
Abstract
The key nanostructural changes occurring in a series of alkali-activated materials (AAM) based on blends of slag and fly ash precursors during exposure to temperatures up to 1000 °C are investigated. The main reaction product in each AAM is a crosslinked sodium- and aluminium-substituted calcium silicate hydrate (C-(N)-A-S-H)-type gel. Increased alkali content promotes the formation of an additional sodium aluminosilicate hydrate (N-A-S-(H)) gel reaction product due to the structural limitations on Al substitution within the C-(N)-A-S-H gel. Heating each AAM to 1000 °C results in the crystallisation of the disordered gels and formation of sodalite, nepheline and wollastonite. Increased formation of N-A-S-(H) reduces binder structural water content after thermal treatment and correlates closely with previous observations of improved strength retention and reduced microcracking in these AAM after heating to 1000 °C. This provides new insight into thermally induced changes to gel atomic structure and thermal durability of C-(N)-A-S-H/N-A-S-H gel blends which are fundamental for the development of new fire-resistant construction materials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alkali-activated materials (AAM) have shown extensive promise as sustainable alternatives to Portland cement (PC), exhibiting desirable technical and environmental characteristics for a large array of applications [1]. AAM have received particular attention from academia and industry due to observations of excellent thermal performance, retaining strength after exposure to the temperatures in excess of 1000 °C which commonly result from structural fires [2,3,4,5,6,7]. This contrasts with PC which is susceptible to severe loss of strength and/or spalling at high temperatures [8, 9], particularly for low-porosity, high-performance concretes. The superior thermal performance of AAM is generally attributed to the differences in the structure of these materials when compared to that of hydrated PC, which rapidly loses strength when exposed to temperatures above 300 °C due to loss of structural water from hydrate phases [7].
The main binding phase in an alkali-activated cement based on blast furnace slag is a calcium (alkali) aluminosilicate hydrate (C-(N)-A-S-H) gel with structural similarity to substituted tobermorite-group minerals, displaying crystallinity ranging from X-ray amorphous to poorly crystalline [10,11,12]. In contrast, the main binding phase in an alkali-activated cement based on fly ash is an alkali aluminosilicate hydrate (N-A-S-(H)) gel framework with a highly polymerised, disordered pseudo-zeolitic structure containing low amounts of structural water [1]. The N-A-S-(H) gel is X-ray amorphous but has been shown to exhibit short-range ordering and to be comprised of nanocrystalline zeolites compacted by an amorphous gel phase [13].
Blended slag/fly ash-based AAM can exhibit advantages, in terms of the balance between reaction kinetics and durability, over those produced from sole precursors [14,15,16], but the coexistence of C-(N)-A-S-H and N-A-S-(H) gels results in complex thermodynamic and chemical interactions which dictate material properties and performance [17, 18]. Coexistence of these gels has been observed in alkali-activated cement based on blast furnace slag [11, 19] and blends of low-Ca and high-Ca precursors [18, 20,21,22]. The stability of this coexistence remains to some degree unclear [23,24,25], with the C-(N)-A-S-H gel framework observed to be the dominant gel in synthetic C-(N)-A-S-H/N-A-S-(H) gel blends [25].
The gel nanostructure of AAM plays an important role in thermal resistance. High-performance concrete based predominantly on hydrated PC exhibits dense binder structures and tortuous pore networks, and consequently experience explosive spalling at high temperatures due to the generation of high internal water vapour pressure [26, 27]. The pore network structure within AAM is dictated by gel nanostructure and has been shown to control internal water vapour pressure at elevated temperatures, with increased porosity and decreased tortuosity in the pore network providing improved resistance to fire spalling due to easier release of trapped water vapour and reduction in internal water vapour pressure [4,5,6,7, 28]. Increased alkali and Al content has been shown to improve strength retention and reduce microcracking in AAM exposed to temperatures in excess of 1000 °C [6]. Additional observations have suggested that the gel Si/Al ratio [5, 28,29,30] and particle size distribution [7] of the precursor material influence thermal performance of AAM systems; however, this influence is likely to be indirect as both of these parameters influence the degree of reaction, structural water content of the gel [17, 31] and pore network structure [32] of the binder.
Despite the extensive focus on the mechanical properties and microstructure of AAM exposed to high temperatures and demanding thermal environments (i.e. those with rapid changes in temperature), little attempt has been made to examine thermally induced changes to gel nanostructure and chemistry. These changes are of particular importance as they ultimately dictate the structural water content, pore network structure and consequently the thermal performance of these materials. Understanding nano- and atomic-scale phase and structural evolution during exposure to elevated temperatures is crucial for formulation and design of AAM optimised for high durability, strength and thermal performance. Here we investigate the composition–structure–property relationships in a series of AAM based on blends of slag and fly ash precursors by identifying the key nanostructural changes occurring during exposure to temperatures up to 1000 °C. This provides new insight into thermally induced changes to gel atomic structure and thermal durability of C-(N)-A-S-H/N-A-S-H gel blends.
Experimental procedures
Alkali-activated cement synthesis
Ground granulated blast furnace slag (GGBFS) and fly ash were purchased from Cement Australia; the chemical composition as determined by X-ray fluorescence (XRF) analysis is shown in Table 1. A commercial blend (‘Melbourne Ash’) of moderate-calcium fly ashes based on fly ashes from Gladstone and Callide power stations, Australia, was used.
Solid precursors were produced by blending the fly ash and slag precursors in a 4:1 ratio. Activating solutions were prepared by dissolution of solid sodium hydroxide (99.9% purity, analytical grade, Chem-Supply, Australia) in water followed by addition of sodium silicate solution (molar ratio SiO2/Na2O = 2, Grade D, PQ Australia). Six AAM samples (each with a different composition) were made by mixing the activating solutions with the solid precursors.
Compositions were chosen to exhibit chemistry within the quaternary CaO-Na2O-Al2O3-SiO2 system which are important for studying N-A-S-(H)/C-(N)-A-S-H-type gel blends [17, 18]. Previous work has shown that the alkali content (Na/Si ratio) of the reaction mixture dictates the amount of each gel formed in N-A-S-(H)/C-(N)-A-S-H-type gel blends [33,34,35,36,37]. Alkali content was therefore varied in the samples used in the work presented here to examine how this affects thermally induced nanostructural changes. Nominal reaction mix compositions are shown in Fig. 1 and Table 2 (w/s = 0.35 for all samples). All samples were mixed with a Hobart mixer for 10 min and subsequently cast into moulds. Samples were then vibrated for 5 min using a vibrating table to compact the paste and release any entrained air and subsequently sealed and cured at ambient temperature (~ 23 °C) for 14 days. This period was chosen to examine the thermally induced nanostructural changes of these samples during the early stages of curing.

Projection of nominal alkali-activated material chemistry onto the ternary CaO–Al2O3–SiO2 system (neglecting Na2O content) for samples A–F as marked. Na/Al ratios for each sample are indicated, and w/s = 0.35 for all samples. Approximate region for C-(N)-A-S-H determined from [38]
Samples to be thermally treated were heated to 1000 °C in an electric furnace at a rate of 6.6 °C/min, held at 1000 °C for 1 h and then cooled to room temperature at a rate of 6.6 °C/min. This temperature was chosen to simulate air temperatures which occur in residential and commercial building fire scenarios (based on Australian Standard AS 1530.4 and American Standard ASTM E119) [2]. Six 50-mm cubic samples which had been cured for 14 days were placed in the furnace with only the bottom surface contacted, with heat exposure to the other surfaces via air convection. A detailed microstructural and mechanical analysis of these samples has been discussed previously [6].
Characterisation
With the exception of scanning electron microscopy, the hardened binders were hand ground using a pestle and mortar, immersed in acetone for 15 min to remove loosely bound water (thereby halting the alkali-activation reaction without significantly altering the AAM gel structure [39]), and subsequently filtered prior to storing in a sealed desiccator.
A Spectro ‘Xepos’ energy-dispersive x-ray fluorescence (XRF) spectrometer was used to obtain chemical composition data of the precursors. Samples fused with a 12:22 lithium tetraborate:metaborate flux in platinum crucibles at 1050 °C for 15 min prior to analysis.
X-ray diffraction (XRD) data were obtained across a 2θ range of 5°–70° using a Bruker D8 Advance instrument with Cu Kα radiation (1.54 Å), a nickel filter, a step size of 0.020°, 1 s/step and rotating at 15 rpm. Diffracted background intensity at low angles was reduced using an anti-scatter blade, and an incident beam divergence of 1.0 mm and a 2.5° Soller slit in the diffracted beam were used. Phase identification was performed using Diffrac.EVA V4.1 software with the ICDD PDF4+ 2015 database.
Scanning electron microscopy (SEM) was performed using a Hitachi TM 3030 instrument with a 15 kV accelerating voltage, a working distance of 8 mm and a backscatter detector. An evenly distributed selection of points across a representative 400 µm × 400 µm section of the sample was analysed using a Bruker Quantax 70 X-ray energy-dispersive spectroscopy (EDX) detector to determine chemical compositions.
An Agilent VNMRS-600 spectrometer (B0 = 14.1 T) and a 4.0-mm triple resonance bioMAS probe were used to obtain solid-state 29Si, 27Al, and 23Na MAS NMR spectra (see Table 3 for experimental parameters). All data were processed using NMRPipe [40]. Gaussian peak profiles were used to deconvolute the 29Si MAS NMR spectra [41]. The minimum number of peaks possible were fitted, and peak full width at half height was restricted to ≤ 5 ppm for C-(N)-A-S-H gel products and ≤ 6 ppm for N-A-S-(H) gel products. Peak intensities were required to be consistent with the structural constraints described by the ‘Crosslinked Substituted Tobermorite Model’ (CSTM) for C-(N)-A-S-H gel products [11], and the thermodynamics of a statistical distribution of Si and Al sites within a Q4 aluminosilicate network for N-A-S-(H) gel products [42]. Previous work utilising 29Si and 27Al MAS NMR and Fourier transform infrared spectroscopy has shown that these models best describe the environments in which Si and Al exist in C-(N)-A-S-H [10,11,12, 17] and N-A-S-(H) [31, 42,43,44,45] gels.
The contribution from unreacted precursor particles was accounted for by fitting a scaled component spectrum calculated from the spectra of the slag and fly ash precursors. The minimum number of peaks necessary were fitted to account for the intensity within the region where C-(N)-A-S-H Si sites are expected to resonate, subject to the structural constraints described by the CSTM [11]. The minimum number of peaks necessary were then fitted to account for the remaining intensity within the region where N-A-S-(H) Si sites are expected to resonate, subject to the structural constraints described the thermodynamics of a statistical distribution of Si and Al sites within a Q4 aluminosilicate network for N-A-S-(H) gel products [42].
Results and discussion
X-ray diffraction
X-ray diffraction data for the precursors, as-cured AAM and thermally treated (to 1000 °C) AAM are shown in Figs. 2, 3, and 4, respectively. Both the slag and fly ash precursors exhibit a dominant broad feature characteristic of their amorphous content. Crystalline phases identified in the fly ash are quartz (PDF # 00-001-0649), mullite (PDF # 00-015-0776), hematite (PDF # 00-033-0664), magnetite (PDF # 04-007-1060) and gypsum (PDF # 00-033-0311), while the slag also contains gypsum as a grinding aid, as well as an identifiable quantity of tricalcium silicate (identified as hatrurite, PDF # 04-011-1393) and tricalcium aluminate (PDF # 01-074-7039), likely to have been introduced as Portland cement contamination during blending, grinding and/or transport of the fly ash and slag. The other usual Portland cement clinker constituent dicalcium silicate is not observed but is likely to be present below the detection limit of the technique used here.

X-ray diffractograms of the blast furnace slag and fly ash precursors

X-ray diffractograms of each AAM sample cured for 14 days

X-ray diffractograms of each AAM sample cured for 14 days and heated to 1000 °C
The appearance of a broad feature upon alkali activation of all samples indicates the formation of an amorphous reaction product and the position of this feature at approximately 29° 2θ is consistent with formation of an amorphous N-A-S-(H) phase (Fig. 3). A broad peak at approximately 29° 2θ (overlapping significantly with the broad feature due to N-A-S-(H)) indicates a phase with structural similarity to aluminium-containing tobermorite (PDF # 00-019-0052) and is assigned to a poorly crystalline C-(A)-S-H phase [17]. Reflections due to all crystalline phases identified in the precursors are also present in the diffractogram for each AAM (Fig. 3); quartz, mullite, and iron oxides are unreactive during alkali activation on the timescale investigated here [18, 47].
After thermal treatment, the diffractograms of each AAM show no evidence of the broad feature that was centred at approximately 29° 2θ in the unheated materials, and high-intensity reflections attributed to crystalline phases present in the precursors are observed, suggesting devitrification of unreacted fly ash particles and partial crystallisation of disordered gel reaction products [48]. Reflections attributed to sodalite (Na8[Al6Si6O24]Cl2, PDF # 01-087-0296) and nepheline (NaAl(SiO4), PDF # 01-083-2372) are observed in the diffractogram of each thermally treated AAM. Formation of nepheline from hydroxysodalite has been previously observed between 600 and 800 °C in sodium silicate-activated metakaolin [48] and between 800 and 1000 °C in sodium silicate-activated fly ash [49]; formation of hydroxysodalite has been observed in silicate-activated fly ash cured at 40 °C [50], and this phase dehydrates between 400 and 630 °C [51]. It follows that the formation of both sodalite and nepheline in the samples here is likely to occur via successive dehydration of hydrosodalite and hydroxysodalite. Reflections attributed to wollastonite (CaSiO3, monoclinic polymorph, PDF # 04-010-2581) are present in XRD data for all thermally treated AAM, indicating the formation of this phase due to either decomposition of C-(A)-S-H at approximately 810 °C [52,53,54] or devitrification of unreacted slag particles. Small traces of gehlenite (Ca2Al(AlSiO7), PDF # 04-014-4683) are also observed in each AAM.
Scanning electron microscopy/energy-dispersive X-ray spectroscopy
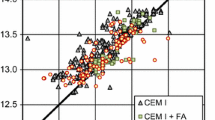
The elemental composition of each sample as determined by SEM–EDX is reported in Fig. 5. The elemental composition of each sample is comparable with that of alkali-activated slag and slag/fly ash blends [18, 21, 38] and exhibits a cluster of points within compositional regions associated with both C-(N)-A-S-H gels containing varying Ca content and N-A-S-(H) gels. This indicates that the main reaction product in all samples is an intimate mixture of both C-(N)-A-S-H and N-A-S-(H) gels. Some data points also exhibit compositions of the precursors, suggesting remnant unreacted precursor particles are also present.


Projection of alkali-activated material chemistry onto the (i) ternary CaO–Al2O3–SiO2 system (neglecting Na2O content) and (ii) ternary Na2O – Al2O3 – SiO2 system (neglecting CaO content) showing elemental composition of AAM cured for 14 days for samples A–F as marked, as determined by SEM–EDX analysis. An evenly distributed selection of points across a representative 400 µm × 400 µm section of the sample were used for analysis. Approximate regions of C-S-H, C-(A)-S-H, C-(N)-A-S-H and N-A-S-(H) determined from [11, 38, 57, 58]. Hypothetical reaction mixture compositions based solely on slag or fly ash, calculated using the composition of the activating solution discussed above, are provided for reference and are indicated by red triangles and green circles, respectively
Increased Si content and increased Na content within the AAM reaction mixture results in increased formation of low-Ca C-(N)-A-S-H and N-A-S-(H) gels; this occurs when either Si or Na content is increased while the other is held constant, and when both Si and Na content are increased at the same time. These trends are consistent with thermodynamic predictions [11, 55]; formation of a low-Ca C-(N)-A-S-H gel will occur to the maximum Al and Na incorporation that is thermodynamically stable, with excess alkali and freely available Si and Al driving formation of N-A-S-(H) gel (observed explicitly by 29Si MAS NMR below).
Figure 5 shows that the elemental composition of each sample generally lies within a compositional region which suggests the formation of a moderate-Ca C-(N)-A-S-H gel reaction product. However, due to the intimate mixtures of C-(N)-A-S-H and N-A-S-(H) gels observed previously in alkali-activated slag/fly ash blends [18, 21, 38] and synthetic analogues [17], it is possible that the elemental composition of each data point represents the bulk composition of a mixture of high-Ca and low-Ca C-(N)-A-S-H, or of high-Ca and N-A-(S)-H, if these phases are mixed on a scale smaller than the depth of X-ray generation for the accelerating voltage used (15 kV) [56]. It is clear that the SEM–EDX analysis cannot resolve the individual C-(N)-A-S-H and N-A-S-(H) gels. Consequently, solid-state NMR spectroscopy has been used, with the results discussed in detail below.
Solid-state magic angle spinning nuclear magnetic resonance spectroscopy
Precursors
27Al, 29Si and 23Na MAS NMR spectra of the slag and fly ash precursors are shown in Fig. 6. 27Al MAS NMR spectra of slag exhibits a broad tetrahedral AlO4 resonance centred at approximately δobs = 63 ppm consistent with the broad amorphous feature identified by XRD, similar to previous 27Al MAS NMR observations [18, 55, 59] and indicating extensive disorder and heterogeneity in the local structure and shielding experienced by Al in slag. A low-intensity shoulder appears on this resonance at approximately 40 ppm, while two low-intensity octahedral AlO6 resonances are observed at δobs = − 2 ppm (broad) and 9 ppm (narrow). These resonances have previously been observed in slag but not attributed to a specific environment [18, 55, 59, 60]. The 29Si MAS NMR spectra of slag exhibit a single broad resonance centred at δiso = − 86 ppm due to a distribution of Qn(mAl) (where 0 ≤ m ≤ n ≤ 4) environments dominated by species with lower polymerisation or higher Al substitution consistent with previous observations [18, 59]. The 23Na MAS NMR spectrum for slag exhibits a single broad resonance centred at δobs = − 12 ppm. The extensive shielding of 23Na in the slag investigated here indicates its incorporation within the slag glass within a phase which exhibits greater electron density than if Na were phase separated.

a 27Al MAS (8 kHz) NMR spectra, b 29Si MAS (10 kHz) NMR spectra and c 23Na MAS (10 kHz) NMR spectra of fly ash and slag precursors
The 27Al MAS NMR spectrum of the fly ash also exhibits a broad tetrahedral AlO4 resonance centred at δobs = 67 ppm (with a low-intensity shoulder at 44 ppm) attributed to Al substituted in silicate glasses [61], as well as a high-intensity octahedral AlO6 resonance at δobs = 0 ppm attributed to Al in disordered mullite-like phases [62]. The 29Si MAS NMR spectrum of the fly ash exhibits a broad resonance centred at approximately δiso = − 112 ppm and attributed to a distribution of Q4(mAl) sites within the amorphous component (consistent with the broad feature identified for this sample by XRD). Si sites within mullite in fly ash (identified by XRD) also resonate at δiso = − 87 ppm and − 106 ppm [62], while those in quartz contribute to the spectrum at approximately δiso = − 112 ppm [63, 64]. 23Na MAS NMR data could not be obtained for fly ash due to the low sodium content (0.81 wt% Na2O) and severe signal dampening caused by the high content of paramagnetic Fe2O3 species (13.16 wt%).
Alkali-activated materials
27Al MAS NMR of as-cured alkali-activated materials
A broad resonance centred at δobs 65 ppm is observed in the 27Al MAS NMR spectra of each AAM (Fig. 7) and is assigned to Al in significantly distorted tetrahedral environments within poorly crystalline C-(N)-A-S-H and amorphous N-A-S-(H) gel products [18, 65, 66] as well as unreacted slag and fly ash. Two distinct octahedral Al environments are indicated by broad overlapping resonances at approximately δobs = 14 and 0 ppm in all AAM samples. The resonance at 0 ppm is attributed to octahedral Al in disordered mullite-like phases in remnant fly ash particles, while that at 14 ppm is likely to be due to contributions from AlVI sites within remnant slag particles. Small variations in the shielding of both tetrahedral and octahedral resonances are observed between samples as a consequence of structural differences due to variations in composition and crystallinity.

27Al MAS (8 kHz) NMR spectra of a each AAM sample cured for 14 days and b each AAM cured for 14 days and heated to 1000 °C
27Al MAS NMR of thermally treated alkali-activated materials
27Al MAS NMR spectra of each thermally treated AAM (Fig. 7) are similar to those of samples prior to heating, with the broad tetrahedral Al resonance (now centred at δobs = − 59 ppm) sharpening and increasing in intensity. This is attributed to contributions from resonances of tetrahedral Al within sodalite and nepheline [67]. The two distinct octahedral Al resonances present in the 27Al MAS NMR of samples prior to heating can no longer be observed, replaced by a single broad octahedral resonance of lower intensity at approximately δobs = 8 ppm. The lineshape of this resonance suggests it could contain contributions from both octahedral Al resonances observed in the 27Al MAS NMR of samples prior to heating, while the sharpening of these resonances is consistent with partial crystallisation of the amorphous material in the AAM observed by XRD. No significant variation in the spectra is observed between samples indicating strong structural similarity in Al-containing sites.
29Si MAS NMR of as-cured alkali-activated materials
The 29Si MAS NMR spectra for each AAM before and after heat treatment are shown in Fig. 8. A broad resonance at approximately δiso = − 83 ppm is observed in the spectra of all samples. Chemical shifts of Qn(mAl) species in these samples are expected to overlap significantly due to shielding by network modifying cations [68, 69] and the presence of Al(VI) species [70].

29Si MAS (10 kHz) NMR spectra of a each AAM sample cured for 14 days and b each AAM cured for 14 days and heated to 1000 °C
Spectral deconvolutions (Fig. 9) are summarised in Table 4. 29Si MAS NMR spectra were deconvoluted by fitting the minimum number of Gaussian peaks to provide an accurate and meaningful interpretation of the spectra and maintain consistency with structural constraints for a C-(N)-(A)-S-H gel described by the ‘Crosslinked Substituted Tobermorite Model’ (CSTM) [11] and a statistical distribution of Si and Al sites within a Q4 aluminosilicate network [18, 42]. Peak positions and widths were held constant throughout all spectral deconvolutions except where noted. Component peaks attributed to remnant precursor particles were rescaled by a single factor, i.e. congruent dissolution of the precursors was assumed in all cases.

29Si MAS (10 kHz) NMR spectra (shown in black) of samples a–f as marked with associated deconvolutions. In each case the fit (shown in red) is the sum of the deconvoluted peaks (peaks attributed to Si sites in C-(N)-A-S-H are shown in blue, peaks attributed to Si sites in N-A-S-(H) are shown in green). The overall contribution of the unreacted precursor is shown in grey (individual peaks attributed to the unreacted precursor are not shown for clarity)
Upon alkali-activation six new Si environments are observed in each sample, indicated by resonances at approximately δiso = − 63.0, − 72.0, − 79.4, − 84.3, − 90.0 and − 95.0 ppm, attributed to Q0, Q1, Q2(1Al), Q2, Q3(1Al), and Q3, respectively. These resonances are attributed to Si environments within a crosslinked C-(N)-A-S-H gel [11, 12, 18, 60]. The peak at − 90 ppm is assigned to both Q3(1Al) and Q4(4Al) sites as both environments exhibit overlapping resonances within this region [71]. Q3 and Q4(3Al) also exhibit overlapping resonances, however, in order to maintain consistency with the structural constraints for a mixed crosslinked/non-crosslinked C-(N)-(A)-S-H gel described by the CSTM a separate peak for Q4(3Al) at − 93 ppm must be fitted to account for the additional intensity above what is provided by the Q3 resonance at − 95 ppm. Consequently, the formation of Q4(4Al) and Q4(3Al) sites is also observed upon alkali activation of all samples and attributed to a polymerised Al-rich aluminosilicate phase. This provides explicit evidence of formation of an additional N-A-S-(H) phase within these blended systems.
Quantification of the intensity of each individual Si site can be used to determine the relative amount of each gel present in each sample, providing insight into the mechanisms which dictate thermally induced nanostructural changes. Quantification of the intensity of the Q4(4Al) site (\( {\text{I}}_{{{\text{Q}}^{4} \left( {4{\text{Al}}} \right)}} \)) can be performed by assuming Q4(mAl) sites are present in an additional Al-rich phase separate from the C-(N)-(A)-S-H gel and using Eq. (1), where \( {\text{I}}_{{{\text{Q}}^{4} \left( {3{\text{Al}}} \right)}} \) is the intensity of the Q4(3Al) resonance determined from deconvolution of the 29Si MAS NMR data, and Si/Al is the Si/Al ratio for the additional Q4(mAl) phase, which is assumed to be 1.2, consistent with the composition of N-A-S-(H) gels in alkali-activated metakaolin [72], statistical thermodynamic model predictions [42] and a generalised structural description of the additional N-A-S-(H) gel reaction product formed in alkali-activated slag containing biphasic C-(N)-A-S-H/N-A-S-(H) gel blends from 29Si and 27Al MAS NMR data [11]. Formation of four new Si environments indicated by peaks at approximately δiso = − 90.0, − 93.0, − 97.0, − 100.0 and − 107.0 ppm, attributed to Q4(4Al), Q4(3Al), Q4(2Al), Q4(1Al) and Q4(0Al), respectively, within a N-A-S-(H) gel are observed. Tricalcium silicate exhibits a 29Si MAS NMR resonance with maximum intensity at approximately δiso = − 73 ppm [73, 74] and will consequently contribute to the 29Si MAS NMR resonance assigned to Q1 species in the as-cured alkali-activated materials.
29Si MAS NMR of thermally treated alkali-activated materials
Heating each AAM sample to 1000 °C results in the formation of a broad, high-intensity resonance with a maximum at approximately δiso = − 88 ppm as well as a low-intensity resonance at approximately δiso = − 73 ppm. Spectral deconvolution was not possible for the 29Si MAS NMR spectra of the thermally treated AAM due to the formation of multiple crystalline phases during thermal treatment and the consequent absence of suitable constraints on the deconvolution. Despite this, important insights may be obtained from the resonances in these spectra.
The broad resonance at − 88 ppm contains contributions from resonances at approximately δiso = − 85, − 92, − 102 and − 100 ppm (indicated by distinct shoulders on the main 29Si MAS NMR resonance). This broad resonance results from contributions from crystalline phases identified by XRD. Nepheline comprises two distinct Q4(4Al) sites (resonating at δiso = − 85 and − 88 ppm, respectively), one Q4(3Al) site resonating at δiso = − 92 ppm and one Q4(0Al) resonating at δiso = − 102 ppm [75, 76]. Sodalite exhibits a single 29Si MAS NMR resonance at δiso = − 85 ppm corresponding to a Q4(4Al) site [77], while tricalcium silicate exhibits a 29Si MAS NMR resonance with maximum intensity at approximately δiso = − 73 ppm [73, 74] and is responsible for the resonance at this position in the 29Si MAS NMR spectra of the thermally treated alkali-activated materials. Si sites within wollastonite are expected to resonate at approximately δiso = − 87.8, − 89.0 and − 89.5 ppm, and will contribute to the 29Si MAS NMR spectra at these positions [78].
As discussed above, Si sites within mullite in remnant fly ash particles (identified by XRD) are expected to resonate at δiso = − 87 and − 106 ppm [62], while those in quartz are expected to contribute to the spectrum at approximately δiso = − 112 ppm [63, 64]. Strong reflections attributed to these phases are identified in XRD data for these samples; these phases are deemed responsible for the resonances in this region of each spectrum.
Transferral of the 29Si MAS NMR signal to spinning sidebands, dampening and broadening of the central resonance occurs to a much greater extent in the spectra of AAM samples prior to heating than those heated to 1000 °C (Fig. 10). This suggests oxidation of paramagnetic Fe(III) in Fe2O3 (identified as hematite in XRD data for AAM samples prior to heating) to ferrimagnetic Fe(II, III) in Fe3O4 (identified as magnetite in XRD data for AAM samples heated to 1000 °C), with the reduction in paramagnetic ions reducing transferral of the signal from the central resonance to spinning sidebands. Beyond this change in oxidation state, however, Fe does not appear to influence the thermal performance of these materials, despite the reasonably high amount of Fe in these materials (~ 8 wt % in the binder).

29Si MAS (10 kHz) NMR spectra showing the spectral region from 100 to − 300 ppm of a each AAM sample cured for 14 days and b each AAM cured for 14 days and heated to 1000 °C. Spinning side bands are indicated by *
Previous work has shown increased alkali content and lower Si/Al ratio results in enhanced strength retention after thermal treatment in the alkali-activated slag/fly ash blends investigated here (i.e. samples of the same composition) [6]. Samples with increased alkali content in the present study exhibited higher amounts of the additional N-A-S-(H) reaction product, due to the structural limitations on Al substitution within C-(N)-A-S-H gels (which can only incorporate Al within bridging tetrahedral sites) [11] and absence of Ca-rich AFm phases [17]. These samples will consequently contain lower amounts of bound structural water, experience reduced water loss and reduced internal water vapour pressure at high temperatures compared to those with lower formation of N-A-S-(H) [8, 9]. These nanostructural features will result in reduced stresses within the AAM at high temperatures and explain the enhanced thermal performance observed for samples exhibiting higher N-A-S-(H) gel formation.
23Na MAS NMR
The 23Na MAS NMR spectra for each AAM exhibit a single broad resonance centred at approximately δobs = − 3 ppm (Fig. 11) attributed to Na+ cations associated with aluminium-centred tetrahedra, playing a charge-balancing role within the C-(N)-A-S-H [66, 79] and N-A-S-(H) gel frameworks [31, 80]. Little variation in Na sites is observed between samples, and the broad nature of the resonance spanning from 20 to − 20 ppm indicates an extensive disorder in Na sites within the AAM. 23Na MAS NMR spectra of each AAM heated to 1000 °C display a single broad resonance centred at approximately δobs = − 16 ppm and attributed to Na sorbed to the surfaces of the nanostructured gels [17] as a consequence of gel dehydration during heating. This resonance also exhibits a low-intensity shoulder at approximately δobs = − 27 ppm that is most pronounced at higher Na content (Na/Al ≥ 1). This broad resonance contains contributions from nepheline at between δobs = − 4 and − 28 ppm, centred at approximately δobs = − 15 ppm (9.4 (T) [81] (the formation of which is expected to be promoted by higher bulk Na content), as well as from sodalite at approximately δobs = − 11 ppm [77, 82].

23Na MAS (10 kHz) NMR spectra of a each AAM sample cured for 14 days and b each AAM cured for 14 days and heated to 1000 °C
Conclusions
The key nanostructural changes occurring in a series of AAM based on blends of slag and fly ash precursors during exposure to temperatures up to 1000 °C were investigated by application of X-ray diffraction (XRD), scanning electron microscopy (SEM) coupled with X-ray energy-dispersive spectroscopy (EDX) and multinuclear solid-state nuclear magnetic resonance (NMR) spectroscopy probing 29Si, 27Al, 23Na nuclei. Each AAM comprised primarily of a disordered C-(N)-A-S-H-type gel, with at least some degree of crosslinking, as well as a disordered N-A-S-(H)-type gel. After heating to 1000 °C each AAM experienced devitrification of these disordered gels and formation of sodalite, nepheline and wollastonite. Samples with increased alkali content exhibit higher formation of an additional N-A-S-(H) reaction product due to the structural limitations on Al substitution within the C-(N)-A-S-H gel and absence of Ca-rich AFm phases [17]. These samples will consequently contain lower amounts of bound structural water, experience reduced water loss and reduced internal water vapour pressure at high temperatures. These nanostructural features will result in reduced stresses within the AAM at high temperatures and explain the enhanced strength after thermal treatment observed for these materials previously. This provides insight into thermally induced changes to gel atomic structure of C-(N)-A-S-H/N-A-S-(H) gel blends which are fundamental for the development of fire-resistant construction materials.
References
Provis JL, Palomo A, Shi C (2015) Advances in understanding alkali-activated materials. Cem Concr Res 78A:110–125
Vickers L, van Riessen A, Rickard WDA (2014) Fire-resistant geopolymers: role of fibres and fillers to enhance thermal properties. Springer, Singapore
Barbosa VFF, MacKenzie KJD (2003) Thermal behaviour of inorganic geopolymers and composites derived from sodium polysialate. Mater Res Bull 38:319–331
Zhao R, Sanjayan JG (2011) Geopolymer and Portland cement concretes in simulated fire. Mag Concr Res 63:163–173
Kong DL, Sanjayan JG (2010) Effect of elevated temperatures on geopolymer paste, mortar and concrete. Cem Concr Res 40:334–339
Kashani A, Ngo TD, Walkley B, Mendis P (2017) Thermal performance of calcium-rich alkali-activated materials: a microstructural and mechanical study. Constr Build Mater 153:225–237
Rivera OG, Long WR, Weiss CA Jr, Moser RD, Williams BA, Torres-Cancel K, Gore ER, Allison PG (2016) Effect of elevated temperature on alkali-activated geopolymeric binders compared to portland cement-based binders. Cem Concr Res 90:43–51
Mendes A, Sanjayan J, Collins F (2008) Phase transformations and mechanical strength of OPC/Slag pastes submitted to high temperatures. Mater Struct 41:345–350
Hertz KD (2005) Concrete strength for fire safety design. Mag Concr Res 57:445–453
Schilling PJ, Butler LG, Roy A, Eaton HC (1994) 29Si and 27AI MAS-NMR of NaOH-activated blast-furnace slag. J Am Ceram Soc 77:2363–2368
Myers RJ, Bernal SA, San Nicolas R, Provis JL (2013) Generalized structural description of calcium-sodium aluminosilicate hydrate gels: the cross-linked substituted tobermorite model. Langmuir 29:5294–5306
Richardson IG (2008) The calcium silicate hydrates. Cem Concr Res 38:137–158
Provis JL, Lukey GC, van Deventer JSJ (2005) Do geopolymers actually contain nanocrystalline zeolites? A reexamination of existing results. Chem Mater 17:3075–3085
Bernal SA, Provis JL, Rose V, de Gutierrez RM (2011) Evolution of binder structure in sodium silicate-activated slag-metakaolin blends. Cem Concr Compos 33:46–54
Yip CK, Lukey GC, Provis JL, van Deventer JSJ (2008) Effect of calcium silicate sources on geopolymerisation. Cem Concr Res 38:554–564
Yip CK, Van Deventer JSJ (2003) Microanalysis of calcium silicate hydrate gel formed within a geopolymeric binder. J Mater Sci 38:3851–3860. https://doi.org/10.1023/A:1025904905176
Walkley B, San Nicolas R, Sani M-A, Rees GJ, Hanna JV, van Deventer JSJ, Provis JL (2016) Phase evolution of C-(N)-A-S-H/N-A-S-H gel blends investigated via alkali-activation of synthetic calcium aluminosilicate precursors. Cem Concr Res 89:120–135
Bernal SA, Provis JL, Walkley B, San Nicolas R, Gehman JD, Brice DG, Kilcullen AR, Duxson P, van Deventer JSJ (2013) Gel nanostructure in alkali-activated binders based on slag and fly ash, and effects of accelerated carbonation. Cem Concr Res 53:127–144
Myers RJ, Bernal SA, Provis JL (2014) A thermodynamic model for C-(N-)A-S-H gel: CNASH_ss. Derivation and validation. Cem Concr Res 66:27–47
Bernal SA, Rodriguez ED, de Gutierrez RM, Gordillo M, Provis JL (2011) Mechanical and thermal characterisation of geopolymers based on silicate-activated metakaolin/slag blends. J Mater Sci 46:5477–5486. https://doi.org/10.1007/s10853-011-5490-z
Ismail I, Bernal SA, Provis JL, San Nicolas R, Hamdan S, van Deventer JSJ (2014) Modification of phase evolution in alkali-activated blast furnace slag by the incorporation of fly ash. Cem Concr Compos 45:125–135
Yip CK, Lukey GC, van Deventer JSJ (2005) The coexistence of geopolymeric gel and calcium silicate hydrate at the early stage of alkaline activation. Cem Concr Res 35:1688–1697
Provis JL, Bernal SA (2014) Geopolymers and related alkali-activated materials. Annu Rev Mater Res 44:299–327
Gartner EM, Macphee DE (2011) A physico-chemical basis for novel cementitious binders. Cem Concr Res 41:736–749
García-Lodeiro I, Palomo A, Fernández-Jiménez A, Macphee DE (2011) Compatibility studies between N-A-S-H and C-A-S-H gels. Study in the ternary diagram Na2O–CaO–Al2O3–SiO2–H2O. Cem Concr Res 41:923–931
Ali F, Nadjai A, Talamona D (2004) Assessment of the susceptibility of normal and high strength concrete for explosive spalling. J Appl Fire Sci 13:79–88
Ali F, O’Connor D, Abu-Tair A (2001) Explosive spalling of high-strength concrete columns in fire. Mag Concr Res 53:197–204
Kong DL, Sanjayan JG, Sagoe-Crentsil K (2007) Comparative performance of geopolymers made with metakaolin and fly ash after exposure to elevated temperatures. Cem Concr Res 37:1583–1589
Kong DL, Sanjayan JG (2008) Damage behavior of geopolymer composites exposed to elevated temperatures. Cem Concr Compos 30:986–991
Kong DL, Sanjayan JG, Sagoe-Crentsil K (2008) Factors affecting the performance of metakaolin geopolymers exposed to elevated temperatures. J Mater Sci 43:824–831. https://doi.org/10.1007/s10853-007-2205-6
Walkley B, San Nicolas R, Sani MA, Gehman JD, van Deventer JS, Provis JL (2016) Phase evolution of Na2O-Al2O3-SiO2-H2O gels in synthetic aluminosilicate binders. Dalton Trans 45:5521–5535
Hajimohammadi A, Provis JL, van Deventer JSJ (2011) Time-resolved and spatially-resolved infrared spectroscopic observation of seeded nucleation controlling geopolymer gel formation. J Colloid Interface Sci 357:384–392
Dai Z, Tran TT, Skibsted J (2014) Aluminum incorporation in the C-S–H phase of white portland cement-metakaolin blends studied by 27Al and 29Si MAS NMR spectroscopy. J Am Ceram Soc 97:2662–2671
Girão AV, Richardson IG, Taylor R, Brydson RMD (2010) Composition, morphology and nanostructure of C–S–H in 70% white Portland cement–30% fly ash blends hydrated at 55 °C. Cem Concr Res 40:1350–1359
Tailby J, MacKenzie KJD (2010) Structure and mechanical properties of aluminosilicate geopolymer composites with Portland cement and its constituent minerals. Cem Concr Res 40:787–794
Buchwald A, Hilbig H, Kaps C (2007) Alkali-activated metakaolin-slag blends—performance and structure in dependence of their composition. J Mater Sci 42:3024–3032. https://doi.org/10.1007/s10853-006-0525-6
Dombrowski K, Buchwald A, Weil M (2007) The influence of calcium content on the structure and thermal performance of fly ash based geopolymers. J Mater Sci 42:3033–3043. https://doi.org/10.1007/s10853-006-0532-7
van Deventer JSJ, San Nicolas R, Ismail I, Bernal SA, Brice DG, Provis JL (2014) Microstructure and durability of alkali-activated materials as key parameters for standardization. J Sustain Cem Based Mater 4:116–128
Ismail I, Bernal S, Provis J, Hamdan S, Deventer JJ (2013) Drying-induced changes in the structure of alkali-activated pastes. J Mater Sci 48:3566–3577. https://doi.org/10.1007/s10853-013-7152-9
Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293
Massiot D, Fayon F, Capron M, King I, Le Calvé S, Alonso B, Durand J-O, Bujoli B, Gan Z, Hoatson G (2002) Modelling one- and two-dimensional solid-state NMR spectra. Magn Reson Chem 40:70–76
Provis JL, Duxson P, Lukey GC, van Deventer JSJ (2005) Statistical thermodynamic model for Si/Al ordering in amorphous aluminosilicates. Chem Mater 17:2976–2986
Fernández-Jiménez A, Palomo A, Sobrados I, Sanz J (2006) The role played by the reactive alumina content in the alkaline activation of fly ashes. Microporous Mesoporous Mater 91:111–119
Lecomte I, Henrist C, Liégeois M, Maseri F, Rulmont A, Cloots R (2006) (Micro)-structural comparison between geopolymers, alkali-activated slag cement and Portland cement. J Eur Ceram Soc 26:3789–3797
Criado M, Fernández-Jiménez A, Palomo A, Sobrados I, Sanz J (2008) Effect of the SiO2/Na2O ratio on the alkali activation of fly ash. Part II: 29Si MAS-NMR Survey. Microporous Mesoporous Mater 109:525–534
Fechtelkord M, Stief F, Buhl J-C (2001) Sodium cation dynamics in nitrate cancrinite: a low and high temperature 23Na and 1H MAS NMR study and high temperature Rietveld structure refinement. Am Miner 86:165–175
Lloyd RR, Provis JL, Deventer JSJ (2009) Microscopy and microanalysis of inorganic polymer cements. 2: the gel binder. J Mater Sci 44:620–631. https://doi.org/10.1007/s10853-008-3078-z
Duxson P, Lukey GC, van Deventer JSJ (2006) Evolution of gel structure during thermal processing of Na-geopolymer gels. Langmuir 22:8750–8757
Bakharev T (2006) Thermal behaviour of geopolymers prepared using class F fly ash and elevated temperature curing. Cem Concr Res 36:1134–1147
Provis JL, Yong CZ, Duxson P, van Deventer JSJ (2009) Correlating mechanical and thermal properties of sodium silicate-fly ash geopolymers. Colloids Surf A 336:57–63
Felsche J, Luger S (1987) Phases and thermal decomposition characteristics of hydro-sodalites Na6+x,[AlSiO4]6,(OH)x·nH2O. Thermochim Acta 118:35–55
Mitsuda T, Taylor HFW (1978) Normal and anomalous tobermorites. Mineral Mag 42:229–235
Kalousek GL (1952) Application of differential thermal analysis in a study of the system lime–silica–water. In: Third international symposium on the chemistry of cement, cement and concrete association, London
Myers RJ, L’Hôpital ÉM, Provis JL, Lothenbach B (2015) Effect of temperature and aluminium on calcium (alumino)silicate hydrate chemistry under equilibrium conditions. Cem Concr Res 68:83–93
Myers RJ, Bernal SA, Gehman JD, van Deventer JSJ, Provis JL (2015) The role of Al in cross-linking of alkali-activated slag cements. J Am Ceram Soc 98:996–1004
Lloyd RR, Provis JL, van Deventer JSJ (2009) Microscopy and microanalysis of inorganic polymer cements. 1: remnant fly ash particles. J Mater Sci 44:608–619. https://doi.org/10.1007/s10853-008-3077-0
Fernández-Jiménez A, Palomo A (2005) Composition and microstructure of alkali activated fly ash binder: effect of the activator. Cem Concr Res 35:1984–1992
Winnefeld F, Leemann A, Lucuk M, Svoboda P, Neuroth M (2010) Assessment of phase formation in alkali activated low and high calcium fly ashes in building materials. Constr Build Mater 24:1086–1093
Bernal SA, San Nicolas R, Myers RJ, Mejía de Gutiérrez R, Puertas F, van Deventer JSJ, Provis JL (2014) MgO content of slag controls phase evolution and structural changes induced by accelerated carbonation in alkali-activated binders. Cem Concr Res 57:33–43
Wang S-D, Scrivener KL (2003) 29Si and 27Al NMR study of alkali-activated slag. Cem Concr Res 33:769–774
Palomo A, Alonso S, Fernández-Jiménez A, Sobrados I, Sanz J (2004) Alkaline activation of fly ashes: NMR study of the reaction products. J Am Ceram Soc 87:1141–1145
Merwin LH, Sebald A, Rager H, Schneider H (1991) 29Si and 27Al MAS NMR spectroscopy of mullite. Phys Chem Miner 18:47–52
Kowalczyk G, Roberts JE (1994) Solid State 29Si NMR determination of crystalline silica in natural iron oxide pigments. Anal Chim Acta 286:25–35
Myers S, Cygan R, Assink R, Boslough M (1998) 29Si MAS NMR relaxation study of shocked Coconino Sandstone from Meteor Crater, Arizona. Phys Chem Miner 25:313–317
Le Saoût G, Ben Haha M, Winnefeld F, Lothenbach B, Jantzen C (2011) Hydration degree of alkali-activated slags: a 29Si NMR study. J Am Ceram Soc 94:4541–4547
Bonk F, Schneider J, Cincotto MA, Panepucci HC (2003) Characterization by multinuclear high-resolution NMR of hydration products in activated blast-furnace slag pastes. J Am Ceram Soc 86:1712–1719
Lippmaa ASE, Mägi M (1986) High-resolution 27Al NMR of aluminosilicates. J Am Chem Soc 108:1730–1735
Moesgaard M, Keding R, Skibsted J, Yue Y (2010) Evidence of intermediate-range order heterogeneity in calcium aluminosilicate glasses. Chem Mater 22:4471–4483
Murdoch JB, Stebbins JF, Carmichael ISE (1985) High-resolution 29Si NMR study of silicate and aluminosilicate glasses: the effect of network-modifying cations. Am Miner 70:370–382
Walkley B, San Nicolas R, Sani M-A, Gehman JD, van Deventer JSJ, Provis JL (2016) Synthesis of stoichiometrically controlled reactive aluminosilicate and calcium-aluminosilicate powders. Powder Technol 297:17–33
Engelhardt G, Michel D (1987) High-resolution solid state NMR of silicates and zeolites. Wiley, Chichester
Duxson P, Provis JL, Lukey GC, Separovic F, van Deventer JSJ (2005) 29Si NMR study of structural ordering in aluminosilicate geopolymer gels. Langmuir 21:3028–3036
Skibsted J, Jakobsen HJ, Hall C (1995) Quantification of calcium silicate phases in Portland cements by 29Si MAS NMR spectroscopy. J Chem Soc Faraday Trans 91:4423–4430
Lippmaa E, Mägi M, Tarmak M, Wieker W, Grimmer AR (1982) A high resolution 29Si NMR study of the hydration of tricalciumsilicate. Cem Concr Res 12:597–602
Stebbins JF, Murdoch JB, Carmichael ISE, Pines A (1986) Defects and short-range order in nepheline group minerals: a silicon-29 nuclear magnetic resonance study. Phys Chem Miner 13:371–381
Lippmaa E, Maegi M, Samoson A, Engelhardt G, Grimmer AR (1980) Structural studies of silicates by solid-state high-resolution silicon-29 NMR. J Am Chem Soc 102:4889–4893
Engelhardt G, Felsche J, Sieger P (1992) The hydrosodalite system Na6+x[SiAlO4]6(OH)x.nH2O: formation, phase composition, and de- and rehydration studied by 1H, 23Na, and 29Si MAS-NMR spectroscopy in tandem with thermal analysis, x-ray diffraction, and IR spectroscopy. J Am Chem Soc 114:1173–1182
Hansen MR, Jakobsen HJ, Skibsted J (2003) 29Si chemical shift anisotropies in calcium silicates from high-field 29Si MAS NMR spectroscopy. Inorg Chem 42:2368–2377
Viallis H, Faucon P, Petit JC, Nonat A (1999) Interaction between salts (NaCl, CsCl) and calcium silicate hydrates (C − S−H). J Phys Chem B 103:5212–5219
Duxson P, Lukey GC, Separovic F, van Deventer JSJ (2005) Effect of alkali cations on aluminum incorporation in geopolymeric gels. Ind Eng Chem Res 44:832–839
Stebbins JF, Farnan I, Williams EH, Roux J (1989) Magic angle spinning NMR observation of sodium site exchange in nepheline at 500 °C. Phys Chem Miner 16:763–766
Buhl JC, Engelhardt G, Felsche J, Luger S, Foerster H (1988) 23Na MAS-NMR and 1H MAS-NMR studies in the hydro-sodalite system. Ber Bunsenges Phys Chem 92:176–181
Acknowledgements
This project was funded by the Australian Research Council (ARC), including support through an ARC Linkage Project (Grant number: LP140100504) and through the ARC Centre for Advanced Manufacturing of Prefabricated Housing. XRD data were acquired at the Materials Characterisation and Fabrication Platform (MCFP), The University of Melbourne, and the Victorian Node of the Australian National Fabrication Facility (ANFF). The authors thank Professor John L. Provis, The University of Sheffield, for very insightful discussions regarding this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Walkley, B., Kashani, A., Sani, MA. et al. Examination of alkali-activated material nanostructure during thermal treatment. J Mater Sci 53, 9486–9503 (2018). https://doi.org/10.1007/s10853-018-2270-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-018-2270-z