
Abstract
Hydrated aluminosilicates were synthesized with and without aqueous heavy metals (Me), such as cobalt (Co), chromium (Cr), and zinc (Zn), by a sol–gel process at different initial molar ratios of Ca/(Si + Al) (0.6–1.6) and Me/Si (0.0–2.0), and constant Al/Si ratio (0.05) using equilibrium-approaching experiments. The chemical composition of the reactive solutions during aluminosilicate precipitation and maturation was monitored by ICP-OES. The mineralogy, nanostructure, and chemical composition of the precipitates were studied by XRD and high-resolution TEM. At Me/Si ratios ≤ 0.2, calcium–aluminium–silicate–hydrates (C–A–S–H) with a defect 14 Å tobermorite-like structure formed, whereas at a Me/Si ratio of 2.0, either trioctahedral Co- and Zn-smectite or amorphous Cr gels precipitated, independent of the initial Ca/(Si + Al) ratio used for gel synthesis. The immobilization capacities for \( {\text{Co}}^{2 + } \), \( {\text{Cr}}^{3 + } \), and \( {\text{Zn}}^{2 + } \) by C–A–S–H, Cr gel, and trioctahedral smectite are 3–40 mg/g, 30–152 mg/g, and 122–141 mg/g, respectively. The immobilization mechanism of heavy metals is based on a combination of isomorphous substitution, interlayer cation exchange, surface (ad)sorption, and surface precipitation. In engineered systems, such as underground concrete structures and nuclear waste disposal sites, hydrated aluminosilicates should exhibit a high detoxication potential for aqueous heavy metals.
Similar content being viewed by others

Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Importance of calcium–aluminium–silicate–hydrate phases
Aluminium-substituted calcium-silicate-hydrates (C–A–S–H) are of great technological importance, because they represent the main hydration product in ordinary Portland cement (OPC) and thus significantly contribute to the physicochemical characteristics, mechanical properties, and durability of hardened cementitious materials, such as concrete and shotcrete [1,2,3,4,5,6,7]. Apart from being the most important construction material of our time, concrete is nowadays applied in special wastewater treatment technologies and in the remediation of (sub)soil structures that are contaminated with hazardous components, like toxic heavy metal ions [4, 8,9,10,11,12]. Moreover, C–A–S–H is becoming increasingly more important as adsorbent, catalysis material, ion exchanger, waste stabilizer, and heat insulator [13,14,15,16,17].
Natural and synthetic C–A–S–H phases are often poorly crystallized and have a highly variable structure and chemical composition, which can be expressed by the Ca/(Si + Al) ratio and subordinate by the water content. Specifically, the Ca/(Si + Al) ratio ranges from 1.5 to 1.9 in hydrated OPC to 1.5–0.7 or lower in hydrated cement binders blended with supplementary cementitious materials (SCMs), such as ground granulated blast furnace slag (GGBFS), fly ash, or metakaolin [2, 3, 18, 19]. Besides the pronounced positive effects of new cement blends containing SCMs on the technical and durability properties, and on ecological aspects, i.e. reduction of the global \( {\text{CO}}_{2} \) emissions from the cement industry sector [20], their increasing production may also pose an environmental risk: SCMs usually contain higher contents of heavy metals than pure OPC [21]. Thus, it is important to know how these critical elements are then fixed in the hardened cement paste.
The structure of ideal calcium–silicate–hydrates (C–S–H) can be described by a calcium oxide (Ca-O) layer having silicate tetrahedra attached on both sides, which are organized in a “dreierketten structure”, that is, a repeating chain of three tetrahedra (Fig. 1). Two of these tetrahedra (the “pairing tetrahedra” or \( {\text{Q}}_{\text{p}}^{2} \) site) are connected to the Ca–O layer, while the third tetrahedron (the “bridging tetrahedron” or \( {\text{Q}}_{\text{b}}^{2} \) site) is bonded to the adjacent \( {\text{Q}}^{3} \) sites [4, 22,23,24,25]. Few tetrahedra, mainly of the \( {\text{Q}}^{1} \) and \( {\text{Q}}^{2} \) sites, are also linked to an interlayer site (Fig. 1), which typically contains weakly bound \( {\text{H}}_{2} {\text{O}} \) molecules, as well as \( {\text{Ca}}^{2 + } \), alkalis, and other ions.

Schematic structure of C–A–S–H (modified from [2] with potential immobilization effects indicated in red (adapted from [29]). \( {\text{Q}}_{{{\text{p}},{\text{b}}}}^{\text{n}} \left( {\text{mAl}} \right) \): Q = \( {\text{SiO}}_{4} \) tetrahedron, n = number of neighbouring \( {\text{SiO}}_{4} \) tetrahedra, m = number of neighbouring \( {\text{AlO}}_{4} \) tetrahedra, p = pairing position, b = bridging position; \( - \): negative charge: H–H: hydrogen bridge bond. See text for further explanations
In the case of high substitution of Al for Si in the \( {\text{Q}}^{2} \) or \( {\text{Q}}^{3} \) sites of C–A–S–H (e.g. Al/Si ratio ≥ 0.1), the relative amount of tetrahedral Al decreases, while the amount of octahedral coordinated Al increases. In the latter case, Al is most likely present either in the form of an amorphous aluminium hydroxide layer or a calcium aluminate hydrate layer at the C–A–S–H surface, or as an aluminium hydrate complex in the interlayer within the C–A–S–H structure [26]. In addition, about 10% of the total structural Al associated with C–A–S–H is present as penta-coordinated Al, regardless of the Ca/Si ratio [25]. Thus, with increasing substitution of Al for Si in C–A–S–H, a negative charge is generated [27, 28], which has to be compensated through a coupled substitution of type: \( \square + {\text{OH}}^{ - } \to {\text{Ca}}^{2 + } + {\text{O}}^{2 - } \), where \( \square \) represents an “empty” position or a defect site in the C–A–S–H structure [29]. This defect site is occupied mainly by \( {\text{Na}}^{ + } \), \( {\text{K}}^{ + } \) and \( {\text{Ca}}^{2 + } \) ions in “traditional” C–A–S–H, or by other metal ions, such as \( {\text{Pb}}^{2 + } \), \( {\text{Cd}}^{2 + } \), \( {\text{Co}}^{2 + } \), \( {\text{Ni}}^{2 + } \), and \( {\text{Zn}}^{2 + } \) (hereafter called “Me” ions) in synthetic C–A–S–H gels, which serve as charge compensators.
C–A–S–H phases share many structural similarities with phyllosilicates. Smectite group minerals, for instance, have a 2:1 layer structure comprising negatively charged octahedral and tetrahedral sheets that are connected to an interlayer site, where hydrated and partly exchangeable cations are intercalated [30,31,32]. The unique structure, besides a small particle size (< 100 nm), a large specific surface area (> 10 m2/g), and presence of surface functional groups (i.e. ≡ Al–OH and ≡ Si–OH°) are the grounds for the high immobilization potential of C–A–S–H and smectite minerals for various Me ions [33], rendering such materials suitable for wastewater treatment or chemical barrier applications. However, contrasting to smectite the role and fate of Me ions during co-precipitation with C–A–S–H is still poorly understood.
Removal of heavy metals by calcium–aluminium–silicate–hydrates
It is well known that pollution of groundwater and drinking water by aqueous heavy metals is increasingly becoming a major environmental issue worldwide, causing hazardous effects on the aquatic and terrestrial ecosystems, and in particular to human health [34,35,36,37]. High concentrations of heavy metals are often released to the environment by the oxidative weathering of ultramafic rocks and minerals, ore bodies and accessory sulphide minerals, like arsenopyrite (FeAsS), galena (PbS), cobaltite (CoAsS), chromitite (\( {\text{CaCrO}}_{4} \)), and sphalerite (α-ZnS), as well as from a range of human activities, such as agriculture, fossil fuel burning, manufacturing, transportation, mining, and waste disposal [38, 39]. In the last five decades, a wealth of improvements in wastewater treatment applications has been found. This includes, for example, the utilization of highly efficient and reclaimable activated carbon components as sorbents, and optimized techniques for Me ion uptake, such as bioremediation, chemical precipitation, adsorption/ion exchange, ultrafiltration, and electrochemical methods [40, 41]. Despite of these improvements, there is still an increasing demand for the development of tailored, low-cost composites to be used in wastewater processing technologies.
Several studies have been carried out in order to describe and to quantify ion exchange and adsorption properties of natural and synthetic C–A–S–H [13, 14, 42] and clay minerals [33, 43]. Especially, smectite minerals have been shown to be good adsorbent materials for the removal of Me ions from aqueous solutions, owning to their worldwide occurrence, low mining costs, and unique physicochemical and surface (charge) properties, as indicated before. The uptake mechanism of Me ions by the various smectites is governed by complex adsorption processes, including physical attachment of Me ions onto charged clay surfaces, surface complexation, and chemical ion exchange, etc. [43]. For C–A–S–H phases basically the same uptake mechanisms of Me ions have been considered, but experimental studies are still scarce and/or lack control of the nature of the precipitates.
Zhao et al. [29] have recently shown that synthetic aluminium- and sucrose-substituted tobermorite (\( {\text{Ca}}_{5} {\text{Si}}_{6} {\text{O}}_{16} \left( {\text{OH}} \right)_{2} \)·4\( {\text{H}}_{2} {\text{O}} \)), that is, a common C–A–S–H component, can exhibit adsorption capacities for \( {\text{Cr}}^{6 + } \) ions as high as ~ 30 mg/g. Based on spectroscopic evidence, Żak and Deja [44] have presented removal efficiencies for \( {\text{Cd}}^{2 + } \), \( {\text{Zn}}^{2 + } \), \( {\text{Pb}}^{2 + } \), and \( {\text{Cr}}^{3 + } \) ions of 99.96% or higher after reaction with freshly prepared C–S–H, and Giergiczny and Król [45] have reported very high immobilization degrees for \( {\text{Pb}}^{2 + } \), \( {\text{Cu}}^{2 + } \), \( {\text{Zn}}^{2 + } \), \( {\text{Cr}}^{6 + } \), \( {\text{Cd}}^{2 + } \), and \( {\text{Mn}}^{2 + } \) ions, from ~ 86 to 99.99%, after reaction with mortars made of OPC and different levels of GGBFS and fly ash. The principle mechanism proposed for the removal of these Me ions by C–A–S–H or C–S–H was structural incorporation, though co-precipitation of discrete Me-oxyhydrates was frequently observed [29, 44, 45], suggesting that the removal mechanism(s) are more complex than previously thought.
To the best of our knowledge, no study yet exists that systematically investigates the relations between mineralogy, structure, and crystal chemistry of C–A–S–H containing low to extremely high loads of Me ions, like \( {\text{Co}}^{2 + } \), \( {\text{Cr}}^{3 + } \), and \( {\text{Zn}}^{2 + } \). Therefore, it remains highly questionable as to whether the removal of heavy metals from solution by C–A–S–H follows isomorphous substitution, ion exchange, physical/chemical adsorption, or a combination of these processes (Fig. 1). This paper reports on the synthesis and on the characterization of C–A–S–H and trioctahedral smectite precipitated at different molar ratios of Ca/(Si + Al) (0.6 to 1.6) and Me/Si (0.0 to 2.0), and at a molar Al/Si ratio of 0.05. The uptake mechanism(s) and the environmental implications of the removal of heavy metals by C–A–S–H and smectite are discussed in relation to phase composition and atomic structure. The importance of these nanocomposites in different engineered systems is highlighted.
Experimental section and methods
Gel synthesis with/without heavy metal ions
Hydrated aluminosilicates were precipitated in batch experiments at distinct molar ratios of Me/Si (0.02, 0.2 and 2.0) and Ca/(Si + Al) (0.6, 1.0 and 1.6), and at a constant molar Al/Si ratio of 0.05, in order to study the effect of gel composition, initial Me concentration, and pH on the removal efficiency of aqueous Co, Cr, and Zn by co-precipitation with C–A–S–H and trioctahedral smectite. In total, 27 experiments were conducted with heavy metals (see Table 1). These experiments are labelled as \( {\rm C}_{\rm x} {\rm Me}_{\rm x} \), where \( {\rm C}_{\rm x} \) and \( {\rm Me}_{\rm x} \) represent the initial molar ratios of Ca/(Si + Al) and Me/Si. In addition, three reference experiments were carried out without heavy metals (\( {\text{RefC}}_{0.6} \), \( {\text{RefC}}_{1.0} \) and \( {\text{RefC}}_{1.6} \)). The molar ratios of Ca/(Si + Al) and Al/Si were chosen to be typical of C–A–S–H commonly found in the hydration products of OPC-based cements [46]. The Me/Si ratios reflect wastewater with very low to extremely high pollution with heavy metals. The Al/Si ratio was set to be low, because at Al/Si molar ratios of ≥ 0.1, other hydrated cement phases such as katoite \( ({\text{Ca}}_{3} {\text{Al}}_{2} ({\text{SiO}}_{4} )_{3 - x} \left( {\text{OH}} \right)_{x} ,\,x = 1.5 - 3.0 \)) or stratlingite \( ({\text{Ca}}_{4} {\text{Al}}_{2} \left( {\text{OH}} \right)_{12} \left[ {{\text{AlSi}}\left( {\text{OH}} \right)_{8} ]_{2} \cdot2{\text{H}}_{2} {\text{O}}} \right) \) can form [2].
In detail, stock solutions (7 times á 1.0 L; pH ~ 12.5) containing 94.5 ± 1 mM \( {\text{Si}}\left( {\text{OH}} \right)_{4} \) (\( {\text{Na}}_{2} {\text{SiO}}_{3} \)·2\( {\text{H}}_{2} {\text{O}} \) from Roth) were prepared in 1.5 L high-density polyethylene (HD-PE) reactors. Ultrapure (Milli-Q Plus UV, Millipore, 18.2 MΩ at 25 °C) and de-carbonated water (prepared by bubbling with \( {\text{N}}_{2} \) gas; ≥ 99,999%, AirLiquide) was used throughout. Then, 200 mL of the \( {\text{Si}}\left( {\text{OH}} \right)_{4} \) solution was transferred into 250 mL HD-PE reactors, followed by the (< 1 min) addition of adequate amounts of analytical grade chemicals of \( {\text{Ca}}^{2 + } \) (\( {\text{CaCl}}_{2} \), Roth), \( {\text{Al}}^{3 + } \) (\( {\text{AlCl}}_{3} \)·5\( {\text{H}}_{2} {\text{O}} \), Roth) and \( {\text{Me}}^{2 + /3 + } \) (\( {\text{CoCl}}_{2} \)·6\( {\text{H}}_{2} {\text{O}} \), Roth; \( {\text{CrCl}}_{3} \)·6\( {\text{H}}_{2} {\text{O}} \), Sigma Aldrich; \( {\text{ZnCl}}_{2} \), Merck) to the reactors. The reactors were sealed and stirred at 200 rpm using magnetic stirrers. All experiments were carried out in a thermostatic room at 24 ± 1 °C.
In order to track temporal changes of the solution composition, the reactive fluid (~ 1.2 mL) was sampled after 0.5, 1, 5, 10, 30 and 60 min after salt addition using a syringe (B. Braun, Omnifix® Solo). The reactive fluid was filtered through a 0.45 µm cellulose acetate membrane filter (Sartorius). An aliquot was acidified to a 2% \( {\text{HNO}}_{3} \) matrix for chemical analyses using HNO3 of suprapure grade (Roth, ROTIPURAN®). Another aliquot was kept non-acidified for pH measurements. Such throughout hydrochemical monitoring was carried out over the duration of the control experiments and for all experiments conducted at initial molar Me/Si and Ca/Si ratios of 0.2 and 1.0, respectively. For the other experiments, a fluid sample (~ 1.2 mL) was taken at the beginning (~ 30 s) and at the end (~ 1 h) of the experiments. The fluid sampling caused < 3% change of the initial volume in all experiments; thus, no corrections for the elemental concentration in solution were made. After ~ 60 min of reaction time, the experiments were terminated. Such a short duration was used to minimize potential changes in the mineralogical and structural composition of the precipitating aluminosilicate hydrates due to recrystallization and to avoid carbonate formation. The solids were separated by centrifugation (Mikro 185, Hettich) at 4500 rpm for 2 min. Then, the supernatant water was decanted and the remaining solids were washed with ethanol to remove electrolytes (3 times). Afterwards, the sample was transferred into a 50 mL PE jar, which was stored in a closed desiccator for drying until weight constancy was reached, which required circa 1–2 months. A saturated \( {\text{CaCl}}_{2} \) solution, which provides a ~ 33% relative humidity atmosphere, was used as the drying agent to ensure “gentle” drying of the reaction products.
Fluid-phase characterization
The pH of the reactive solutions was measured with a SenTix 41 glass electrode connected to a WTW Multi 350i pH-meter, which was calibrated against NIST buffer standard solutions at pH 4.01, 7.00 and 10.00 at 25 °C. The analytical precision is ± 0.03 pH units at pH ≤ 10.0 and increases to ± 0.17 pH units at pH ≥ 12.0, based on replicate analyses of buffers and samples.
The total concentrations of aqueous Na, Al, Ca, Co, Cr, Si and Zn were analysed in acidified aliquots (2% \( {\text{HNO}}_{3} \)) by inductively coupled plasma optical emission spectroscopy (ICP-OES) using a PerkinElmer Optima 8300 DV. The analytical precision (2σ, 3 replicates) is better than ± 2% for Co and Si analyses and ± 4% for Al, Ca, Cr and Zn analyses, respectively, relative to replicate measurements of NIST 1640a, in-house and SPS-SW2 Batch 130 standards [31].
The removal efficiency of aqueous heavy metals is expressed by the percentage of Me ions removed from solution at equilibrium (%removal), according to Eq. (1):
where \( C_{0} \) and \( C_{\text{e}} \) are the initial and equilibrium concentrations of the respective Me ion (in mg L−1) in the experimental solution. In this study, the term “equilibrium” is referred to the attainment of a constant chemical composition of the solution, which is reached after few minutes of reaction time.
Solid-phase characterization
Powder X-ray diffraction (XRD) patterns were recorded on dried samples using a PANalytical X’Pert PRO diffractometer operated at 40 kV and 40 mA (Co-Kα) and outfitted with a spinner stage, 0.5° antiscattering and divergence slits, primary and secondary Soller and a high-speed Scientific X’Celerator detector. The samples were prepared using the top loading technique and examined in the range 4–85° 2θ with a step size of 0.008° 2θ and a count time of 40 s per step. Mineral identification was realized using the PANalytical X’Pert Highscore Plus software (version 2.2e) and a pdf-4 database [47], as well as by comparison with XRD data collected from synthetic C–S–H gels [44].
The particle form, nanostructure, and chemical composition of the precipitated matter from experiments \( {\text{RefC}}_{1.0} , {\text{C}}_{1.0} {\text{Co}}_{0.2} , {\text{C}}_{1.0} {\text{Cr}}_{0.2} , {\text{C}}_{1.0} {\text{Zn}}_{0.02} , {\text{C}}_{1.0} {\text{Zn}}_{0.2} , \) and \( {\text{C}}_{1.0} {\text{Zn}}_{2.0} \) were analysed by transmission electron microscopy (TEM) on a FEI Tecnai F20 operated at an acceleration voltage of 200 kV. The TEM is outfitted with a single-crystal LaB6 Schottky Field Emitter, an UltraScan CCD camera for acquisition of high-resolution images, and an EDAX Sapphire Si(Li) detector for energy-dispersive X-ray spectroscopy (EDX) analysis. The TEM–EDX spectra were acquired using a count time of 30 s in order to reduce element migration and element loss during the measurements. The elemental k-factors were obtained from phyllosilicate standards [48,49,50]. The standard deviations were app. < 30% for Al, < 10–15% for Me, and < 5% for Ca and Si, respectively, which is equivalent to an analytical uncertainty of the TEM–EDX results of ± 2 at.% for most of the major elements.
Results
Chemical evolution of the experimental solutions
Temporal changes in the chemical composition of the reactive fluids are displayed in Fig. 2 for experiments conducted at initial molar Me/Si ratios of 0.2 and Ca/(Si + Al) ratios of 1.0. For comparison of all experimental data, the pH values and the total concentrations of aqueous Ca, Al, Si, and Me ions, obtained at the beginning and at the end of the individual experiments, are reported in Table 1, together with the calculated removal efficiencies of the elements of interest.

Temporal evolution of the aqueous concentrations of Ca, Al, Si, and Me ions, as well as of solution pH for experiments conducted at initial molar ratios of Me/Si of 0.2 and Ca/(Si + Al) of 1.0, respectively. a\( {\text{RefC}}_{1.0} \). b\( {\text{C}}_{1.0} {\text{Cr}}_{0.2} \). c\( {\text{C}}_{1.0} {\text{Co}}_{0.2} \). d\( {\text{C}}_{1.0} {\text{Zn}}_{0.2} \). The analytical uncertainty is included in the symbol size. Chemical steady-state has been attained after ~ 5 to 10 min in all experiments. See text for further explanations
From Fig. 2 and Table 1, it becomes clear that the pH of the experimental solutions decreased systematically as a function of the initial molar ratios of Ca/(Si + Al) and Me/Si, and type of heavy metals used for the C–A–S–H gel synthesis. Three distinct, but different effects on the evolution of solution pH were recognized. Firstly, an increase in the initial molar Ca/(Si + Al) ratio, from 0.6 and 1.0 to 1.6, resulted in a relatively slight pH decrease, from 11.9–12.1 and 11.0–11.2 to 10.7–10.9, in the experiments conducted at initial molar Me/Si ratios of 0.02. The pH values of the control experiments (12.2, 11.3, and 11.1) were found to be only ~ 0.1 to 0.3 pH units lower than those of the experimental series conducted at low Me concentrations. This suggests that the initial molar Ca/(Si + Al) ratio is much more important on controlling the evolution of pH than the type of Me ion added for the synthesis, especially at very low Me concentrations. Secondly, a similar trend towards decreasing pH values at increasing initial molar Ca/(Si + Al) ratios was observed in the series ran at initial molar Me/Si ratios of 0.2. However, the observed pH decrease was much more pronounced (e.g. 10.6–11.6, 10.2–11.0, and 10.0–10.6), which indicates that the type of Me ions used is becoming more important in these experiments (Fig. 2). Thirdly, no effect of the initial molar Ca/(Si + Al) ratio was recognized in experiments conducted at initial molar Me/Si ratios of 2.0. The type of Me ion used was the main influencing factor on controlling the evolution of pH, as it is seen by lower final pH values (e.g. 6.7–6.8, 5.7–6.0, and 3.4–3.5), when using aqueous Co, Zn, and Cr at very high concentrations (Table 1).
The total concentrations of aqueous Ca, Al, Si, and Me were significantly reduced at the end of the experiments after ~ 1 h of reaction time (Table 1). Noteworthy, the decrease in the elemental concentrations was extremely fast, which reflects the rapid formation of hydrated aluminosilicates (see below for solid-phase characterization). Chemical steady-state between the precipitating aluminosilicate phase and the reactive fluid was reached within less than 10 min in all experiments (Fig. 2), irrespective of the initial molar Ca/(Si + Al) ratio and Me concentration used. This finding matches the results of Pardal et al. [28], who have reported that the formation of a tobermorite-like C–A–S–H structure typically requires minutes to less than a few hours. Indeed, at the end of all experimental series, constant chemical conditions (or near-equilibrium conditions) between reactive fluid and precipitant phase were achieved. The attainment of thermodynamic equilibrium between a given mineral phase and reactive fluid is a prerequisite for the calculation of thermodynamic properties, such as solubility products, according to Purgstaller et al. [49]. However, such considerations are far beyond the scope of the present study.
The net removal of aqueous Ca (0.40–99.90%removal), Al (36.38–99.99%removal), Si (15.53–99.47%removal), and Me (22.46–99.99%removal) ions from solution depended on the experimental conditions initially used for aluminosilicate synthesis (Table 1). In contrast, the concentration of aqueous Na remained unchanged in all experiments (< 0.5%removal). Indeed, in all experiments conducted at initial molar Me/Si ratios of ≤ 0.2, the net removal of aqueous Ca (46.01–99.90%removal) and Si (74.38–99.47%removal) was moderate to high, and for aqueous Al and Me, it was very high (99.30–99.99%removal), which underpins the formation of C–A–S–H with incorporated Me. On the contrary, the experiments conducted at initial molar Me/Si ratios of 2.0 yielded generally lower removal efficiencies for aqueous Me (22.46–6.46%removal), Ca (0.40–51.38%removal), Al (36.38–99.92%removal) and Si (15.54–99.37%removal), which indicates that solid product(s) other than C–A–S–H formed.
Mineralogical characterization of precipitates with/without heavy metals
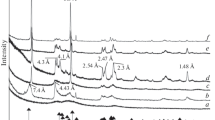
A summary of the mineralogical composition of the precipitates is provided in Table 2. Figure 3 shows the XRD patterns of precipitates with and without \( {\text{Zn}}^{2 + } \) (for experiments with Co and Cr similar XRD patterns were obtained, but are not shown). XRD analyses confirmed the formation of C–A–S–H in all experiments conducted at an initial molar Me/Si ratio ≤ 0.02 (see Fig. 3a, b for the control experiments and for Zn series, respectively). Irrespective of the initial molar Ca/(Si + Al) ratio used for the synthesis, the C–A–S–H phase exhibited sharp reflections at 3.03 Å and 1.82 Å, and diffuse reflections at ~ 14 Å, 5.33 Å and 2.80 Å. Such patterns are indicative of the orthorhombic crystal structure of (fully hydrated) 14 Å tobermorite (\( {\text{Ca}}_{5} {\text{Si}}_{6} {\text{O}}_{16} \left( {\text{OH}} \right)_{2} \)·7\( {\text{H}}_{2} {\text{O}} \)) [51,52,53,54]. No indication of formation of Al-O-OH phases, such as gibbsite or boehmite, and of other C–A–S–H phases, like hydrogarnet group minerals (\( {\text{Ca}}_{3} \left( {{\text{Al}},{\text{Fe}}} \right)_{2} [{\text{SiO}}_{4} ]_{y} \left( {\text{OH}} \right)_{{4\left( {3 - y} \right)}} ;0 < y < 3 \)), was found. However, the presence of trace amounts of amorphous silica and/or poorly crystallized Al-hydroxides cannot be fully excluded.

X-ray diffraction patterns of materials precipitated at initial molar ratios of Ca/(Si + Al) of 0.6, 1.0, and 1.6, Zn/Si of 0.0, 0.02, 0.2 and 2.0, and at a constant Al/Si ratio of 0.05. a, b “Ordered” 14 Å tobermorite (Tb) with stacking disorder along the c-axis. c Disordered tobermorite structure. d Sauconite-like material (Sau; Zn-smectite). Note that some patterns have been shifted vertically for better visibility
In the experiments conducted at an initial molar Zn/Si ratio of 0.2, the characteristic peaks of ordered 14 Å tobermorite disappeared or significantly lost intensity. Instead, broad and very weak peaks centred at ~ 3.0 Å, ~ 2.8 Å and ~ 1.8 Å developed (Fig. 3c). These reflections can be correlated with the Ca–Ca–Me physical repeat distance within the C–A–S–H structure and presence of dreierketten chains [55], features that are indicative of a highly disordered 14 Å (?) tobermorite-like structure. However, in the experimental series carried out with aqueous Co and Cr, no systematic trends between the initial molar ratios of Me/Si and Ca/(Si + Al), and the crystallinity degree of the C–A–S–H were recognized, even though the XRD patterns are generally very similar to those of precipitates from the Zn series.
In experiments carried out with an initial molar Zn/Si ratio of 2.0, broad and asymmetric peaks appeared at ~ 14–15 Å, 7.87 Å, 4.51 Å, 3.21 Å, 2.67 Å and 1.55 Å (Fig. 3d). These XRD peaks likely correspond to the (001), (002), (02 l), (005), (110) and (060) reflections of a sauconite-like phase (\( {\text{Na}}_{0.6} {\text{Zn}}_{3} [{\text{Si}}_{3.4} {\text{Al}}_{0.6} {\text{O}}_{10} ]\left( {\text{OH}} \right)_{2} \)·n\( {\text{H}}_{2} {\text{O}} \)) [56], which is a trioctahedral smectite rich in Zn. In analogy, in the experiments conducted with an initial molar Co/Si ratio of 2.0, a Co-smectite-like phase formed, as it can be seen by diffuse reflections at ~ 14 Å (001), 4.61 Å (02 l), 2.66 Å (110) and 1.56 Å (060) [57]. In contrast, the XRD patterns of solids received from experiments conducted with initial molar Cr/Si ratios of ≥ 0.2 revealed, in any case, formation of an X-ray amorphous reaction product (hereafter called Cr gel).
In some experiments, trace amounts of calcite, vaterite (i.e. \( {\text{CaCO}}_{3} \) polymorphs) and/or monohydrocalcite (\( {\text{CaCO}}_{3} \)·\( {\text{H}}_{2} {\text{O}} \)) were identified (Table 2). The occurrence of carbonates in the final precipitates suggests that, despite of continuous bubbling with \( {\text{N}}_{2} \), small amounts of dissolved inorganic carbon species remained in the reactor during the mineral synthesis. On the other hand, carbonates could have also been formed during washing and/or drying of the solids in the desiccator, through adsorption of \( {\text{CO}}_{2} \) from the atmosphere. It is worthy to note that neither (an)hydrous Me-carbonates nor Co-, Cr- and Zn-oxyhydrates were identified in the precipitates [58], suggesting that the Me ions are in the aluminosilicate hydrates structure.
Particle form and chemical composition of precipitates with/without heavy metals
Representative TEM images and corresponding TEM–EDX spectra of laboratory synthesized aluminosilicate hydrates, like C–A–S–H and Zn-smectite, are shown in Fig. 4. All C–A–S–H precipitated at initial molar Me/Si ratios ≤ 0.2 are mainly composed of nanometre-sized aggregates of clumped particles. The individual particles are < 30 nm in size, have a platy to globular shape with curled edges, a smooth particle surface, and a high inter-particle and surface porosity (Fig. 4a–e), applying the nomenclature of Yang et al. [59]. Generally, no differences were observed between C–A–S–H with and without Me ions incorporated with regard to the particle form and particle size. In contrast, the sample \( C_{1.0} Zn_{2.0} \) was composed of aggregated smectite particles, which are commonly < 10 nm in size and have a veil-like to filmy particle form (Fig. 4f).

Low-resolution TEM images and EDX spectra of synthetic aluminosilicate hydrates. a\( {\text{RefC}}_{1.0} \); b\( {\text{C}}_{1.0} {\text{Co}}_{0.2} \); c\( {\text{C}}_{1.0} {\text{Cr}}_{0.2} \); d\( {\text{C}}_{1.0} {\text{Zn}}_{0.02} \); e\( {\text{C}}_{1.0} {\text{Zn}}_{0.2} \); f\( {\text{C}}_{1.0} {\text{Zn}}_{2.0} \). g EDX spectra of C–A–S–H and smectite with incorporated Me ions (spot positions are marked in a–f). The Cu and C peaks in the EDX spectra stem from the TEM Cu grid and C film, respectively. The small Fe and Co peaks come from the pole piece of the TEM, except for \( {\text{C}}_{1.0} {\text{Co}}_{0.2} \), where the intensity of the Co peaks is higher due to abundant Co in the C–A–S–H structure
The bulk chemical compositions of the precipitates (expressed here as molar ratios of Ca/(Si + Al), Al/Si and Me/Si) are reported in Table 2. These compositions were calculated by subtracting the concentrations of aqueous Ca, Al, Si, and Me ions obtained at the beginning and at the end of a single experiment (Table 1). This simplified approach is valid, because Me, Ca, Al, and Si are homogenously distributed in C–A–S–H and in smectite (Fig. 4g) and the amount of other co-precipitates, like carbonates, is very low (< 5% of the total precipitate mass). Hence, the chemical compositions of ideal (pure) C–A–S–H and trioctahedral smectite are most likely similar to the reported bulk compositions. All C–A–S–H gels formed at initial molar Me/Si ratios ≤ 0.2 exhibited a sum of (Ca + Me)/(Si + Al) approximating 0.98 ± 0.14. Such a composition resembles that of either ordered 14 Å tobermorite at (Ca + Me)/(Si + Al) molar ratios ≤ 0.8–1.1 or defect tobermorite structures at (Ca + Me)/(Si + Al) molar ratios > 0.8–1.1 [2, 46, 52].
Discussion
Fate of Me ions during precipitation of C–A–S–H and trioctahedral smectite
The XRD and TEM results (Figs. 3 and 4) reveal (dis)ordered, tobermorite-type C–A–S–H, amorphous Cr gels and trioctahedral smectite as the main reaction products. No indication for the precipitation of either Me-carbonates or Me-oxyhydrates was found, which suggest that the principle removal mechanism of heavy metals from solution is governed by structural incorporation during co-precipitation with hydrated aluminosilicates. However, it is worthy to note that, from a thermodynamic point of view, the experimental solutions were highly supersaturated with respect to \( {\text{Co}}\left( {\text{OH}} \right)_{2} \), \( {\text{Zn}}\left( {\text{OH}} \right)_{2} \), and \( {\text{Cr}}\left( {\text{OH}} \right)_{3} \), particularly in the experiments carried out at initial molar Me/Si ratios ≤ 0.2, where highly alkaline conditions prevailed throughout (i.e. pH ≥ 10; Table 1). The identification of such precipitates can be challenging, as synthetic Me-oxyhydrates are often poorly crystallized, and the nano-sized particles tend to agglomerate onto previously formed hydrated aluminosilicate surfaces.
Therefore, a high-resolution TEM study was performed (Fig. 5) in order to proof the absence of co-precipitated Me-oxyhydrates. The lattice images of particles from experiments \( {\text{RefC}}_{1.0} \), \( {\text{C}}_{1.0} {\text{Co}}_{0.2} \), \( {\text{C}}_{1.0} {\text{Zn}}_{0.02} \), \( {\text{C}}_{1.0} {\text{Zn}}_{0.2} \), and \( {\text{C}}_{1.0} {\text{Zn}}_{2.0} \) typically show well-defined and almost defect-free stacking sequences with thicknesses between ~ 20 Å and ~ 500 Å (Fig. 5a, b, d–f) [60]. Fast Fourier Transform (FFT) measurements of the noise-filtered TEM fringe images frequently reveal interplanar distances of 3.0 Å, 2.7 Å, and 2.4 Å, which likely correspond to the (020 and − 220), (200 and 107), and (− 127) planes of C–A–S–H (i.e. depending on the particle orientation and applying the structural model for tobermorite of Bonaccorsi et al. [51] and Grangeon et al. [61]). In the case of the trioctahedral Zn-smectite, (001) planes with a ~ 13 Å thickness were frequently observed [32, 48]. In contrast, no ordered stacking sequences (i.e. areas lacking any short range order due to abundant lattice strain and folding) were found within particles obtained from experiment \( {\text{C}}_{1.0} {\text{Cr}}_{0.2} \) (Fig. 5b), which further documents the amorphous nature of the Cr gel.

High-resolution TEM lattice fringe images and fast Fourier transform (FFT) patterns of synthetic C–A–S–H and trioctahedral Zn-smectite. a\( {\text{RefC}}_{1.0} \); b\( {\text{C}}_{1.0} {\text{Co}}_{0.2} \); c\( {\text{C}}_{1.0} {\text{Cr}}_{0.2} \); d\( {\text{C}}_{1.0} {\text{Zn}}_{0.02} \); e\( {\text{C}}_{1.0} {\text{Zn}}_{0.2} \); f\( {\text{C}}_{1.0} {\text{Zn}}_{2.0} \). Note the absence of discrete Me-oxyhydrates on the precipitate surfaces, suggesting that most of the Me ions are structurally incorporated in the aluminosilicate hydrates
Importantly, no evidence for the formation of discrete Me-oxyhydrates was found in the investigated samples, which suggests that the vast majority of heavy metals taken up from the experimental solution (Table 1) occupy structural sites in the synthetic C–A–S–H, Cr gels, and trioctahedral smectite (see below for further discussion). The extremely fast precipitation of aluminosilicate hydrates (i.e. within few minutes, see Fig. 2) under far-from-equilibrium conditions has probably hindered Me-oxyhydrates to form from the beginning of the entire experiments despite of potentially favourable conditions. Indeed, the strong influence of Me ion incorporation during the formation of the aluminosilicate hydrates (through isomorphous substitution) is clearly reflected by the pronounced structural collapse of the crystal lattice of C–A–S–H phases at higher proportions of incorporated Me ions (see Fig. 3): at increasing molar Me/Si ratios in the precipitates a transition from the ordered tobermorite structure to a defect and poorly crystallized tobermorite-type structure is recognizable. Such an impact on the crystal structure of C–A–S–H cannot be explained by a classical adsorption mechanism (i.e. surface complexation, cation exchange, physical attachment of Me ions) nor by the co-precipitation of Me-oxyhydrates onto previously formed and charged aluminosilicate surfaces [43].
Removal capacities of Me ions by C–A–S–H and trioctahedral smectite
In the last five decades, increasingly more studies have been published that aimed at describing and quantifying the reaction pathways and fundamental mechanisms underlying the uptake of aqueous Me ions by natural and synthetic C–A–S–H. These works have shown that C–A–S–H, especially tobermorite, can exhibit sufficiently high sorption properties for a suite of potentially hazardous Me ions, including \( {\text{Cs}}^{ + } \), \( {\text{Rb}}^{ + } \), \( {\text{K}}^{ + } \), \( {\text{Na}}^{ + } \), \( {\text{Ba}}^{2 + } \), \( {\text{Sr}}^{2 + } \), \( {\text{Mg}}^{2 + } \), \( {\text{Cd}}^{2 + } \), \( {\text{Pb}}^{2 + } \), \( {\text{Co}}^{2 + } \), \( {\text{Cu}}^{2 + } \), \( {\text{Ni}}^{2 + } \), \( {\text{Zn}}^{2 + } \), \( {\text{As}}^{3 + /5 + } \), \( {\text{Al}}^{3 + } \), \( {\text{Fe}}^{3 + } \), \( {\text{Cr}}^{3 + /6 + } \), and many others [13,14,15, 17, 29, 62,63,64]. However, a comparative analysis of the removal potential for Me ions based on adsorption data, Me uptake rates, or immobilization degrees etc., obtained from different materials, is difficult [40]. This is mainly because of large differences in the test conditions and test matrices used, and inconsistencies in the literature data. Despite of these problems, some general relations between mineralogy, structure, composition, and Me ion removal capacity of C–A–S–H and smectite can be discerned. From Fig. 6, it is evident that the nature of the reaction product(s) takes a key control on the removal efficiency of Me ions. To ensure a reliable comparison of the Me ion removal potential of C–A–S–H and smectite, individual sorption capacities (\( q_{\text{e}} : {\text{mg}}/{\text{g}} \)) have been calculated, according to Eq. (2):
where m is the total mass of reaction products formed during gel synthesis (in g; reported on a water-free basis), and V refers to the volume of the solution (L). The calculated \( q_{\text{e}} \) values are given in Table 2. Note that the \( q_{\text{e}} \) values do not necessarily represent maximum sorption capacities; hence, comparison with published data has to be made very carefully taking into account the respective experimental conditions.

Removal efficiencies for aqueous Co, Cr, and Zn by C–A–S–H, Cr gel, and trioctahedral smectite
The estimated sorption capacities range from 2.9 to 5.4 mg/g for C–A–S–H formed at rather low Me ion concentrations (~ 1.9 mM). C–A–S–H precipitated at moderately high Me ion concentrations (~ 18.9 mM) exhibited much higher \( q_{\text{e}} \) values in the range from 27.5 to 40.4 mg/g. This is in the narrow range of the maximum adsorption capacities of \( {\text{Cr}}^{6 + } \) by Al- and sucrose-incorporated tobermorite (~ 32 mg/g and ~ 29 mg/g, respectively), according to Zhao et al. [29]. However, chromitite formed as a reaction by-product in their work, which suggests that the reported adsorption capacities for \( {\text{Cr}}^{6 + } \) by these tobermorite materials may be slightly lower. Despite of this constraint, we propose that the maximum sorption capacity of C–A–S–H with a tobermorite-like structure (without surface modification) should be in the same order of magnitude (~ 30–40 mg/g for \( {\text{Co}}^{2 + } \), \( {\text{Cr}}^{3 + } \), and \( {\text{Zn}}^{2 + } \)). Trioctahedral smectite and Cr gel precipitated at extremely high Me ion concentrations (~ 188.5 mM) exhibit higher sorption capacities of 121.9–141.3 and 108.3–151.7 mg/g, respectively.
Interestingly, no effect of the type of Me ion (\( {\text{Co}}^{2 + } \), \( {\text{Cr}}^{3 + } \), and \( {\text{Zn}}^{2 + } \)) and initial molar Ca/Si ratio (at least in the range between 0.6 and 1.6) used for the C–A–S–H and smectite synthesis on the obtained \( q_{\text{e}} \) values is observable (Table 2). This finding shows that the Me ion concentration initially introduced to the reactors as well as the local atomic structure and the mineralogical composition of the precipitates are much more important in regulating the uptake of heavy metals by hydrated aluminosilicates [40]. This is because the isomorphous substitution of Me ions with an ionic radii (i.e. 72 pm for \( {\text{Co}}^{2 + } \), 63 pm for \( {\text{Cr}}^{3 + } \), and 74 pm for \( {\text{Zn}}^{2 + } \)) smaller than that of the \( {\text{Ca}}^{2 + } \) ion (99 pm) induces structural distortion and charge imbalances in the Ca-O layer of C–A–S–H (see below for detailed discussion). In case of trioctahedral smectite and Cr gels, most of the available octahedral sites are occupied entirely by Me ions; thus resulting in higher removal capacities compared to C–A–S–H (Table 2).
Potential removal mechanism(s) for Me ions by C–A–S–H and trioctahedral smectite
The principle removal mechanisms for \( {\text{Co}}^{2 + } \), \( {\text{Cr}}^{3 + } \), \( {\text{Zn}}^{2 + } \), and other ions by synthetic C–A–S–H and trioctahedral smectite include (1) interlayer (cat)ion exchange, (2) isomorphous substitution in the Ca-O layer or in the octahedral sheet, (3) replacement of tetrahedral sites (\( Q^{1} \) to \( Q^{3} \) positions), (4) physisorption or chemical precipitation on the mineral surface, or (5) a combination of the processes (1)–(4) (see Fig. 1 for comparison).
From the obtained XRD data, no significant changes in the (hk0) dimensions of the Al- and Me-incorporated tobermorite-like products were recognized in all experiments (Fig. 2), which implies that Al and Me ions or their aquo-complexes do not occupy the interlayer space of the C–A–S–H gels to a great extent. In this line, Anderson et al. [24] and Richardson [65] have argued that isomorphous substitution of Al for Ca in the Ca-O sheet of C–A–S–H is also highly limited, because of the different ionic radii and charges of Ca and Al, generating charge imbalances and distortion of the C–A–S–H structure. Therefore, it can be concluded that the majority of the structurally bound Al in synthetic tobermorite-like materials obtained in this study should be present as tetrahedral Al, i.e. substituting for Si in the tetrahedral networks [1, 66].
With respect to the (hydrated) Me ions, such an interpretation is even more problematic, because the present data do not allow for a detailed structural analysis of C–A–S–H. However, the lack of Me-(oxy)hydrates or other Me-containing phases in the final precipitates indicates that the vast majority of the Me ions is likely incorporated in the C–A–S–H structure, rather than being co-precipitated onto the C–A–S–H surface. This assertion may be supported by the collapse of the ordered tobermorite-type structure and its subsequent transformation into a poorly crystallized, defect tobermorite-type structure at higher levels of Me ions incorporated (Figs. 2 and 3). The spectroscopic results of Żak and Deja [44] provide further evidence for significant incorporation of Me ions into synthetic C–S–H structures: the substitution of \( {\text{Zn}}^{2 + } \), \( {\text{Cd}}^{2 + } \), \( {\text{Cr}}^{3 + } \), and \( {\text{Pb}}^{2 + } \) ions results in a high degree of de-polymerization of the dreierketten chains, as it can be seen by a breaking of the Si–O–Si bands and neo-formation of Si-\( {\text{O}}^{ - } \) bands. This key observation implies that Me ions can substitute in the Ca–O layer and in the interlayer site of C–S–H [44] and probably C–A–S–H structures. Such an incorporation mechanism would explain the loss of diffracted intensity in C–A–S–H bearing moderately high loads of Me ions, as observed for materials precipitated at initial molar Me/Si ratios of 0.2 (Fig. 3). Because \( {\text{Co}}^{2 + } \) ions exhibit physicochemical properties similar to the other Me ions mentioned above, it is likely that Co occupies the same structural sites in C–A–S–H, i.e. the Ca–O layer and the interlayer position, but not the tetrahedral sheets (Fig. 1). An exception could be the incorporation of \( {\text{Cr}}^{3 + } \) in C–A–S–H at higher concentrations, because the isomorphous substitution of \( {\text{Cr}}^{3 + } \) for \( {\text{Ca}}^{2 + } \) and \( {\text{Al}}^{3 + } \) in the Ca–O layer, interlayer, and possibly dreierketten chains typically leads to the formation of Cr(III)-bearing C–A–S–H with an impaired crystal structure [44]. This behaviour might explain the formation of Cr gels with unusually high Cr/Si molar ratios (1.8 to 3.2) at initial molar Cr/Si ratios of 2.0 (Table 2). In the case of precipitation of trioctahedral smectite (Table 2 and Fig. 3d), most of the structural Al and Si are placed in the tetrahedral sheet, Co and Zn dominate in the octahedral sheet, and Ca is the main interlayer cation [48, 57, 67].
From the present datasets and the knowledge from previously published data, it can be inferred that the majority of Me ions incorporated in C–A–S–H and smectite preferentially occupy the octahedral and interlayer sites. Some chemical environments of 4-coordinated Co, Cr, and Zn may also exist in the tetrahedral sheets [68]. A certain degree of surface adsorption and surface complexation by hydrated Me ions and subsequent precipitation of Me-oxyhydrates onto the C–A–S–H surface cannot be fully excluded, as indicated before, although we found no analytical evidence for such removal mechanism(s) to take place under the given experimental conditions. However, Qi et al. [62] have presented some evidence of coupled chemical precipitation, physisorption and structural incorporation phenomena for the adsorption of \( {\text{Co}}^{2 + } \) ions by mesoporous C–S–H. In conclusion, the removal mechanisms of Me ions by aluminosilicate hydrates are highly complex and include ion exchange in the interlayer, isomorphous substitution in the octahedral and tetrahedral layers, and possibly surface (ad)sorption and chemical precipitation (Fig. 1).
Environmental implications of heavy metals uptake by C–A–S–H and smectite
The removal mechanisms and removal efficiencies of aqueous Me ions by C–A–S–H and trioctahedral smectite depend mainly on the physicochemical, mechanical, and electrokinetic properties, and on the surface charge of the (ad)sorbent material, as well as on chemical key characteristics (pH, temperature, ionic strength etc.) of the interacting fluids [34,35,36,37,38,39,40,41]. From the data reported in Table 1, it becomes clear that the pH value, obtained at nearly chemical steady-state, is a key parameter in controlling the net uptake of Me ions by aluminosilicate hydrates. Importantly, the measured pH values and the Ca/Si ratios calculated for C–A–S–H are in reasonable agreement with published C–S–H gel solubility data (compare Table 1 and Table 2 with Fig. 4a in [46]). This indicates that C–A–S–H obtained in this study should have surface charge and physicochemical properties similar to other natural and synthetic C–S–H and C–A–S–H structures. These similarities exist despite of apparent differences in the experimental conditions used to precipitate C–A–S–H, such as curing times of few minutes to several weeks, curing temperatures from 17 to ≥ 40 °C, and liquid/solid mass ratios from 8 to 4000 [46, 59]. In this framework, the obtained results can provide new insights into the environmental significance of Me ion removal by C–A–S–H and smectite.
Concrete structures
In recent decades, the use of cementitious materials for the detoxication of environments polluted by aqueous heavy metals has been considered more often [8,9,10,11,12,13,14,15,16,17,18, 29, 35, 39, 69]. For example, Svatovskaya et al. [70] reacted powdered cement clinker, slag, and concrete with and without SCMs with an aqueous solution originally containing 0.01 mM of \( {\text{Cd}}^{2 + } \), \( {\text{Cu}}^{2 + } \), \( {\text{Fe}}^{2 + } \), \( {\text{Mn}}^{2 + } \), \( {\text{Ni}}^{2 + } \), or \( {\text{Cr}}^{3 + } \) ions in laboratory-scale column experiments, and reported that the removal of Me ions was extremely fast (i.e. completed within minutes). The obtained removal capacities for the Me ions mentioned above (~ 1 to 5 mg/g [70]) are in the same range than for C–A–S–H formed in this study at initial molar Me/Si ratios of 0.02 (~ 3 to 5 mg/g), which indicates that the same removal mechanisms were active in both studies. These authors have argued further that the secondary hydration products (i.e. C–S–H and C–A–S–H) formed are less soluble, compared to the cement hydrates without Me ions incorporated [70]. This finding supports the idea of using C–S–H and C–A–S–H for the removal of heavy metals from contaminated (sub)soil structures and groundwater, though field reports are scarce, yet.
A high retention potential for Me ions is also of great significance during early cement hydration, and during subsequent exposure of the hardened concrete to aquatic systems, because it is critical to know to which extent Me ions can be incorporated in the hydrated cement binders and later released back into the ecosystem. Specifically, cement blends made with high levels of SCMs, such as GGBFS, metakaolin, fly ash, and limestone fillers, can contain higher concentrations of heavy metals than OPC-type cements (see Table 3 in [21]). Our results indicate that the uptake of Me ions during C–A–S–H formation is very high (Table 1). We therefore propose that the release back of Me ions from hardened concrete structures into the aquatic environment should be small, especially under highly alkaline conditions, where C–S–H and C–A–S–H structures are thermodynamically stable [46]. However, as soon as the pH of the interacting fluids drops below a certain threshold value, i.e. pH ~ 10 or higher for C–S–H phases with a (Ca + Me)/(Si + Al) ratio of ≥ 0.8 and pH ~ 10 for C–S–H phases with a (Ca + Me)/(Si + Al) ratio from 0.3 to 0.8 [46, 71], decomposition of the cement hydrates is likely to occur. This reaction could result in severe contamination of the “outer” aquatic environment by heavy metals. For the latter reason, the content of heavy metals should be monitored in raw materials, as well as in cement clinker and cement paste during production, curing, hydration, hardening, and service life performance.
Nuclear waste disposal sites
Knowledge of the physicochemical interactions between concrete and bentonite, especially the intercalation of C–A–S–H and montmorillonite (\( \left( {{\text{Na}},{\text{Ca}}} \right)_{0.3} \left( {{\text{Al}},{\text{Mg}},{\text{Fe}}} \right)_{2} \left[ {{\text{Si}}_{4} {\text{O}}_{10} } \right]\left( {\text{OH}} \right)_{2} \cdot {\text{nH}}_{2} {\text{O}}) \) at the pore scale, is also important in the disposal of low- to high-level radioactive waste in argillaceous rocks in the geological underground [72]. In contact with the concrete buffer highly alkaline pore solutions (pH > 12.5) are generated, where montmorillonite can rapidly alter to mixed-layer illite–smectite, Na/K-zeolite, and trioctahedral smectite, causing modifications of the mechanical properties (i.e. loss of strength) of the bentonite buffer and progressive lowering of the self-retention potential of the bentonite barrier for radionuclides and heavy metals [73, 74]. Moreover, the simultaneous formation of C–A–S–H in equilibrium with brucite (\( {\text{Mg}}({\text{OH}}_{2} ) \)) and magnesium silicate hydrate (M–S–H) phases can further weaken the concrete and bentonite barriers matrices (i.e. increase in secondary porosity), which can facilitate various other forms of physical and chemical attack, such as the alkali-aggregate reaction, recrystallization of C–A–S–H induced by \( {\text{Ca}}^{2 + } \) leaching, carbonation, formation of salt crusts, sulphate attack, corrosion of steel containers, and ion exchange [7, 75,76,77].
It is clear that the chemical reactivity of the concrete–bentonite buffer system is controlled mainly by the physicochemical conditions that develop at the interface between the newly formed C–A–S–H and clay minerals. For example, at higher temperature (60 °C to 120 °C), the dissolution rate of montmorillonite increases, which can result in a higher availability of \( {\text{Al}}^{3 + } \) ions for the subsequent precipitation of C–A–S–H. Accordingly, secondary C–A–S–H that form from montmorillonite precursors typically show increased Al/Ca and Si/Ca ratios, and a higher crystallinity degree [72]. With respect to heavy metals uptake, such C–A–S–H will probably have higher Me/Al and Me/Si ratios than components originally present in the concrete barrier, taken that Me ions preferentially substitute for \( {\text{Ca}}^{2 + } \) ions in the C–A–S–H structure. This process (i.e. isomorphous substitution) is therefore suggested to significantly affect the transport properties of Me ions in nuclear waste disposal sites. The present work reveals a high Me ion removal capacity for neo-formed C–A–S–H, Cr gels, and trioctahedral smectite in the magnitude of ~ 30 up to ~ 150 mg/g. This suggests that secondary hydrated aluminosilicates can scavenge even extreme loads of heavy metals (up to few hundreds of ppm), thus having the potential to prevent the outer environment from contamination.
Conclusions and perspective
Nanoparticulate C–A–S–H (tobermorite-type) phases, Cr gels, and trioctahedral smectite were synthesized by the reaction of an alkaline \( {\text{Si}}\left( {\text{OH}} \right)_{4} \) solution with Ca, Al, and Me salts. The removal efficiencies and uptake mechanisms of \( {\text{Co}}^{2 + } \), \( {\text{Cr}}^{3 + } \), and \( {\text{Zn}}^{2 + } \) ions by the hydrated aluminosilicates were studied at initial molar Me/Si ratios from 0.02 to 2.0 and Ca/(Si + Al) ratios from 0.6 to 1.6, at 24 ± 1 °C. At molar Me/Si ratios ≤ 0.2, 14 Å tobermorite-like structures formed, independent of the Ca/(Si + Al) ratio used for the synthesis. These C–A–S–H phases have a (Ca + Me)/(Si + Al) ratio equal to ~ 1 and are composed of porous spherules, < 30 nm in size. Formation of X-ray amorphous Cr gels was observed at molar Cr/Si ratios ≥ 0.2. The Cr gels have higher Al/Si and Cr/Si ratios and highly variable Ca/Si ratios from 0.5 to 1.3, compared to C–A–S–H, and are composed of porous spherical particles, 5–10 nm in diameter. Trioctahedral Co- and Zn-smectite with a low crystallinity degree formed at molar Me/Si ratios of 2.0. The smectites have lower Ca/Si and Al/Si ratios and higher Me/Si ratios than the Cr gels and C–A–S–H and are composed of < 50 nm sized, filmy to veil-like particles. The metal ion removal efficiencies obtained for different aluminosilicate hydrates depended mainly on the molar Me/Si ratio used for the synthesis, but generally increased in the order C–A–S–H < Cr gel < trioctahedral smectite. Precipitation of discrete Me-oxyhydrates was not observed under the experimental conditions used in this study despite of potentially favourable conditions. The removal mechanisms of Me ions during the precipitation and maturation of C–A–S–H, Cr gels and trioctahedral smectites are highly complex and include isomorphous substitution in octahedral and tetrahedral positions, ion exchange in the interlayer sites, and possibly surface (ad)sorption, and surface precipitation. Under highly alkaline conditions, the detoxication potential for Me ions by secondary aluminosilicate hydrates is considered to be very high, likely exceeding that of available (cementitious) adsorbent materials.
More studies on the crystallization paths and rates of hydrated aluminosilicate precipitation from solutions containing different Me ions are required to resolve and quantify details on crystal chemistry, thermodynamic properties, and (meta)stability of these important silicate minerals. Future experimental work should focus on elucidating the solubility and reactivity of C–A–S–H structures to demonstrate their high metal ion retention potential under varying environmental conditions (temperature, pH, ionic strength, redox conditions, etc.). In this framework, leaching scenarios and regeneration studies have to be performed.
References
Sun GK, Young JF, Kirkpatrick RJ (2006) The role of Al in C–S–H: NMR, XRD, and compositional results for precipitated samples. Cem Concr Res 36:18–29
L’Hôpital E, Lothenbach B, Le Saout G, Kulik D, Scrivener K (2015) Incorporation of aluminium in calcium-silicate-hydrates. Cem Conc Res 75:91–103
Love CA, Richardson IG, Brough AR (2007) Composition and structure of C–S–H in white Portland cement—20% metakaolin pastes hydrated at 25 °C. Cem Concr Res 37:109–117
Richardson IG (2008) The calcium silicate hydrates. Cem Concr Res 38:137–158
Mittermayr F, Rezvani M, Baldermann A, Hainer S, Breitenbücher P, Juhart J, Graubner C-A, Proske T (2015) Sulfate resistance of cement-reduced eco-friendly concretes. Cem Concr Comp 55:364–373
Chen JJ, Thomas JJ, Taylor HFW, Jennings HM (2004) Solubility and structure of calcium silicate hydrate. Cem Concr Res 34:1499–1519
Galan I, Baldermann A, Kusterle W, Dietzel M, Mittermayr F (2019) Durability of shotcrete for underground support—review and update. Constr Build Mater 202:465–493
van Jaarsveld JGG, van Deventer JSJ (1996) The potential use of geopolymeric materials to immobilize toxic metals: part I. Theory and applications. Miner Eng 10:659–669
Gougar MLD, Scheetz BE, Roy DM (1996) Ettringite and C–S–H Portlandite cement phases for waste ion immobilization: a review. Waste Manage 16:295–303
Dermatas D, Meng X (2003) Utilization of fly ash for stabilization/solidification of heavy metal contaminated soils. Eng Geol 70:377–394
Liu X, Zhao X, Yin H, Chen J, Zhang N (2018) Intermediate-calcium based cementitious materials prepared by MSWI fly ash and other solid wastes: hydration characteristics and heavy metals solidification behavior. J Hazard Mater 349:262–271
Ma J, Qin G, Zhang Y, Sun J, Wang S, Jiang L (2018) Heavy metal removal from aqueous solutions by calcium silicate powder from waste coal fly-ash. J Clean Prod 182:776–782
Coleman NJ, Brassington DS (2003) Synthesis of Al-substituted 11 Å tobermorite from newsprint recycling residue: a feasibility study. Mater Res Bull 38:485–497
Coleman NJ (2006) Interactions of Cd(II) with waste-derived 11 Å tobermorites. Sep Purif Technol 48:62–70
Mostafa NY, Kishar EA, Abo-El-Enein SA (2009) FTIR study and cation exchange capacity of Fe3+- and Mg2+-substituted calcium silicate hydrates. J Alloy Compd 473:538–542
Guan W, Ji FY, Chen QK, Yan P, Zhang Q (2013) Preparation and phosphorus recovery performance of porous calcium–silicate–hydrate. Ceram Int 39:1385–1391
Tsutsumi T, Nishimoto S, Kameshima Y, Miyake M (2014) Hydrothermal preparation of tobermorite from blast furnace slag for Cs+ and Sr2+ sorption. J Hazard Mater 266:174–181
Girão AV, Richardson IG, Taylor R, Brydson RMD (2010) Composition, morphology and nanostructure of C–S–H in 70% white Portland cement—30% fly ash blends hydrated at 55 °C. Cem Concr Res 40:1350–1359
Lothenbach B, Scrivener K, Hooton RD (2011) Supplementary cementitious materials. Cem Concr Res 41:1244–1256
Baldermann A, Rezvani M, Proske T, Grengg C, Steindl F, Sakoparnig M, Baldermann C, Galan I, Emmerich F, Mittermayr F (2018) Effect of very high limestone content and quality on the sulfate resistance of blended cements. Constr Build Mater 188:1065–1076
Achternbosch M, Bräutigam K-R, Gleis M, Hartlieb N, Kupsch C, Richers U, Stemmermann P (2003) Heavy metals in cement and concrete resulting from the co-incineration of wastes in cement kilns with regard to the legitimacy of waste utilisation. Wissenschaftliche Berichte, FZKA, Karlsruhe, p 6923
Komarneni S, Roy R, Roy DM, Fyfe CA, Kennedy GJ, Bothnerby AA, Dadok J, Chesnick AS (1985) Al-27 and Si-29 magic angle spinning nuclear magnetic-resonance spectroscopy of Al-substituted tobermorites. J Mater Sci 20:4209–4214. https://doi.org/10.1007/BF00552416
Richardson IG, Groves GW (1993) The incorporation of minor and trace elements into calcium silicate hydrates (C–S–H) gel in hardened cement paste. Cem Concr Res 23:131–138
Andersen MD, Jakobsen HJ, Skibsted J (2003) Incorporation of aluminum in the calcium silicate hydrate (C–S–H) of hydrated portland cements: a high-field 27Al and 29Si MAS NMR investigation. Inorg Chem 42:2280–2287
Renaudin G, Russias J, Leroux F, Cau-dit-Coumes C, Frizon F (2009) Structural characterization of C–S–H and C–A–S–H samples-Part II: local environment investigated by spectroscopic analyses. J Solid State Chem 182:3320–3329
Shaw S, Clark SM, Henderson CMB (2000) Hydrothermal formation of the calcium silicate hydrates, tobermorite (Ca5Si6O16(OH)2·4H2O) and xonotlite (Ca6Si6O17(OH)2): an in situ synchrotron study. Chem Geol 167:129–140
Faucon P, Charpentier T, Nonat A, Petit JC (1998) Triple-quantum two-dimensional Al-27 magic angle nuclear magnetic resonance study of the aluminum incorporation in calcium silicate hydrates. J Am Chem Soc 120:12075–12082
Pardal X, Pochard I, Nonat A (2009) Experimental study of Si-Al substitution in calcium–silicate–hydrate (C–S–H) prepared under equilibrium conditions. Cem Concr Res 39:637–643
Zhao Z, Wei J, Li F, Qu X, Shi L, Zhang H, Yu Q (2017) Synthesis, characterization and hexavalent chromium adsorption characteristics of aluminum and sucrose-incorporated tobermorite. Materials 10:597
Wu PX, Liao ZW, Zhang HF, Guo JG (2001) Adsorption of phenol on inorganic–organic pillared montmorillonite in polluted water. Environ Inter 26:401–407
Baldermann A, Grießbacher AC, Baldermann C, Purgstaller B, Letofsky-Papst I, Kaufhold S, Dietzel M (2018) Removal of barium, cobalt, strontium, and zinc from solution by natural and synthetic allophane adsorbents. Geosciences 8:309
Baldermann A, Dohrmann R, Kaufhold S, Nickel C, Letofsky-Papst I, Dietzel M (2014) The Fe-Mg- saponite solid solution series—a hydrothermal synthesis study. Clay Miner 49:391–415
Wu P, Wu W, Li S, Xing N, Zhu N, Li P, Wu J, Yang C, Dang Z (2009) Removal of Cd2+ from aqueous solution by adsorption using Fe-montmorillonite. J Hazard Mater 169:824–830
Crévecoeur S, Debacker V, Joaquim-Justo C, Gobert S, Scippo ML, Dejonghe W, Martin P, Thomé JP (2011) Groundwater quality assessment of one former industrial site in Belgium using a TRIAD-like approach. Environ Pollut 159:2461–2466
Malik QA, Khan MS (2016) Effect on human health due to drinking water contaminated with heavy metals. J Pollut Eff Control 5:1000179
Chabukdhara M, Gupta SK, Kotecha Y, Nema AK (2017) Groundwater quality in Ghaziabad district, Uttar Pradesh, India: multivariate and health risk assessment. Chemosphere 179:167–178
Wei J, Yang Z, Sun Y, Wang C, Fan J, Kang G, Zhang R, Dong X, Li Y (2019) Nanocellulose-based magnetic hybrid aerogel for adsorption of heavy metal ions from water. J Mater Sci. https://doi.org/10.1007/s10853-019-03322-0
Bhuiyan MAH, Islam MA, Dampare SB, Parvez L, Suzuki S (2010) Evaluation of hazardous metal pollution in irrigation and drinking water systems in the vicinity of a coal mine area of northwestern Bangladesh. J Hazard Mater 179:1065–1077
Burakov AE, Galunin EV, Burakova IV, Kucherova AE, Agarwal S, Tkachev AG, Gupta VK (2018) Adsorption of heavy metals on conventional and nanostructured materials for wastewater treatment purposes: a review. Ecotoxicol Environ Saf 148:702–712
De Gisi S, Lofrano G, Grassi M, Notarnicola M (2016) Characteristics and adsorption capacities of low-cost sorbents for wastewater treatment: a review. Sust Mater Technol 9:10–40
Carolin CF, Kumar PS, Saravanan A, Joshiba GJ, Naushad M (2017) Efficient techniques for the removal of toxic heavy metals from aquatic environment: a review. J Environ Chem Eng 5:2782–2799
Coleman NJ (2005) Synthesis, structure and ion exchange properties of 11 Å tobermorites from newsprint recycling residue. Mater Res Bull 40:2000–2013
Uddin MK (2017) A review on the adsorption of heavy metals by clay minerals, with special focus on the past decade. Chem Eng J 308:438–462
Żak R, Deja J (2015) Spectroscopy study of Zn, Cd, Pb and Cr ions immobilization on C–S–H phase. Spectrochim Acta A 134:614–620
Giergiczny Z, Król A (2008) Immobilization of heavy metals (Pb, Cu, Cr, Zn, Cd, Mn) in the mineral additions containing concrete composites. J Hazard Mater 160:247–255
Walker CS, Sutou S, Oda C, Mihara M, Honda A (2016) Calcium silicate hydrate (C–S–H) gel solubility data and a discrete solid phase model at 25 °C based on two binary non-ideal solid solutions. Cem Concr Res 79:1–30
Richoz S, Baldermann A, Frauwallner A, Harzhauser M, Daxner-Höck G, Klammer D, Piller WE (2017) Geochemistry and mineralogy of the Oligo-Miocene sediments of the Valley of Lakes, Mongolia. Palaeobio Palaeoenv 97:233–258
Baldermann A, Warr LN, Letofsky-Papst I, Mavromatis V (2015) Substantial iron sequestration during green-clay authigenesis in modern deep-sea sediments. Nat Geosci 8:885–889
Purgstaller B, Dietzel M, Baldermann A, Mavromatis V (2017) Control of temperature and aqueous Mg2+/Ca2+ ratio on the (trans-)formation of ikaite. Geochim Cosmochim Acta 217:128–143
Baldermann A, Mavromatis V, Frick PM, Dietzel M (2018) Effect of aqueous Si/Mg ratio and pH on the nucleation and growth of sepiolite at 25 °C. Geochim Cosmochim Acta 227:211–226
Bonaccorsi E, Merlino S, Kampf AR (2005) the crystal structure of tobermorite 14 Å (plombierite), a C–S–H Phase. J Am Ceram Soc 88:505–512
Richardson IG (2004) Tobermorite/jennite- and tobermorite/calcium hydroxide-based models for the structure of C–S–H: applicability to hardened pastes of tricalcium silicate, β-dicalcium silicate, Portland cement, and blends of Portland cement with blast-furnace slag, metakaolin, or silica fume. Cem Concr Res 34:1733–1777
Dilnesa BZ, Lothenbach B, Renaudin G, Wichser A, Kulik D (2014) Synthesis and characterization of hydrogarnet Ca3(AlxFe1−x)2(SiO4)y(OH)4(3−y). Cem Concr Res 59:96–111
Walkley B, Kashani A, Sani MA (2018) Examination of alkali-activated material nanostructure during thermal treatment. J Mater Sci 53:9486–9503. https://doi.org/10.1007/s10853-018-2270-z
Vieland D, Li J, Yuan L, Zhengkui X (1996) Mesostructure of calcium silicate hydrate (C–S–H) gels in portland cement pastes: short-range ordering, nanocrystallinity, and local compositional order. J Am Ceram Soc 79:1731–1744
Zhou R, Basu K, Hartman H, Matocha CJ, Sears SK, Vali H, Guzman MI (2017) Catalyzed synthesis of zinc clays by prebiotic central metabolites. Sci Rep 7:533
Bruce LA, Sanders JV, Turney TW (1986) Hydrothermal synthesis and characterization of cobalt clays. Clays Clay Miner 34:25–36
Bucca M, Dietzel M, Tang J, Leis A, Köhler SJ (2009) Nucleation and crystallization of otavite, witherite, calcite, strontianite, hydrozincite, and hydrocerussite by CO2 membrane diffusion technique. Chem Geol 266:143–156
Yang J, Li D, Fang Y (2017) Synthesis of nanoscale CaO–Al2O3–SiO2–H2O and Na2O–Al2O3–SiO2–H2O using the hydrothermal method and their characterization. Materials 10:695
Paradiso P, Santos RL, Horta RB, Lopes JNC, Ferreira PJ, Colaço R (2018) Formation of nanocrystalline tobermorite in calcium silicate binders with low C/S ratio. Acta Mater 152:7–15
Grangeon S, Claret F, Linard Y, Chiaberge C (2013) X-ray diffraction: a powerful tool to probe and understand the structure of nanocrystalline calcium silicate hydrates. Acta Cryst B69:465–473
Qi G, Lei X, Li L, Yuan C, Sun Y, Chen J, Chen J, Wang Y, Hao J (2015) Preparation and evaluation of a mesoporous calcium-silicate material (MCSM) from coal fly ash for removal of Co(II) from wastewater. Chem Eng J 279:777–787
Moon DH, Wazne M, Cheong KH, Chang Y-Y, Baek K, Ok YS, Park J-H (2015) Stabilization of As-, Pb-, and Cu-contaminated soil using calcined oyster shells and steel slag. Environ Sci Pollut Res 22:11162–11169
You W, Hong M, Zhang H, Wu Q, Zhuang Z, Yu Y (2016) Functionalized calcium silicate nanofibers with hierarchical structure derived from oyster shells and their application in heavy metal ions removal. Phys Chem Chem Phys 18:15564–15573
Richardson IG (2014) Model structures for C–(A)–S–H(I). Acta Crystallogr B B70:903–923
Kupwade-Patil K, Chin S, Ilavsky J, Andrews RN, Bumajdad A, Büyüköztürk O (2018) Hydration kinetics and morphology of cement pastes with pozzolanic volcanic ash studied via synchrotron-based techniques. J Mater Sci 53:1743–1757. https://doi.org/10.1007/s10853-017-1659-4
Yokoyama S, Tamura K, Hatta T, Nemoto S, Watanabe Y, Yamada H (2006) Synthesis and characterization of Zn-substituted saponite (sauconite). Clay Sci 13:75–80
Rose J, Moulin I, Masion A, Bertsch PM, Wiesner MR, Bottero J-Y, Mosnier F, Haehnel C (2001) X-ray absorption spectroscopy study of immobilization processes for heavy metals in calcium silicate hydrates. 2. Zinc. Langmuir 17:3658–3665
Weng CH, Huang CP (1994) Treatment of metal industrial wastewater by fly ash and cement fixation. J Environ Eng 120:1470–1487
Svatovskaya L, Shershneva M, Baydarashvily M, Sychova A, Sychov M, Gravit M (2015) Geoecoprotective properties of cement and concrete against heavy metal ions. Procedia Eng 117:345–349
Boch R, Dietzel M, Reichl P, Leis A, Baldermann A, Mittermayr F, Pölt P (2015) Rapid ikaite (CaCO3·6H2O) crystallization in a man-made river bed: hydrogeochemical monitoring of a rarely documented mineral formation. Appl Geochem 63:366–379
Fernández R, Ruiz AI, Cuevas J (2016) Formation of C–A–S–H phases from the interaction between concrete or cement and bentonite. Clay Miner 51:223–235
Gaucher EC, Blanc P (2006) Cement/clay interactions: a review—experiments, natural analogues, and modeling. Waste Manag 26:776–788
Sánchez L, Cuevas J, Ramírez S, Ruiz de León D, Fernández R, Vigil de la Villa R, Leguey S (2006) Reaction kinetics of FEBEX bentonite in hyperalkaline conditions resembling the cement–bentonite interface. Appl Clay Sci 33:125–141
Brew DRM, Glasser FP (2005) Synthesis and characterisation of magnesium silicate hydrate gels. Cem Concr Res 35:85–98
Albinsson Y, Andersson K, Börjesson S, Allard B (1996) Diffusion of radionuclides in concrete and concrete–bentonite systems. J Contam Hydrol 21:189–200
Sato I, Maeda K, Suto M, Osaka M, Usuki T, Koyama S-I (2015) Penetration behavior of water solution containing radioactive species into dried concrete/mortar and epoxy resin materials. J Nucl Sci Technol 52:580–587
Acknowledgements
Open access funding provided by Graz University of Technology. The authors are grateful to M. Hierz and A. Wolf (Graz University of Technology), who assisted us with the synthesis and the characterization of the C–A–S–H phases with/without heavy metals. The thoughtful comments of the three anonymous reviewers and the editor, Chris Blanford, are highly appreciated.
Funding
The research was financially supported by the Austrian Society for Construction Technology and the Austrian Research Promotion Agency (FFG, 856080), as well as by the NAWI Graz Geocenter.
Author information
Authors and Affiliations
Contributions
A.B. contributed to conceptualization, investigation, writing—original draft preparation, and project administration; A.B., A.L., and F.S. were involved in experimental; A.L., F.S., and I.L.P. contributed to methodology, data acquisition, and curation; A.B., I.L.P., I.G., F.M., and M.D. were involved in data analysis and validation; F.M. and M.D. contributed to resources and acquisition of funding; A.B. supervised the study.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Baldermann, A., Landler, A., Mittermayr, F. et al. Removal of heavy metals (Co, Cr, and Zn) during calcium–aluminium–silicate–hydrate and trioctahedral smectite formation. J Mater Sci 54, 9331–9351 (2019). https://doi.org/10.1007/s10853-019-03541-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-019-03541-5