped with a refrigerated cooling system. The analysis of the DSC curves was performed by means of the Universal V4.5A software supplied by TA Instruments. Based on the curves, the crystallization and melting peaks temperature and the enthalpy of crystallization were determined.
POM
The investigations of polypropylene crystallization in non-isothermal conditions were performed in the polarized light. The samples of polypropylene doped with GO/PA and rGO/PA were melted in 230 °C and then cooled at the cooling rate of 5 K/min. During the cooling, the progress of crystallization was observed. For the examinations, the heated stage Mettler Toledo FP82HT (Mettler, Greifensee, Switzerland) coupled with a control unit Mettler Toledo FP90 mounted on a polarizing microscope (Reichert Austria) was applied. The progress of crystallization was registered using a video camera (Moticam 2300).
WAXS
The crystalline structure of GO and rGO as well as polypropylene doped with GO/PA and rGO/PA was examined using the wide-angle X-ray scattering (WAXS). The WAXS measurements were carried out with the URD 6 Seifert diffractometer equipped in a copper target X-ray tube (λ = 0.154 nm) operated at 40 kV and 30 mA. Cu Kα radiation was monochromatized with Ni filter and pulse-height analyser. WAXS curves were recorded in the symmetric mode in the angular range 3°–40°, with a step of 0.1 and the registration time of 20 s per step. The investigated samples were powdered and pressed into a sample holder. Samples with the radius of 12 mm and the thickness of 2 mm were prepared. The diffraction curves of samples were analysed by means of WAXSFIT computer program [44, 45].
Results
SEM and EDS of not functionalized and functionalized GO and rGO
The representative SEM micrographs of GO and rGO before functionalization are presented in Fig. 1. For GO, some irregular aggregates with the external dimensions from 75 to 220 μm are observed. The aggregates reveal crumpled and rippled structure resulting from the introduction of the oxygen-containing functional groups between regularly stacked graphene sheets.

SEM microphotographs of: a rippled GO aggregates; b exfoliated rGO aggregates
In the thermally reduced rGO, the aggregates have significantly lower dimensions (Fig. 1b). Due to a micro-explosion during their thermal reduction, the aggregates reveal vermiculite morphology similar to graphene oxide exfoliated by hydrogen arc discharge or microwave irradiation [46, 47]. The aggregates are built from highly wrinkled stacks of ultra-thin nanosheets. The thickness of particular sheets equals a few μm, which is several times larger than the thickness of a single graphene layer. The aggregates possess highly porous structures with interconnected pores extending from one side of the stack to another.
The EDS analysis reveals that for the GO aggregates the weight concentration of carbon reaches 53% and an average atomic ratio C/O equals 1.5. Reduced oxygen has led to the elimination of the functional groups and a considerable decrease in the oxygen content. Consequently, rGO contains 76% of carbon and the C/O atomic ratio exceeds 4.
The SEM micrographs of functionalized GO and rGO are presented in Fig. 2.

SEM microphotographs of GO and rGO functionalized with pimelic acid: a particles of calcium pimelate attached to the surface of GO aggregates; b particles of calcium pimelate supported on rGO aggregates
On the micrographs, numerous particles attached to the surface of GO aggregates are observed (Fig. 2a). The particles are formed from more or less regular tiny granules. The EDS analysis reveals that the average calcium content in granules achieves approximately 5% (Fig. 3).

EDS spectra for functionalized GO aggregates
Calcium is distributed according to the distribution of granules, which clearly indicates that the granules are built from calcium pimelate formed in the reaction of calcium hydroxide and pimelic acid during the GO functionalization. The dimensions of single granules are about 1 μm, much greater than the dimensions of singular calcium pimelate molecule. Similarly to GO on rGO aggregates, the irregular particles of calcium pimelate are supported (Fig. 2b). The particles of calcium pimelate are irregularly distributed among rGO aggregates. Most particles are attached to the side edges of wrinkled layers of the stacked graphene sheets. The dimensions of the attached particles are relatively small and do not exceed 1 μm.
WAXS of graphene oxide and reduced graphene oxide
WAXS patterns of GO and rGO are presented in Fig. 4. For the GO on the WAXS curve, the intensive diffraction peak (001) at 2θ = 11.25° is visible. For the rGO, the diffraction peak is much broader and exhibits much lower intensity. The position of the peak moves more towards the higher angles to 2θ = 23.3°.

WAXS patterns of GO and rGO
Based the on WAXS patterns, the interlayer distance and the number of graphene layers in stacks were determined. The interlayer distance was calculated according to Bragg’s law on the basis of the diffraction peaks position. The average number of layers in stacks was determined as the ratio of the crystallite size calculated from the Scherrer’s equation to interlayer distance [48, 49]. The calculated parameters for GO and rGO are presented in Table 1.
For GO, the interlayer distance equals 0.786 nm. The distance is almost two times greater than the typical interlayer distance in graphite [50, 51]. The increase in the distance in GO indicates a high degree of exfoliation caused by the introduction of oxygen-containing functional groups between graphene layers. As a result of oxidation and exfoliation, the average number of graphene layers in stacks decreased to 17 layers.
As for rGO, the interlayer distance decreased to 0.381 nm. The decrease in the distance after thermal reduction was repeatedly observed and is caused by the removal of oxygen-containing functional groups from the interlayer area. The distance in rGO is only slightly higher in comparison with the native graphite. As a result of the reduction, the average number of graphene layers in stacks decreases significantly to 6 sheets.
FTIR of graphene oxide/pimelic acid and reduced graphene oxide/pimelic acid
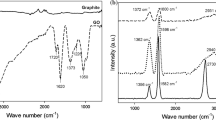
The FTIR spectra of GO, GO/PA and PA are presented in Fig. 5. The spectrum of GO in the range 3800–2200 cm−1 exhibits a set of overlapping bands assigned to O–H stretching vibrations of adsorbed water molecules and a structural hydroxyl group. At 1740 cm−1, strong band related to stretching motions of carbonyl groups situated at the edges of the GO sheets was observed. Then, at 1620 cm−1, there is a visible band with the shoulder at lower-frequency side at 1580 cm−1 assigned to either the oxygen surface compounds or ring vibrations of the C=C bonds of the basal plane in the unoxidized aromatic regions. In addition to the above-mentioned bands, in the fingerprint region below 1500 cm−1 a large number of convoluted bands related to oxygen functional groups are visible on the spectrum [52, 53].

FTIR spectra of GO, GO/PA and PA
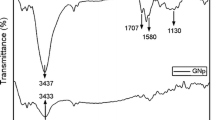
On the GO/PA spectrum, in the range 3800–2200 cm−1 one intense band at 3640 cm−1 appeared. According to the literature, the band corresponds to the OH groups of unreacted calcium hydroxide [54,55,56]. Other bands in this range assigned to water molecules and structural OH groups are not visible. In the range of 2939 and 2861 cm−1, we can see strong bands assigned to asymmetric and symmetric stretching vibrations of carbon–hydrogen bonds in aliphatic chains of the pimelic acid. The presence of these bands indicates the attachment of acid molecules to the surface of the GO crystals. In addition to the aliphatic bands, several characteristic bands attributed to pure pimelic acid are visible on the spectra registered for GO/PA. At 1706 cm−1, the band corresponding to the stretching vibration of the carbonyl group C=O in the pimelic acid is pronounced. The next detected bands are at 1435 and 1272 cm−1 attributed to oscillation vibrations in the plane of hydroxyl group O–H and stretching of C–O bond, as well as the strong band at 937 cm−1 assigned to bending vibrations of the plane of O–H bond.
The FTIR spectra of rGO, rGO/PA and PA are presented in Fig. 6. In the rGO spectrum, in contrary to spectrum registered for GO, only a few bands are visible. In the range 3800–2200 cm−1, the line is flat and no bands assigned to OH group are noticeable. The first relatively strong band assigned to the carbonyl groups appears at 1750 cm−1. The next strong band is at the 1600 cm−1, corresponding to C=C bonds in the ring structure. The third noticeable band related to carboxyl groups occurs at 1280 cm−1.

FTIR spectra of rGO, rGO/PA and PA
The FTIR spectrum of rGO evidently indicates that during the reduction of GO thermal decomposition of the oxygen-containing functional groups occurs. Reduction eliminates less stable and more easily removable hydroxyl and epoxy groups [57]. Other oxygen-containing groups, carbonyl or carboxylic, are more resistant to reducing treatment, and they have not been completely removed.
The rGO/PA spectrum is very similar to the GO/PA spectrum. Like for GO/PA, the characteristic bands related to pure pimelic acid can be distinguished in the spectra. Apart from the many bands related to the oxygen functional groups at 3640 cm−1, the band assigned to OH group of calcium hydroxide is observed. Moreover, at 2939 and 2861 cm−1 two characteristic bands assigned to C–H bonds in aliphatic chains are visible. The appearance of these bands proves that PA molecules are bonded also to the rGO surface.
In both, GO/PA and rGO/PA spectra in addition to bands visible for pure PA at 1586 and 1450 cm−1 two new strong bands have emerged. The bands belong to two ranges 1560–1600 cm−1 and 1410–1450 cm−1 which are characteristic for aliphatic carboxylates and assigned to the symmetric and asymmetric stretching vibration of the carboxyl group in the metal salts [19]. In previous investigations carried out for various compounds functionalized with PA, a double band at 1580 and 1540 cm−1 was registered in the first range [58,59,60]. The 1540 cm−1 band was assigned to the symmetric vibration of carbonyl group within the calcium pimelate structure [19, 60]. During the investigation on titanium dioxide (TiO2), the appearance of the double band was interpreted as the proof that an additional bond between PA and titanium ion was being formed and as the evidence of successful attachment of the PA molecule to the TiO2 surface [61]. In other investigations too, the double band was interpreted as a confirmation of supporting calcium pimelate on the surface of montmorillonite [58, 62].
In our investigations, one band at 1586 cm−1 has been detected. For both GO and rGO, the band is relatively broad and may result from overlapping of the two nearby bands. The appearance of the band indicates the formation of new links involving calcium atom, which connect carboxylic groups within the calcium pimelate structure and groups from PA with groups attached to GO and rGO layers. The possible connections are presented in Fig. 7.

Bridging link between GO and PA
The band assigned to the symmetric vibration is approximately 140 cm−1 separate from the band of asymmetric vibration. The separation of asymmetric and symmetric bands indicates the nature of the coordination between the metal atom and carboxylic ion. According to literature, the distance of 140 cm−1 indicates the chelate coordination [19].
POM
POM photographs registered between 130 and 118 °C during non-isothermal crystallization are presented in Fig. 8.

Polarized optical micrographs during non-isothermally crystallization of pure polypropylene, polypropylene + GO/PA and polypropylene + rGO/PA
During cooling of the melt of pure polypropylene, the self-assembled chains form individual nuclei. As a result of the homogenous nucleation at 128 °C, the first sporadic spherulites appear. The spherulites are well separated from each other and possess clear outer boundaries. The spherulites represent typical spherulites of the α-form. During further cooling, simultaneously with the growth of the previously formed spherulites, the new spherulites are formed. The number of the homogenous nuclei formed in pure polypropylene is relatively lower. Consequently, the number of spherulites formed at the completion of the crystallization is fewer and their sizes are relatively big.
For the polypropylene doped with GO/PA, the crystallization begins at a higher temperature. Heterogeneous nucleation on the crystals of GO/PA leads to the rapid increase in the nucleation density. Consequently, a big number of tiny crystals of the β-form are observed already at 130 °C. Unlike the big separate spherulites grown in pure polypropylene, the crystallites are densely packed and their boundaries are difficult to distinguish. Due to the high nuclei density, the crystals cannot grow freely and they are much smaller than pure polypropylene.
Similarly, for the polypropylene doped with rGO/PA numerous tiny β-crystals are formed. In comparison with the polypropylene doped with GO/PA at 130 °C, the number of the nuclei in polypropylene doped with rGO/PA is slightly lower. The crystallization begins by higher supercooling at a little lower temperature. Then, at lower temperatures, the nucleation density increases and becomes comparable with that of the polypropylene doped with GO/PA. With high nucleation density, the densely packed fine-grained structure is formed.
DSC
The DSC melting curves registered in the non-isothermal conditions are presented in Fig. 9. At 165.6 °C, we can see a strong peak attributed to the melting of α-form on the melting curve for pure polypropylene. On the curves registered for specimens containing GO/PA and rGO/PA, except the melting peak attributed to the α-form, in a lower temperature two less intensive peaks are visible. For the specimen containing GO/PA, well-distinguished sharp peaks occur at 142.5 and 150.7 °C. For the specimen doped with rGO/PA the peaks are a little less intense and their position is minimally moved towards the higher temperatures of 142.9 and 151.5 °C.

DSC curves during melting of: a pure polypropylene; b polypropylene + GO/PA; c polypropylene + rGO/PA
The double peak visible for specimens containing GO/PA and rGO/PA is attributed to the β-form of polypropylene. The similar peak was repeatedly observed for samples containing β-form with higher structural instability [63]. According to Varga, a doubled melting peak results from two kinds of recrystallization taking place during the heating of samples rich in the β-form, ββ-recrystallization within the β-phase leading to the perfection of the β-form crystal structure and βα-recrystallization which involved the transition of the β-form into more stable α-form [64].
The DSC non-isothermal crystallization curves for pure polypropylene and polypropylene doped with GO and rGO are presented in Fig. 10. With the addition of the GO and rGO, the position of the crystallization peak is shifted towards higher temperatures.

DSC curves during cooling of: a pure polypropylene; b polypropylene + GO/PA; c polypropylene + rGO/PA
For pure polypropylene, the crystallization starts at 122.8 °C (Table 2). Adding the functionalized GO and rGO significantly increases the nucleation temperature which reaches higher values of 9.7 °C for GO and 11.7 °C for rGO. Simultaneously, the crystallization temperature for sample doped with GO and rGO is higher by 15.5 and 16.6 °C, respectively. For the samples doped with GO and rGO, the width of crystallization peak is almost twice as small. The addition of GO and rGO results in the decrease in the enthalpy of crystallization.
WAXS
WAXS patterns of pure polypropylene and polypropylene doped with GO/PA and rGO/PA are presented in Fig. 11. The curve registered for pure polypropylene consists of diffraction peaks characteristic for the α-form. For the polypropylene containing GO/PA, besides the peaks of the α-form on the pattern at 16.1°, a strong peak typical for the β-form is visible. Similarly, for polypropylene doped with rGO/PA peak characteristics for two polymorphic forms of polypropylene are observed. In this case, the peaks of the α-form are weaker. In the same time, the peak for the β-form exhibits high intensity, corresponding to samples with very high β-crystals content.

X-ray diffraction patterns of: 1 pure polypropylene; 2 PP + GO/PA; 3 PP + rGO/PA
According to the known equation of Turner Jones on the base of the WAXS patterns, the relative content of β-form crystals Kβ was determined (1).
where Iβ(300) is the intensity of (300) diffraction peak of the β-form and Iα(110), Iα(040) and Iα(130) the intensities of (110), (040) and (130) peaks of α-form [8].
The intensity of the particular peaks was determined on the basis of the peak separation procedure by constructing a theoretical curve best fitted to the experimental pattern. For the purposes of the analysis, the shape of peaks and amorphous halo was approximated with a superposition of Gauss and Cauchy functions. A modified Rosenbrock’s optimization method was applied for curves fitting. The example of the fitting procedure for the polypropylene doped with rGO/PA is presented in Fig. 12.

Analysis of WAXS diffraction pattern for polypropylene doped with rGO/PA (The peaks of modification are drawn with a solid line, peaks of β modification with a dotted line and the amorphous halo with dash-dot line)
The calculated Kβ parameter for polypropylene containing GO/PA amounts up to 0.74. The value corresponds to the relatively high β-form content obtained in the samples by application of classic and new effective β-nucleating agents, for example: 0.8 for quinacridone [65], 0.74 for zinc phthalate [31] and 0.82 for potassium 7-oxabicyclo [2.2.1] hept-5-ene-2,3-dicarboxylate [66]. For the polypropylene doped with rGO/PA, the relative content of the β-form is much lower and the Kβ equals 0.30.
Conclusions
During the functionalization of GO and rGO, calcium hydroxide reacts with pimelic acid and forms a salt calcium pimelate. The calcium pimelate formed during the reaction is attached to the surface of GO and rGO aggregates in the form of irregular lumpish particles with the dimensions of approximately 1 μm.
In the process, calcium atoms participate in the formation of bonds linking carboxylic groups. Some bonds are formed between the adjacent molecules of pimelic acid, as internal bonds in calcium pimelate crystals. Other bonds link carboxylic groups of pimelic acid with carboxylic groups placed on the side edges of the graphene layers. The bonds can be formed for both GO and rGO. GO obtained by oxidation of graphite possesses numerous groups capable of forming such bonds. Despite the reduction process eliminating oxygen functionalized groups, rGO still contains sufficient groups to attach calcium pimelate molecules.
Calcium pimelate is attached only to the surface of GO and rGO aggregates. There is no evidence that the pimelic acid diffuses between graphene layers and causes the exfoliation of the graphene layers stack.
The functionalized GO and rGO added to crystallising isotactic polypropylene generate the formation of the β-form. Both compounds functionalized graphene oxide and reduced graphene oxide have a nucleating ability towards polypropylene crystallization. Both compounds cause a significant shift in the nucleation and non-isothermal crystallization temperatures towards higher values. In the presence of both compounds, the structure with a high content of the β-form is formed. The content of the β-form formed during crystallization in non-isothermal conditions is comparable to the content obtained by the application of efficient β-nucleating agents.
References
Bruckner S, Meille SV, Petraccone V, Pirozzi B (1991) Polymorphism in isotactic polypropylene. Prog Polym Sci 16:361–403
Lotz B, Wittman JC, Lovinger AJ (1996) Structure and morphology of poly(propylenes): a molecular analysis. Polymer 37:4979–4992
Natta G, Corradini P (1960) Structure and properties of isotactic polypropylene. Nuovo Cimento Suppl 15:40–51
Meille SV, Ferro DR, Brückner S, Lovinger A, Padden FJ (1994) Structure of β-isotactic polypropylene: a long-standing structural puzzle. Macromolecules 27:2615–2622
Leugering VHJ (1967) Einfluβ der kristallstruktur und der überstruktur auf einige eigenschaften von polypropylene. Macromol Chem Phys 109:204–216
Varga J (2002) β-modification of isotactic polypropylene: preparation, structure, processing, properties, and application. J Macromol Sci Part B 41:1121–1171
Papageorgiou DG, Chrissafis K, Bikiaris DN (2015) β-nucleated polypropylene: processing, properties and nanocomposites. Polym Rev 55:596–629
Turner Jones A, Aizlewood JM, Beckett DR (1964) Crystalline forms of isotactic polypropylene. Die Makromol Chem 75:134–158
Lovinger AJ, Chua JO, Gryte CC (1977) Studies on the α and β forms of isotactic polypropylene by crystallisation in a temperature gradient. J Polym Sci Part B Polym Phys 15:641–656
Dragaun H, Hubeny H, Muschik H (1977) Shear-induced β-form crystallisation in isotactic polypropylene. J Polym Sci Polym Phys Ed 15:1779–1789
Zhang C, Wang B, Yang J, Ding D, Yan X, Zheng G, Dai K, Liu C, Guo Z (2015) Synergies among the self-assembled β-nucleating agent and the sheared isotactic polypropylene matrix. Polymer 60:40–49
Garbarczyk J, Paukszta D (1981) Influence of additives on the structure and properties of polymers: 2. Polymorphic transitions of isotactic polypropylene caused by aminosulphur compounds. Polymer 22:562–564
Tjong SC, Shen JS, Li RKY (1996) Morphological behaviour and instrumented dart impact properties of β-crystalline-phase polypropylene. Polymer 37:2309–2316
Li JX, Wl C (1999) Conversion of growth and recrystallisation of β-phase in doped iPP. Polymer 40:2085–2088
Dou Q (2008) Effect of the composition ratio of pimelic acid/calcium stearate bicomponent nucleator and crystallisation temperature on the production of β crystal form in isotactic polypropylene. J Appl Polym Sci 107:958–965
Varga J, Mudra I, Ehrenstein GW (1999) Highly active thermally stable β-nucleating agents for isotactic polypropylene. J Appl Polym Sci 74:2357–2368
Kotek J, Raab M, Baldrian J, Grellmann W (2002) The effect of specific β-nucleation on morphology and mechanical behaviour of isotactic polypropylene. J Appl Polym Sci 85:1174–1184
Marco C, Gomez MA, Ellis G, Arribas JM (2002) Activity of a β-nucleating agent for isotactic polypropylene and its influence on polymorphic transitions. J Appl Polym Sci 86:531–539
Li X, Hu K, Ji M, Huang Y, Zhou G (2002) Calcium dicarboxylates nucleation of β-polypropylene. J Appl Polym Sci 86:633–638
Dong M, Guo Z, Yu J, Su Z (2008) Crystallisation behavior and morphological development of isotactic polypropylene with an aryl amide derivative as β-form nucleating agent. J Polym Sci Part B Polym Phys 46:1725–1733
Huang MR, Li XG, Fang BR (1995) β-nucleators and β-crystalline form of isotactic polypropylene. J Appl Polym Sci 56(1995):1323–1337
Marcincin A, Ujhelyiova A, Marcincin K, Alexy P (1996) Nucleation of the β-modification of isotactic polypropylene. J Therm Anal 46:581–595
Broda J (2003) Nucleating activity of the quinacridone and phthalocyanine pigments in polypropylene crystallisation. J Appl Polym Sci 90:3957–3964
Fujiyama M (1996) Structures and properties of injection moldings of β-crystal nucleator-added polypropylenes. Int Polym Process 11:271–274
Fujiyama M, Kawamura Y, Wakino T (1988) Study on rough-surface biaxially oriented polypropylene film. IV. Influence of addition of crystal nucleator. J Appl Polym Sci 36:1025–1034
Radhakrishnan S, Tapale M, Shah N, Rairrkar E, Shirodkar V, Natu HP (1997) Effect of β-phase nucleating additives on structure and properties of blow extruded polypropylene. J Appl Polym Sci 64:1247–1253
Broda J (2003) Polymorphism in polypropylene fibers. J Appl Polym Sci 89:3364–3370
Broda J (2004) Polymorphic composition of colored polypropylene fibers. Cryst Growth Des 4:1277–1282
Broda J (2004) Influence of processing on structure of β-nucleated poly(propylene) fibers. J Appl Polym Sci 91:1413–1418
Zhao S, Liu K, Zhou S, Shi Y, Xin Z (2017) A novel self-dispersed β nucleating agent for isotactic polypropylene and its unique nucleation behaviour and mechanism. Polymer 132:69–78
Qin W, Xin Z, Pan C, Sun S, Jiang X, Zhao S (2019) In situ formation of zinc phthalate as a highly dispersed β-nucleating agent for mechanically strengthened isotactic polypropylene. Chem Eng J 358:1243–1252
Kalaitzidou K, Fukushima H, Lawrence PA, Drzal T (2008) The nucleating effect of exfoliated graphite nanoplatelets and their influence on the crystal structure and electrical conductivity of polypropylene nanocomposites. J Mater Sci 43:2895–2907. https://doi.org/10.1007/s10853-007-1876-3
Ferreira CI, Dal Castel C, Oviedo MAS, Maulera RS (2013) Isothermal and non-isothermal crystallisation kinetics of polypropylene/exfoliated graphite nanocomposites. Thermochim Acta 553:40–48
Xu JZ, Liang YY, Huang HD, Zhong GJ, Lei J, Li ZM (2012) Isothermal and nonisothermal crystallisation of isotactic polypropylene/graphene oxide nanosheet nanocomposites. J Polym Res 19:9975
Yun YS, Bae YH, Kim DH, Lee JY, Chin IJ, Jin HJ (2011) Reinforcing effects of adding alkylated graphene oxide to polypropylene. Carbon 49:3553–3559
Wang H, Ren PG, Chen YH, Yan DX, Li ZM, Xu L (2014) Effects of dodecyl amine functionalized graphene oxide on the crystallisation behavior of isotactic polypropylene. J Appl Polym Sci 131:40000
Long J, Li S, Liang J, Wang Z, Liang B (2019) Preparation and characterization of graphene oxide and it application as a reinforcement in polypropylene composites. Polym Compos 40:723–729
Bo Y, Zhaoyi H, Lu L, Xingyue S, Zengheng H (2019) Effects of β-nucleating agent and graphene oxide on the crystallisation and polymorphic composition of isotactic polypropylene/graphene oxide composites for bridge pavement. J Polym Res 26:9
Yu Y, Zeng F, Chen J, Kang J, Yang F, Cao Y, Xiang M (2019) Regulating polycrystalline behavior of the b-nucleated isotactic polypropylene/graphene oxide composites by melt memory effect. Polym Compos 40:E440–E448
Yu Y, Zeng F, Chen J, Kang J, Yang F, Cao Y, Xiang M (2018) Isothermal crystallisation kinetics and subsequent melting behaviour of β-nucleated isotactic polypropylene/graphene oxide composites with different ordered structure. Polym Int 67:1212–1222
Bao RY, Cao J, Liu ZY, Yang W, Xie BH, Yang MB (2014) Towards balanced strength and toughness improvement of isotactic polypropylene nanocomposites by surface functionalized graphene oxide. J Mater Chem A 2:3190–3199
Anastacio-Lopez ZS, Gonzalez-Calderon JA, Saldivar-Guerrero R, Velasco-Santos C, Martınez-Hernandez AL, Carlos Fierro-Gonzalez J, Almendarez-Camarillo A (2019) Modification of graphene oxide to induce beta crystals in isotactic polypropylene. J Mater Sci 54:427–443. https://doi.org/10.1007/s10853-018-2866-3
Hummers WS, Offeman RE (1958) Preparation of graphitic oxide. J Am Chem Soc 80:1339
Rabiej M (2003) Application of multicriterial optimization for determination of crystallinity degree of semicrystalline polymers. Polimery 48:288–295
Rabiej M, Rabiej S (2006) Analiza rentgenowskich krzywych dyfrakcyjnych polimerów za pomocą programu komputerowego WAXSFIT. Publishing House ATH, Bielsko Biala
Ali G, Mehmood A, Ha HY, Kim J, Chung KY (2017) Reduced graphene oxide as a stable and high-capacity cathode material for Na-ion batteries. Sci Rep 7:40910. https://doi.org/10.1038/srep40910
Wu ZS, Ren W, Gao L, Zhao J, Chen Z, Liu B, Tang D, Yu B, Jiang C, Cheng HM (2009) Synthesis of graphene sheets with high electrical conductivity and good thermal stability by hydrogen arc discharge exfoliation. ACS Nano 3(2):411–417
Botas C, Alvarez P, Blanco C, Santamarıa R, Granda M, Dolores Gutierrez M, Rodrıguez-Reinoso F, Menendez R (2013) Critical temperatures in the synthesis of graphene-like materials by thermal exfoliation–reduction of graphite oxide. Carbon 52:476–485
Alexander LE (1969) X–ray diffraction methods in polymer science. Wiley, New York
Saha U, Jaiswal R, Singh JP, Goswami TH (2014) Diisocyanate modified graphene oxide network structure: steric effect of diisocyanates on bimolecular cross-linking degree. J Nanoparticle Res 16:2404
Stankovich S, Dikin DA, Dommett GHB, Kohlhaas KM, Zimney EJ, Stach EA, Piner RD, Nguyen ST, Ruoff RS (2006) Graphene-based composite materials. Nature 442:282–286
Szabo T, Berkesi O, Dekany I (2005) DRIFT study of deuterium-exchanged graphite oxide. Carbon 43:3186–3189
Frogley MA, Wang C, Cinque G, Barber AH (2014) Polarised infrared microspectroscopy of edge-oriented graphene oxide papers. Vib Spectrosc 75:178–183
Asikin-Mijan N, Taufiq-Yap YH, Lee HV (2015) Synthesis of clamshell derived Ca(OH)2 nano-particles via simple surfactant-hydration treatment. Chem Eng J 262:1043–1051
Poggi G, Toccafondi N, Chelazzi D, Canton P, Giorgi R, Baglioni P (2016) Calcium hydroxide nanoparticles from solvothermal reaction for the deacidification of degraded waterlogged wood. J Colloid Interface Sci 473:1–8
Darroudia M, Bagherpourc M, Ali Hosseinie H, Ebrahimi M (2016) Biopolymer-assisted green synthesis and characterization of calcium hydroxide nanoparticles. Ceram Int 42:3816–3819
Dung T, Han D, Jeong M (2015) Graphene prepared by thermal reduction–exfoliation of graphite oxide: effect of raw graphite particle size on the properties of graphite oxide and graphene. Mater Res Bull 70:651–657
Dai X, Zhang Z, Wang C, Ding Q, Jiang J, Mai K (2013) A novel montmorillonite with β-nucleating surface for enhancing β crystallisation of isotactic polypropylene. Compos Part A 49:1–8
Zhang Z, Wang C, Junping Z, Mai K (2012) β-nucleation of pimelic acid supported on metal oxides in isotactic polypropylene. Polym Int 61:818–824
Gonzalez-Calderon JA, Castrejon-Gonzalez EO, Medellin-Rodriguez FJ, Stribeck N, Almendarez-Camarillo A (2015) Functionalization of multi-walled carbon nanotubes (MWCNTs) with pimelic acid molecules: effect of linkage on b-crystal formation in an isotactic polypropylene (iPP) matrix. J Mater Sci 50:1457–1468. https://doi.org/10.1007/s10853-014-8706-1
Gonzalez-Calderon JA, Vallejo-Montesinos J, Mata-Padilla JM, Perez E, Almendarez-Camarillo A (2015) Effective method for the synthesis of pimelic acid/TiO2 nanoparticles with a high capacity to nucleate β-crystals in isotactic polypropylene nanocomposites. J Mater Sci 50:7998–8006. https://doi.org/10.1007/s10853-015-9365-6
Cao J, Wen N, Zheng Y (2018) The preparation of calcium pimelate modified OMMT from natural montmorillonite and its application as β-nucleating agent for polypropylene. Polym Test 65:352–359
Varga J, Garzo G, Ille G (1986) Kristallisation, umkristallisation und schmelzen der β-modifikation des polypropylens. Angew Makromol Chem 142:171–181
Varga J (1995) Crystallisation, melting and supermolecular structure of isotactic polypropylene. In: Karger Kocsis J (ed) Polypropylene: structure, blends and composites. Chapman & Hall, London, pp 56–115
Jacoby P, Bersted BH, Kissel WJ, Smith CE (1986) Studies on the β-crystalline form of isotactic polypropylene. J Polym Sci Part B Polym Phys 24:461–491
Pan C, Qin W, Chen L, Xin Z, Zhao S, Ye C (2018) A novel β-nucleating agent for isotactic polypropylene. J Therm Anal Calorim 134:2029–2040
Author information
Authors and Affiliations
Contributions
JB was involved in conception, experimental design and manuscript composition; MB carried out measurements and analysed the results; JF carried out measurements and analysed the results; DB carried out measurements; RF was involved in preparation of samples.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Broda, J., Baczek, M., Fabia, J. et al. Nucleating agents based on graphene and graphene oxide for crystallization of the β-form of isotactic polypropylene. J Mater Sci 55, 1436–1450 (2020). https://doi.org/10.1007/s10853-019-04045-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-019-04045-y