
Abstract
Nickel sulfide thin films and nanocrystals stabilized with oleylamine ligands are prepared from two different nickel xanthates as single-source precursors, which decompose at approx. 180 °C, and are thereby converted into nickel sulfide. These nickel xanthates comprise branched alkyl moieties allowing the choice of a wide range of nonpolar organic solvents for the processing to nickel sulfide thin films as well as to nanoparticles. The crystal structures of both compounds show a typical square-planar coordination of the sulfur atoms of both xanthates to the nickel central atom. The thermal decomposition via the Chugaev reaction forming nickel sulfide was studied by thermal gravimetric analysis showing the reaction taking place at about 180 °C. Consequently, by thermally converting spin-coated metal xanthate films directly on the substrates in the solid state, thin films consisting of hexagonal nickel sulfide are formed. If the nickel xanthates are heated in an oleylamine solution, oleylamine-capped nickel sulfide nanocrystals are obtained, exhibiting hexagonal NiS as main phase and Ni3S4 as secondary phase. This is also reflected in a sulfur-rich composition of the synthesized nickel sulfide nanocrystals as observed from TEM–EDX analyses.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The transition metal chalcogenide compound nickel sulfide exhibits a multiplicity of phases and stoichiometries, such as NiS (α-NiS and β-NiS), NiS2, Ni3S2, Ni3S4 and Ni7S6 [1]. Depending on the phase and the morphology, these compounds possess unique chemical and physical properties, which give nickel sulfides a wide applicability in numerous technologies ranging from catalysis to energy conversion and storage applications [2,3,4,5,6,7,8,9,10,11,12,13,14].
Nickel-(II)-sulfide (NiS) possesses two polymorphs, the low-temperature rhombohedral phase (β-NiS, R3 m, millerite) and the high-temperature hexagonal phase (α-NiS, P63/mmc, NiAs structure). Depending on the composition, the α- to β-phase transition can take place between 282 and 379 °C and is accompanied with a volume change about 4% [1]. Millerite β-NiS has metallic properties [15, 16], whereas the α-phase undergoes a phase transition from a semiconducting antiferromagnetic to a metallic paramagnetic phase at a temperature of about −8 °C [17, 18]. NiS has shown potential as an efficient counter electrode for dye-sensitized solar cells [2], as a cathode material in rechargeable lithium batteries [3] and as a catalyst for the hydrogen evolution reaction [4].
Cubic pyrite NiS2 (vaesite), suggested as a Mott–Hubbard insulator, exhibits semiconductor behavior with interesting electrical, optical and magnetic properties [19] and is explored for applications in hydrogen production [5] and batteries [6]. Ni3S2 (heazlewoodite) is a Pauli paramagnetic metal [20] with a stable rhombohedral crystal structure [21] and displays catalytic activity in water splitting reactions [7, 8]. Metallic Ni3S4 (polydymite) crystallizes in the cubic spinel structure and shows itinerant electron ferrimagnetism [22]. However, the synthesis of single-phase metastable Ni3S4 is challenging, because it decomposes at higher temperatures (356 °C) to form NiS and NiS2 [1]. Ni3S4 has been shown to be an efficient electrode material for high-performance supercapacitors [9, 10] and lithium ion batteries [11]. Ni7S6 (godlevskite) has an orthorhombic structure [23] and revealed excellent catalytic activity in hydrogenation reactions [12] and electrochemical sensors [13].
For the preparation of nickel sulfide nanomaterials with different morphologies and phases, various methods have been already successfully applied. Among these are solid-state reactions [24, 25], physical vapor and atomic layer depositions [14, 26], laser-induced reactions [27], electrochemical processes [8], solvo- and hydrothermal routes [2, 4, 11,12,13], spray pyrolysis [16], solution-based processes at room temperature [6, 22], or microwave-assisted syntheses [3, 5].
In recent years, single-source precursors (SSPs) have received significant attention since they can often give a better control over stoichiometry and offer the ability to tune their properties through ligand design, which granted in many cases a good control over phase and morphology of the nanostructures, which is very important in the view of using these nickel sulfide nanostructures for specific applications. Numerous single-source precursor compounds, which already contain a nickel sulfur bond, such as nickel dithiocarbamate [28,29,30,31,32,33,34], dithiocarbonate (xanthate) [35,36,37,38,39,40], thiolate [10, 41], dithiooxamide [42], thiourea [43], thiobenzoate [44], thiobiuret [45], mercaptobenzothiazole [46], thiosemicarbazide [47], polysulfide [48], imidodiphosphinate [49] or dithiophosphate [50], have already been investigated for the fabrication of various nickel sulfide nanostructures.
The attractive properties of metal xanthates, like their usually good stability (also in solution), relatively low decomposition temperature (below 200 °C) and simplicity to tailor solubility and decomposition behavior by variation of their organic side chains, have ensured that they are widely employed in the preparation of various metal sulfide nanomaterials [35,36,37,38,39, 51,52,53,54,55,56,57,58,59,60].
A variety of binary (Bi2S3 [52, 61], CdS [53], CuS [36], HgS [36], MnS [36], ZnS [36], PbS [60], PdS [35], SnS [59], NiS [35,36,37,38,39]), ternary (copper indium sulfide [51], copper antimony sulfide [54]) and quaternary (copper zinc tin sulfide [55]) metal sulfide nanomaterials have already been synthesized via a thermally induced decomposition of the metal xanthates, proceeding via the Chugaev elimination mechanism [36, 51]. Far less explored is the conversion of metal xanthates to metal sulfides at room temperature by photochemically induced decomposition using UV irradiation [56, 57] or direct chemical reaction with primary amines. In this latter study, we have recently demonstrated that the conversion of copper and indium xanthates into CuInS2 nanocrystals at room temperature in the presence of oleylamine proceeds via a reaction mechanism different to the Chugaev elimination [58].
Considering the attractive potential of metal xanthates as SSPs for the production of metal sulfide nanomaterials, it is surprising that there are only a few reports dealing with the thermal conversion of nickel xanthate complexes [35,36,37,38,39]. Therefore, in this contribution we demonstrate the potential of the two novel nickel xanthate precursors, nickel (II) O-3,3-dimethylbutan-2-yl dithiocarbonate (NiXa-C6) and nickel (II) O-2,2-dimethylpentan-3-yl dithiocarbonate (NiXa-C7), as SSP for the preparation of nickel sulfide nanostructures. Thereby, we cover the synthesis and characterization of both precursor compounds, discuss three routes to convert them into nickel sulfide thin films and nanocrystals and investigate the properties of the obtained nickel sulfide materials.
Experimental
Materials and methods
All chemicals were used as received without further purification and purchased from commercial sources as follows: nickel (II) chloride (NiCl2, 98%, Sigma-Aldrich), oleylamine (OLA, 96.0% min. primary amine, C18-content 80–90%, ACROS Organics), chloroform (CHCl3, puriss. p.a., Sigma-Aldrich), deuterated chloroform (CDCl3, 99.8 atom % D, VWR Chemicals), methanol (MeOH, puriss., ≥99.7%, Sigma-Aldrich), 2-propanol (99.9%, Sigma-Aldrich), toluene (≥99.0%, VWR Chemicals). Potassium O-3,3-dimethylbutan-2-yl dithiocarbonate (KXa-C6) and potassium O-2,2-dimethylpentan-3-yl dithiocarbonate (KXa-C7) were prepared by the following previously published procedures [51, 55].
NMR spectra were acquired on a Bruker Ultrashield 300 MHz (1H NMR: 300 MHz, 13C NMR: 75 MHz) spectrometer at ambient temperatures in CDCl3 solutions. Chemical shifts (δ) are quoted in parts per million (ppm) and referenced to the residual solvent peaks according to values given in the literature (1H NMR: 7.26 ppm, 13C NMR: 77.16 ppm) [62]. Coupling constants (J) are reported in Hertz (Hz), and splitting patterns are indicated by the following abbreviations: s (singlet), d (doublet), q (quadruplet), m (multiplet).
FTIR spectra were recorded on a Bruker Alpha FTIR spectrometer with a spectral resolution of 4 cm−1 in the range between 4000 and 400 cm−1 with 24 scans. Films on silicon wafers were measured in conventional transmission mode. All other FTIR measurements were taken in attenuated total reflection (ATR) mode using the ALPHA’s Platinum ATR single-reflection diamond ATR module.
Thermal gravimetric measurements were taken on a Netzsch Jupiter STA 449C thermogravimetric analyzer in aluminum oxide crucibles using helium as purge gas with a flow rate of 50 mL/min. All measurements were taken in the temperature range of 20–550 °C with a heating rate of 10 °C/min. The decomposition temperature T d is quoted as the temperature where 5% mass loss occurs.
Surface profile measurements were acquired on a Bruker Dektak XT stylus profilometer equipped with a 12.5-µm-radius stylus tip. Line scans were performed over a length of 2000 µm with a stylus force of 3 mg, a vertical bit resolution of 0.1 nm and a horizontal distance of 0.667 µm between data points. Film thickness and surface roughness were determined by means of arithmetic averaging calculations from the obtained two-dimensional profile using the software Vision 64.
UV–Vis absorption spectra of solutions were measured on a Shimadzu UV-1800 spectrophotometer using a quartz cuvette of 1 cm path length with increments of 1 nm. Transmittance and reflectance spectra of thin films (substrate: glass slide) as well as absorption spectra of nanoparticle solutions (solvent: chloroform) were obtained using a PerkinElmer Lambda 950 UV–Vis–NIR spectrophotometer equipped with a 150-mm integrating sphere. Measurements were taken in the wavelength range from 400 to 1600 nm in 1-nm steps with a scan speed of 281 nm/min and a slit width of 2 nm.
SEM–EDX analyses were performed at an acceleration voltage of 3 kV on a Zeiss Ultra 55 fitted with a thermal field emission gun and an EDAX Trident EDS detector. An Everhart–Thornley secondary electron (SE) detector and an energy-selective backscattered electron (EsB) detector featured with an integrated filtering grid were used for imaging.
Specimens for transmission electron microscopy (TEM) observations were prepared by depositing a drop of a nanoparticle solution (solvent: chloroform) on a copper TEM grid supported with a Quantifoil holey carbon film, followed by solvent evaporation at room temperature. TEM images were obtained on a FEI Tecnai F20 microscope equipped with a Schottky field emission gun (FEG) fitted with a monochromator and a high-resolution Gatan imaging filter (HR-GIF) with an UltraScan CCD camera. Energy-dispersive X-ray (EDX) spectra were recorded with an EDAX Sapphire Si(Li) detector, which was attached to the Tecnai F20 microscope.
X-ray diffraction measurements were taken on a PANalytical Empyrean diffractometer in Bragg–Brentano configuration operated at 40 kV and 40 mA using CuKα radiation (λ = 1.5418 Å).
For single-crystal X-ray diffractometry, suitable crystals were selected and removed from the mother liquor and immediately covered with silicone oil. A single crystal was selected, mounted on a glass rod on a copper pin, and placed in the cold N2 stream provided by an Oxford Cryosystems Cryostream. XRD data collection was performed for the compounds nickel (II) O-3,3-dimethylbutan-2-yl dithiocarbonate (NiXa-C6) and nickel (II) O-2,2-dimethylpentan-3-yl dithiocarbonate (NiXa-C7) on a Bruker APEX II diffractometer with use of MoKα radiation (λ = 0.71073 Å) and a CCD area detector. Empirical absorption corrections were applied using SADABS (Bruker (2012) Bruker AXS Inc., Madison, Wisconsin, USA) [63]. The structures were solved with the use of the intrinsic phasing option in SHELXT and refined by the full-matrix least-square procedures in SHELXL [64, 65]. The space-group assignments and structural solutions were evaluated using PLATON [66]. Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were located in calculated positions corresponding to standard bond lengths and angles. Disorder, as observed for compound NiXa-C6, was handled by modeling the occupancies of the individual orientations using free variables to refine the respective occupancy of the affected fragments [67]. Disordered positions for the S2CORR’ ligands in NiXa-C6 were refined using 75/25 split positions. Intermolecular interactions are interpreted based on a Cambridge Structural Database [68] search. Hydrogen bond and anagostic interaction values fall within expected ranges [69,70,71,72,73,74,75,76]. Table 1 contains crystallographic data and details of measurements and refinement for compounds NiXa-C6 and NiXa-C7. CCDC 1520119 and 1520120 contain the supplementary crystallographic data for compounds NiXa-C6 and NiXa-C7, respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Synthesis of nickel (II) O-3,3-dimethylbutan-2-yl dithiocarbonate (NiXa-C6)
Nickel (II) chloride (1.05 g, 8.1 mmol, 1.0 eq.) was weighed into a Schlenk flask and dissolved in 40 ml deionized water. A separately prepared solution of potassium O-3,3-dimethylbutan-2-yl dithiocarbonate (3.83 g, 17.7 mmol, 2.2 eq.) in 120 mL distilled water was added dropwise to the nickel chloride solution at room temperature under vigorous stirring. A greenish-yellow precipitate was formed immediately, and the reaction mixture was stirred for further 3 h. The resulting precipitate was isolated by filtration, washed several times with distilled water and subsequently dried under vacuum. The crude solid was dissolved in 10 mL chloroform, filtrated through a syringe filter to remove insoluble side products and precipitated in 100 mL methanol. The precipitate was separated by filtration, washed with chilled methanol and finally dried under vacuum to yield NiXa-C6 as green powder (yield: 2.8 g, 84%). Single crystals suitable for X-ray diffractometry were obtained by slow diffusion of methanol into a chloroform solution of NiXa-C6 at 4 °C.
1H NMR (300 MHz, CDCl3): δH/ppm: 5.06 (q, J = 6.3 Hz, 1H, CH), 1.31 (d, J = 6.3 Hz, 3H, CH3), 0.96 (s, 9H, C(CH3)3). 13C NMR (75 MHz, CDCl3): δC/ppm: 230.8 (CS2O), 88.4 (CH), 35.0 (C(CH3)3), 25.7 (3C, C(CH3)3), 14.9 (CH3).
FTIR (on Si wafer): ν/cm−1: 2964, 2868, 1467, 1440, 1377, 1367, 1357, 1281, 1216, 1118, 1073, 1044, 1020, 1000, 981.
Thermal gravimetric analysis: T d = 187 °C, mass loss = 72% (calculated for NiS: 78%).
Synthesis of nickel (II) O-2,2-dimethylpentan-3-yl dithiocarbonate (NiXa-C7)
The reaction was performed in an analogous manner to the described synthesis procedure of NiXa-C6, with the only difference that potassium O-2,2-dimethylpentan-3-yl dithiocarbonate (4.08 g, 17.7 mmol, 2.2 eq.) was used instead of potassium O-3,3-dimethylbutan-2-yl dithiocarbonate. NiXa-C7 was obtained as a green powder (yield: 2.6 g, 73%).
1H NMR (300 MHz, CDCl3): δH/ppm: 5.07–4.92 (m, 1H, CH), 1.79–1.63 (m, 2H, CH2), 0.96 (s, 12H, CH3). 13C NMR (75 MHz, CDCl3): δC/ppm: 231.9 (CS2O), 93.8 (CH), 35.7 (C(CH3)3), 26.1 (3C, C(CH3)3), 23.0 (CH2), 11.1 (CH3).
FTIR (on Si wafer): ν/cm−1: 2968, 2874, 1475, 1459, 1397, 1369, 1355, 1306, 1279, 1255, 1216, 1130, 1079, 1053, 1028, 951.
Thermal gravimetric data: T d = 176 °C, mass loss = 73% (calculated for NiS: 79%).
Preparation of nickel sulfide thin films
Nickel sulfide thin films were produced through solid-state thermal decomposition of precursors NiXa-C6 and NiXa-C7 on silicon wafers (for FTIR measurements) or glass slides (for UV–Vis–NIR analysis and XRD measurements) with sizes of 25 mm × 25 mm. Silicon and glass substrates were cleaned by sonication in isopropanol and distilled water each for 30 min and immediately before the coating process, by treatment with an oxygen plasma for 5 min (Femto low-pressure plasma system, Diener electronic). Precursor solutions in chloroform of NiXa-C6 and NiXa-C7, respectively, with a concentration of 100 mg/mL were deposited on glass slides via spin coating at 1800 rpm for 30 s and on silicon wafers via drop coating. Obtained precursor layers were converted to nickel sulfide films by annealing in a preheated tube furnace (Nabertherm R 50/500/12) at 200 °C for 30 min under nitrogen atmosphere. Layer thicknesses of the films deposited on the glass substrates were found to be 60 and 40 nm for the films prepared from the NiXa-C6 and NiXa-C7 precursors, respectively.
Preparation of nickel sulfide nanoparticles
Nickel sulfide nanoparticles were prepared by a solvothermal method and a synthesis route at room temperature. In the first approach, a green-black slurry of the precursor compound NiHex and NiHep, respectively, in oleylamine with a concentration of 0.063 mmol/mL was prepared and transferred into a round-bottom Schlenk flask. The slurry was heated to 200 °C through a preheated oil bath (210 °C) under nitrogen atmosphere, and the reaction was allowed to proceed for 1 h under continuous stirring before it was cooled down to room temperature. Subsequent separation and purification steps were performed under ambient conditions. The black colored nickel sulfide nanoparticles were precipitated by adding methanol and collected by centrifugation at 2500 rpm for 5 min. To remove by-products and unreacted precursors, as-prepared nanoparticles were re-dissolved in chloroform, precipitated with methanol, separated by centrifugation and finally dried under vacuum.
In an analogous manner to our recently published procedure [58], nanoparticles were also prepared at room temperature under nitrogen atmosphere in a glovebox (LABmaster DP, MBraun) by dissolving NiXa-C6 in toluene with a concentration of 0.063 mmol/mL. Decomposition of NiXa-C6 was induced by adding oleylamine (OLA) in a NiXa-C6:OLA molar ratio of 1:4 under vigorous stirring, whereby the green solution turned brown. The reaction mixture was stirred for 72 h before the obtained black colored nickel sulfide nanoparticles were separated and purified in the same way as described above.
Results and discussion
Synthesis and structural analysis
The precursor synthesis and the subsequent preparation of nickel sulfide thin films and nanocrystals via three different routes from the metal xanthates are schematically displayed in Fig. 1. Nickel xanthate precursors nickel (II) O-3,3-dimethylbutan-2-yl dithiocarbonate (NiXa-C6) and nickel (II) O-2,2-dimethylpentan-3-yl dithiocarbonate (NiXa-C7) were synthesized analogously to our previously published procedures for metal xanthates [51, 55] and isolated in high yields. The obtained green powders are stable over an extended period of time under ambient conditions and exhibit high solubility in nonpolar organic solvents such as chloroform, toluene or chlorobenzene due to the branched alkyl side chains. NMR spectra, confirming the proposed structures and the purity of these two precursor compounds, show the typical high chemical shift of 230.8 ppm (NiXa-C6) and 231.9 ppm (NiXa-C7) for the carbon of the dithiocarbonate group. Also, the IR spectra are in accordance with the proposed structure and will be discussed in the next section.

Synthesis of the nickel xanthate precursors (NiXa-C6, NiXa-C7) and schematic illustration of the preparation of nickel sulfide thin films and nanoparticles (stabilized by oleylamine ligands)
Single-crystal X-ray diffraction analyses reveal that the nickel atoms lie on a center of symmetry in both prepared nickel xanthates. Each nickel atom is four coordinate with two S2COR ligands bonded through the sulfur atoms giving rise to a square-planar arrangement, which is typical for this type of nickel compounds (Fig. 2). A similar square-planar arrangement was, for example, also found for other nickel alkyl xanthates [39, 69]. In both compounds, the S–C bond lengths are nearly the same, expressing the bidentate nature of the S2CORR’ ligands (Table 2). However, each bidentate ligand displays a slight distortion to square planarity through narrow bite angles (S1–Ni–S2) of 79.58(3)° for NiXa-C6 and 79.64(6)° for NiXa-C7. With respect to Ni–S bonds, an increased averaged length is observed when comparing NiXa-C6 (2.082(8) Å) and NiXa-C7 (2.210(2) Å). Upon comparison of all other bond lengths between the compounds, no other deviations are observed, and thus, the increase in bond length can be attributed to the increased steric bulk afforded by the ethyl substituted S2COEttbu ligand.

a Crystal structure of NiXa-C6. b Crystal structure of NiXa-C7. All non-carbon atoms shown as 30% shaded ellipsoids. Hydrogen atoms removed for clarity
The most distinct difference between these compounds is how ligand frameworks and presence of hydrogen bonds affect the crystal packing in the extended solid state for both of these compounds. Both NiXa-C6 and NiXa-C7 display intra- and intermolecular hydrogen bonds through the sulfur atoms of neighboring molecules (C–H···S) (Table 3). In both compounds, the intramolecular hydrogen bond occurs from both methylene hydrogens of each S2CORR’ ligand and a sulfur atom of the same S2CORR’ moiety. The values for these intramolecular C–H···S hydrogen bonds are comparable with a length of 2.59 Å and angle of 116° for NiXa-C6 and a length of 2.55 Å and angle of 115° for NiXa-C7. In NiXa-C6, the same hydrogen is then involved in a second intermolecular C–H···S hydrogen bond (2.96 Å, 142°) with a sulfur atom from the S2COMetbu ligand of a neighboring molecule propagating 1D chains (Fig. 3). However, the same intermolecular interaction is not observed in NiXa-C7. Here, a hydrogen from the ethyl chain (2.99 Å, 152°) is responsible for the intermolecular interaction, which results in a larger gap between neighboring molecules in the 1D chain (Fig. 4). The most striking feature of NiXa-C7 is the presence of an anagostic interaction (C–H···Ni) occurring between the neighboring and coplanar 1D chains creating 2D sheets. These are occurring through the nickel atoms and a hydrogen from one of the methyl groups from the tbutyl substituent on the S2COEttbu ligand. These anagostic interactions (2.86 Å, 142°) occupy the vacant axial sites on the metal centers providing a pseudo-octahedral coordination environment further expressed with a H···M···H calculated angle of 180° relative to the NiS4 plane. Anagostic interactions often occur in square-planar d8 systems and typically display H···M distances of 2.3–2.9 Å, and C–H···M angles are usually in the range of 130–170° [69,70,71,72,73,74,75,76,77,78]. The closest possible C–H···Ni interaction (3.09 Å) of NiXa-C6 is above accepted values. No close S···S interactions were observed for NiXa-C6 (3.81 Å) or for NiXa-C7 (3.84 Å) due to the values being above the sum of van der Waals radii (3.60 Å).

a View of the crystal packing diagram of NiXa-C6 along the a-axis. b View of the crystal packing diagram of NiXa-C6 along the 1D chain propagated by C–H···S hydrogen bonds. C–H···S hydrogen bonds highlighted by dashed bonds. All non-carbon atoms shown as 30% shaded ellipsoids. Hydrogen atoms not involved in interactions removed and carbon atoms grayed for clarity

a View of the crystal packing diagram of NiXa-C7 along the a-axis. b View of the crystal packing diagram of NiXa-C7 along the 2D sheets propagated by C–H···S hydrogen bonds and anagostic interactions (C–H···Ni). C–H···S hydrogen bonds and anagostic interactions (C–H···Ni) highlighted by dashed bonds. All non-carbon atoms shown as 30% shaded ellipsoids. Hydrogen atoms not involved in interactions removed and carbon atoms grayed for clarity
Preparation of nickel sulfide thin films
Nanocrystalline nickel sulfide thin films were successfully prepared on silicon wafers and glass substrates by spin coating of the precursor solution onto the substrates followed by thermal decomposition of the xanthate precursors NiXa-C6 and NiXa-C7 in a nitrogen environment at 200 °C resulting in the conversion to the sulfide. The resulting films from both precursor compounds showed good adherence to the substrates and exhibited a uniform appearance with a black-gray color.
Thermal decomposition behavior of the nickel xanthate precursors was investigated by thermal gravimetric measurements and FTIR spectroscopy. Thermogravimetric curves of both precursor compounds, as displayed in Fig. 5a, show a sharp single-step conversion to the nickel sulfide with a rapid mass loss between 170 and 210 °C for NiXa-C7 and between 180 and 210 °C for NiXa-C6, respectively. The thermally induced decomposition of the nickel xanthates is assumed to proceed according to the Chugaev elimination reaction, in which volatile decomposition products such as the respective alkene and COS are formed to yield residue-free nickel sulfide [36, 51]. NiXa-C7 exhibits a slightly lower decomposition onset temperature at 176 °C (determined at a mass loss of 5%) compared with NiXa-C6, having its onset at 187 °C. The observed final residual weights of 27 and 28% are slightly higher than the theoretically expected ones for the formation of stoichiometric NiS from NiXa-C7 to NiXa-C6, respectively. However, this difference might be caused due to small amounts of residual decomposition compounds or due to uncertainties in the thermogravimetric measurement setup. Additionally, FTIR spectroscopy measurements were taken (see Fig. 5b), which could not detect any organic residues in the converted NiS thin films. The comparison of the spectra of NiXa-C6 and NiXa-C7 with the corresponding NiS thin films shows that the characteristic vibrational bands of the nickel xanthates, such as asymmetric C–O–C stretching vibrations (at 1281 cm−1 for NiXa-C6 and 1279 cm−1 for NiXa-C7) and C–S stretching vibrations (at 1044/1020 cm−1 for NiXa-C6 and 1053/1028 cm−1 for NiXa-C7), have completely vanished, and no other remaining IR bands stemming from organic residues can be identified. A detailed assignment of the IR absorption bands of the NiXa-C6 and NiXa-C7 precursor films is given in Table S1 in the supporting information.

a Thermogravimetric analysis of NiXa-C6 and NiXa-C7. b FTIR spectra of the NiXa-C6 and NiXa-C7 precursor films and the corresponding thermally converted NiS thin films deposited on silicon wafers
For further characterization of the NiS thin films with regard to their phase composition and crystallinity, X-ray diffraction measurements were taken. The XRD patterns, as depicted in Fig. 6a, reveal that in the NiS-C7 sample pure α-NiS phase (reference pattern for hexagonal α-NiS: PDF 01-075-0613) with main diffraction peaks at 30.1 (100), 34.6 (101), 45.9 (102) and 53.5° 2θ (110) is formed. In the NiS-C6 sample, additional minor diffraction peaks at, for example, 18.5°, 32.2°, 35.8°, 40.6°, 48.9° and 50.2° 2θ, are present, which can be ascribed to the trigonal β-NiS phase (PDF 01-086-2280). The average size of crystalline domains, estimated by the Scherrer equation using the FWHM of the 102 reflection (45.9°2θ), is approx. 28 nm for the NiS-C6 sample and about 22 nm for the NiS-C7 sample.

a X-ray diffraction patterns of the NiS thin films on glass fabricated from the precursors NiXa-C6 and NiXa-C7 with reference patterns for hexagonal NiS (PDF 01-075-0613) and trigonal NiS (PDF 01-086-2280). b Tauc plots for the NiS thin films of the films deposited onto glass substrates
SEM–EDX measurements also confirmed the formation of an almost stoichiometric NiS phase with a Ni:S ratio of approx. 53:47 at % (films prepared from NiXa-C6) and approx. 49:51 at % (films prepared from NiXa-C7). The formation of a mixture of α-NiS and β-NiS phase by thermal decomposition of nickel xanthates and dithiocarbamates is common and has also already been reported in other publications [30, 33, 37, 39].
Moreover, it should be mentioned that the nature of the substrate surface as well as changes in processing steps can also influence the phase composition of the films, e.g., trigonal β-NiS was formed as majority phase by conversion of the precursors on glass substrates covered with mesoporous TiO2 films instead on plain glass (see Figure S1, supporting information).
Figure 6b shows the Tauc plots for the prepared NiS thin films. The absorption data have been determined from transmission and reflection spectra (see Figure S2). By extrapolating the linear part of the data in the Tauc plots, optical bandgap values of about 0.7 eV were obtained for both thin films [79].
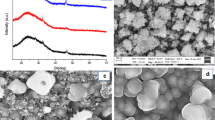
As can be seen from the SEM images depicted in Fig. 7, the precursor structure influences the film morphology significantly. NiXa-C7 produces homogeneous and porous, but crack-free films containing densely packed nanosized grains with a high apparent coverage. In contrast, thin films prepared from NiXa-C6 as precursor exhibit a less homogeneous appearance. This observation is in concordance with the higher roughness value (35 nm for NiXa-C6 and 10 nm for NiXa-C7, measured using a surface profiler) of the film prepared from the NiXa-C6 precursor and is also reflected in the optical appearance and the reflection spectra (NiXa-C6-derived films show a matte surface, whereas films prepared from NiXa-C7 exhibit a metallic reflective surface). The observed changes in film morphology may be derived from several factors such as the different primary crystallite sizes (28 nm for NiXa-C6, 22 nm for NiXa-C7), differences in crystallization behavior of the precursors during deposition of the precursor films or different precursor reactivities, which have important implications on nucleation and growth kinetics. Regarding this latter issue, MacLachlan et al. [53] and Lewis et al. [60] have already demonstrated that the morphology of metal sulfide nanocomposites can be efficiently tuned by the chemical structure of xanthate or dithiocarbamate complexes. Similar as in the work of Lewis et al., the observed differences in film morphology in this study might also originate from changes in precursor reactivity.

Secondary electron (SE) SEM images showing the surface of the nickel sulfide thin films on glass prepared from the precursors NiXa-C6 (a) and NiXa-C7 (b)
Preparation of nickel sulfide nanoparticles
Nickel sulfide nanoparticles (NPs) capped with oleylamine ligands were prepared via two different approaches: (1) by reaction of the NiXa-C6 precursor dissolved in toluene with oleylamine at room temperature and (2) by a solvothermal conversion of the precursors NiXa-C6 and NiXa-C7 in oleylamine at 200 °C.
For the room-temperature synthesis, NiXa-C6 was dissolved in toluene and the reaction was started by the addition of oleylamine, according to a recently published procedure for the preparation of CuInS2 and CdS nanocrystals [58]. Within a few hours after the addition of the amine, the color of the solution changes from green, via brown to black, indicating the decomposition of the greenish nickel xanthates and the formation of nickel sulfide nanoparticles. After purification of the nanoparticles by precipitation in methanol and dissolution in chloroform, a UV–Vis–NIR spectrum was recorded (see Fig. 8a), which shows a steady increase in absorption in the measured range from 1600 to 400 nm. The X-ray diffraction pattern (Fig. 8b) of the nanoparticle powder reveals just one broad amorphous peak around 20° 2θ, which is characteristic for oleylamine [80]. This indicates that the nanoparticles formed at room temperature are not crystalline but have an amorphous nature.

a UV–Vis–NIR spectra of the oleylamine-stabilized nickel sulfide NPs in chloroform solutions prepared by the solvothermal (200 °C) route and the room-temperature (RT) synthesis and a UV–Vis spectrum of the precursor compound NiXa-C6. b X-ray diffraction patterns of the oleylamine-capped nickel sulfide NPs with reference patterns for hexagonal NiS (PDF 01-075-0613) and cubic Ni3S4 (PDF 01-076-1813)
Therefore, in a second approach, nickel sulfide nanoparticles were also prepared by conversion of the xanthates at 200 °C in oleylamine as solvent and capping ligand. During the reaction time of one hour, the color of the greenish-black slurry turned into black. Afterward, the nanoparticles were isolated by repeated precipitation in methanol and dissolution in chloroform or toluene. The UV–Vis–NIR absorption spectra of the nickel sulfide nanoparticles prepared from NiXa-C6 and NiXa-C7 precursors are depicted in Fig. 8a, and their shape is very similar to the spectrum of the nickel sulfide nanoparticles prepared at room temperature. The estimated absorption edges of the nanoparticles, which form brown colored solutions in chloroform, are found to be in the near-infrared range (1200–1000 nm/1.0–1.2 eV). In addition, it should be mentioned that the long absorption tails extending over the near-infrared region and the absence of a steep absorption onset makes an exact determination of the band gap of the prepared nanoparticles difficult and furthermore indicates a broad particle size distribution of the NPs.
When looking at the X-ray diffraction patterns (Fig. 8b), a significant difference between the nanoparticles prepared at room temperature and at 200 °C is revealed. The diffraction patterns show that at 200 °C crystalline NPs with a hexagonal α-NiS phase and a metastable cubic Ni3S4 as secondary phase were obtained. The nanoparticle samples fabricated by the solvothermal conversion of NiXa-C6 and NiXa-C7, respectively, exhibit quite similar diffraction patterns with reflexes coinciding with the reference pattern for hexagonal NiS (PDF 01-075-0613) at 30.4°, 34.9°, 46.2° and 53.9° (overlapped by the reflex at 54.6°) 2θ, and reflexes attributable to the reference pattern for cubic Ni3S4 (PDF 0-076-1813) at 16.2°, 26.6°, 31.2°, 37.8°, 49.9° and 54.6° (overlapped by the reflex at 53.9°) 2θ. The broadening of the peaks reveals a nanosized nature of the samples with average crystallite sizes (estimated by Scherrer equation from the peaks at 37.8° and 46.2° 2θ) of about 10–15 nm. These results are consistent with reports in which solvothermal decomposition of dithiocarbamates, in the presence of primary amines, led to the formation of the sulfur-rich metastable Ni3S4 phase [30, 33, 34]. In these studies, the role of the primary amine as reaction component, besides its function as solvent and stabilizing agent, is demonstrated, whereby the decomposition mechanism or rather phase composition can be tuned by the nature of the amine.
The prepared nickel sulfide NPs were further investigated by FTIR spectroscopy and thermogravimetric measurements to study the organic ligand shell of the NPs. FTIR spectra, as shown in Fig. 9a, confirm that the NPs are functionalized with oleylamine ligands, since characteristic vibrational bands, which can be associated with the amine group, such as the asymmetric and symmetric N–H (R–NH2) stretching modes (C6, 200 °C: 3248 cm−1; C7, 200 °C: 3270 cm−1), the C–H (C=C–H) stretching vibration (C6, 200 °C: 3003 cm−1; C7, 200 °C: 3001 cm−1) and the C–H (–CH2–, R–CH3) deformation vibration (C6, 200 °C: 1457 cm−1; C7, 200 °C: 1457 cm−1), are present in the spectra [81]. These vibrational bands show a shift toward lower wavenumbers compared with those of pure oleylamine (3333, 3005 and 1464 cm−1), indicating that the amine ligand is bound to the NP surface through coordination of the electron-donating nitrogen atom. FTIR spectra also confirm the purity of the synthesized NPs since besides the vibrational modes of the oleylamine ligand no additional IR bands, which can be ascribed to the xanthate precursors or other organic by-products, can be recognized.

a FTIR spectra of the oleylamine-capped nickel sulfide NPs and pure oleylamine. b Thermogravimetric analysis of the nickel sulfide NPs
The amount and the thermal stability of the oleylamine ligand shells were determined from thermogravimetric data displayed in Fig. 9b. The NPs produced by the solvothermal route exhibit quite similar thermogravimetric curves, comparable with curves previously obtained from oleylamine-capped NPs [58], with a gradual mass loss (total mass loss: 29% for C6, 200 °C and 27% for C7, 200 °C) over a wide temperature range (180–480 °C).
Bright-field TEM images of the nickel sulfide NPs prepared via the solvothermal synthesis route (as depicted in Fig. 10) confirm the broad particle size distribution in the sample. Besides small nanoparticles with sizes ranging from 5 to 20 nm, also larger disk-shaped particles with sizes between 40 and 60 nm are observed in both samples. In addition, TEM–EDX analyses of the nanoparticles reveal a Ni:S ratio of approx. 42:58 at %, which matches well with the formation of Ni3S4 as secondary phase as it was observed in the X-ray diffraction measurements.

Bright-field TEM images of the nickel sulfide NPs synthesized by a solvothermal conversion of the NiXa-C6 (a) and NiXa-C7 (b) precursor in oleylamine at 200 °C
Conclusion
In conclusion, we presented three different methods to prepare nickel sulfide thin films or nanocrystals from two nickel xanthate precursors. The used nickel xanthates only differ in one additional CH2 group; however, significant differences in their crystal structure, in the thermal decomposition behavior as well as in the final nickel sulfide thin films and nanocrystals were observed. Nanocrystalline thin films with a primary crystallite size of 20–30 nm could be conveniently prepared by a solid-state reaction directly on the substrate. Nickel sulfide nanocrystals were prepared by heating the xanthates dissolved in a toluene/oleylamine mixture to 200 °C. The obtained NiS nanoparticles are capped with oleylamine ligands and are well soluble in nonpolar organic solvents. X-ray diffraction measurements revealed that the nanoparticles consist of hexagonal NiS and cubic Ni3S4 as secondary phase. Thus, the herein presented new nickel xanthates are interesting single-source precursors for nanocrystalline nickel sulfide materials, and due to the good solubility in nonpolar solvents and compatibility with organic polymers, they could also be applied for the preparation of hybrid NiS/polymer nanocomposites.
References
Kullerud G, Yund RA (1962) The Ni-S system and related minerals. J Petrol 3:126–175
Wang X, Batter B, Xie Y, Pan K, Liao Y, Lv C, Li M, Sui S, Fu H (2015) Highly crystalline, small sized, monodisperse α-NiS nanocrystal ink as an efficient counter electrode for dye-sensitized solar cells. J Mater Chem A 3:15905–15912
Idris NH, Rahman MM, Chou S-L, Wang J-Z, Wexler D, Liu H-K (2011) Rapid synthesis of binary α-NiS–β-NiS by microwave autoclave for rechargeable lithium batteries. Electrochim Acta 58:456–462
Pan Y, Chen Y, Li X, Liu Y, Liu C (2015) Nanostructured nickel sulfides: phase evolution, characterization and electrocatalytic properties for the hydrogen evolution reaction. RSC Adv 5:104740–104749
Pang H, Wei C, Li X, Li G, Ma Y, Li S, Chen J, Zhang J (2014) Microwave-assisted synthesis of NiS2 nanostructures for supercapacitors and cocatalytic enhancing photocatalytic H2 production. Sci Rep 4:3577
Liu Z, Zheng X, Luo S-L, Xu S-Q, Yuan N-Y, Ding J-N (2016) High performance Li–S battery based on amorphous NiS2 as the host material for the S cathode. J Mater Chem A 4:13395–13399
Feng LL, Yu G, Wu Y, Li GD, Li H, Sun Y, Asefa T, Chen W, Zou X (2015) High-index faceted Ni3S2 nanosheet arrays as highly active and ultrastable electrocatalysts for water splitting. J Am Chem Soc 137:14023–14026
Falkowski JM, Concannon NM, Yan B, Surendranath Y (2015) Heazlewoodite, Ni3S2: a potent catalyst for oxygen reduction to water under benign conditions. J Am Chem Soc 137:7978–7981
Zhang Y, Sun W, Rui X, Li B, Tan HT, Guo G, Madhavi S, Zong Y, Yan Q (2015) One-pot synthesis of tunable crystalline Ni3S4@amorphous MoS2 core/shell nanospheres for high-performance supercapacitors. Small 11:3694–3702
Gupta R, Fisher TS (2017) Scalable coating of single-source nickel hexadecanethiolate precursor on 3D graphitic petals for asymmetric supercapacitors. Energy Technol 5:740–746. doi:10.1002/ente.201600475
Ji S, Zhang L, Yu L, Xu X, Liu J (2016) In situ carbon-coating and Ostwald ripening-based route for hollow Ni3S4@C spheres with superior Li-ion storage performances. RSC Adv 6:101752–101759
Cao F, Liu R, Zhou L, Song SY, Lei YQ, Shi WD, Zhao F, Zhang H (2010) One-pot synthesis of flowerlike Ni7S6 and its application in selective hydrogenation of chloronitrobenzene. J Mater Chem 20:1078–1085
Wu W, Li Y, Jin J, Wu H, Wang S, Xia Q (2016) A novel nonenzymatic electrochemical sensor based on 3D flower-like Ni7S6 for hydrogen peroxide and glucose. Sens Actuator B Chem 232:633–641
Li H, Shao Y, Su Y, Gao Y, Wang X (2016) Vapor-phase atomic layer deposition of nickel sulfide and its application for efficient oxygen-evolution electrocatalysis. Chem Mater 28:1155–1164
Krishnakumar SR, Shanthi N, Sarma DD (2002) Electronic structure of millerite NiS. Phys Rev B 66:115105
Boughalmi R, Rahmani R, Boukhachem A, Amrani B, Driss-Khodja K, Amlouk M (2015) Metallic behavior of NiS thin film under the structural, optical, electrical and ab initio investigation frameworks. Mater Chem Phys 163:99–106
Sparks JT, Komoto T (1967) Metal-to-semiconductor transition at the magnetic ordering temperature of NiS. Phys Lett A 25:398–399
Sparks JT, Komoto T (1968) Metal-to-semiconductor transition in hexagonal NiS. Rev Mod Phys 40:752–754
Kautz RL, Dresselhaus MS, Adler D, Linz A (1972) Electrical and optical properties of NiS2. Phys Rev B 6:2078–2082
Metcalf PA, Crooker BC, McElfresh M, Kakol Z, Honig JM (1994) Low-temperature electronic and magnetic properties of single-crystal Ni3S2. Phys Rev B 50:2055–2060
Parise JB (1980) Structure of hazelwoodite (Ni3S2). Acta Cryst B 36:1179–1180
Manthiram A, Jeong YU (1999) Ambient temperature synthesis of spinel Ni3S4: an itinerant electron ferrimagnet. J Solid State Chem 147:679–681
Kulagov EA, Evstigneeva TL, Yushko-Zakharova OE (1969) The new nickel sulphide godlevskite. Geol Rudnykh Mestorozhderii 11:115–121
Bonneau PR, Shibao RK, Kaner RB (1990) Low-temperature precursor synthesis of crystalline nickel disulfide. Inorg Chem 29:2511–2514
An G, Chenguang L, Hou Y, Zhang X, Liu Y (2008) Transition metal dichalcogenide materials: solid-state reaction synthesis of nanocrystalline nickel disulfide. Mater Lett 62:2643–2646
Nozaki H, Aizawa T, Kosuda K (2005) Stabilized epitaxial films of hexagonal Ni1−xS on MgO(001). J Solid State Chem 178:2001–2007
Anischik VM, Markevich MI, Piskunov FA, Yanushkevich VA (1995) Pulsed-laser-induced synthesis of nickel sulphides in sulphur-containing liquids. Thin Solid Films 261:183–185
O’Brien P, Park JH, Waters J (2003) A single source approach to deposition of nickel sulfide thin films by LP-MOCVD. Thin Solid Films 431–432:502–505
Chen X, Wang Z, Wang X, Wan J, Liu J, Qian Y (2004) A single-source approach to metastable Ni3S4 crystallites and their optical properties. Chem Lett 33:1294–1295
Hollingsworth N, Roffey A, Islam H-U, Mercy M, Roldan A, Bras W, Wolthers M, Catlow CRA, Sankar G, Hogarth G, de Leeuw NH (2014) Active nature of primary amines during thermal decomposition of nickel dithiocarbamates to nickel sulfide nanoparticles. Chem Mater 26:6281–6292
Prakasam BA, Lahtinen M, Peuronen A, Muruganandham M, Kolehmainen E, Haapaniemi E, Sillanpää M (2015) Spectral and structural studies on Ni(II) dithiocarbamates: nickel sulfide nanoparticles from a dithiocarbamate precursor. Inorg Chim Acta 425:239–246
Sathiyaraj E, Gurumoorthy G, Thirumaran S (2015) Nickel(II) dithiocarbamate complexes containing the pyrrole moiety for sensing anions and synthesis of nickel sulfide and nickel oxide nanoparticles. New J Chem 39:5336–5349
Roffey A, Hollingsworth N, Islam H-U, Mercy M, Sankar G, Catlow CRA, Hogarth G, de Leeuw NH (2016) Phase control during the synthesis of nickel sulfide nanoparticles from dithiocarbamate precursors. Nanoscale 8:11067–11075
Gervas C, Mlowe S, Akerman MP, Ezekiel I, Moyo T, Revaprasadu N (2017) Synthesis of rare pure phase Ni3S4 and Ni3S2 nanoparticles in different primary amine coordinating solvents. Polyhedron 122:16–24
Cheon J, Talaga DS, Zink JI (1997) Laser and thermal vapor deposition of metal sulfide (NiS, PdS) films and in situ gas-phase luminescence of photofragments from M(S2COCHMe2)2. Chem Mater 9:1208–1212
Pradhan N, Katz B, Efrima S (2003) Synthesis of high-quality metal sulfide nanoparticles from alkyl xanthate single precursors in alkylamine solvents. J Phys Chem B 107:13843–13854
Alam N, Hill MS, Kociok-Köhn G, Zeller M, Mazhar M, Molloy KC (2008) Pyridine adducts of nickel(II) xanthates as single-source precursors for the aerosol-assisted chemical vapor deposition of nickel sulfide. Chem Mater 20:6157–6162
Barry L, Holmes JD, Otway DJ, Copley MP, Kazakova O, Morris MA (2010) Unusual magnetism in templated NiS nanoparticles. J Phys Condens Matter 22:076001
Akhtar M, Revaprasadu N, Malik MA, Raftery J (2015) Deposition of phase pure nickel sulfide thin films from bis(O-alkylxanthato)–nickel(II) complexes by the aerosol assisted chemical vapour deposition (AACVD) method. Mater Sci Semicond Process 30:368–375
Macreadie LK, Maynard-Casely HE, Batten SR, Turner DR, Chesman ASR (2015) Soluble xanthate compounds for the solution deposition of metal sulfide thin films. ChemPlusChem 80:107–118
Ghezelbash A, Sigman MB, Korgel BA (2004) Solventless synthesis of nickel sulfide nanorods and triangular nanoprisms. Nano Lett 4:537–542
Abboudi M, Mosset A (1994) Synthesis of d transition metal sulfides from amorphous dithiooxamide complexes. J Solid State Chem 109:70–73
Mgabi LP, Dladla BS, Malik MA, Garje SS, Akhtar J, Revaprasadu N (2014) Deposition of cobalt and nickel sulfide thin films from thio- and alkylthio-urea complexes as precursors via the aerosol assisted chemical vapour deposition technique. Thin Solid Films 564:51–57
Tian L, Yep LY, Ong TT, Yi J, Ding J, Vittal JJ (2009) Synthesis of NiS and MnS nanocrystals from the molecular precursors (TMEDA)M(SC{O}C6H5)2 (M=Ni, Mn). Cryst Growth Des 9:352–357
Abdelhady AL, Malik MA, O’Brien P, Tuna F (2012) Nickel and iron sulfide nanoparticles from thiobiurets. J Phys Chem C 116:2253–2259
Geng B, Liu X, Ma J, Du Q (2007) A new nonhydrolytic single-precursor approach to surfactant-capped nanocrystals of transition metal sulphides. Mater Sci Eng B 145:17–22
Sobhani A, Salavati-Niasari M (2013) Synthesis, characterization, optical and magnetic properties of a nickel sulfide series by three different methods. Superlattices Microstruct 59:1–12
Beal JHL, Etchegoin PG, Tilley RD (2010) Transition metal polysulfide complexes as single-source precursors for metal sulfide nanocrystals. J Phys Chem C 114:3817–3821
Babashkina MG, Safin DA, Garcia Y (2012) Influence of the coordination mode in [Ni{RC(S)NP(S)(OiPr)2}2] for the formation of nickel-containing nanoparticles. Dalton Trans 41:2234–2236
Yadav R, Singh AK, Waghadkar Y, Kociok-Köhn G, Kumar A, Chauhan R, Rane S, Gosavi S (2017) 1,2-Bis(diphenylphosphino)ethane nickel(II) O, O’-dialkyldithiophosphates as potential precursors for nickel sulfides. New J Chem 41:1327–1333
Rath T, Edler M, Haas W, Fischereder A, Moscher S, Schenk S, Trattnig R, Sezen M, Mauthner G, Pein A, Meischler D, Bartl K, Saf R, Bansal N, Haque SA, Hofer F, List EJW, Trimmel G (2011) A direct route towards polymer/copper indium sulfide nanocomposite solar cells. Adv Energy Mater 1:1046–1050
Kaltenhauser V, Rath T, Haas W, Torvisco A, Müller SK, Friedel B, Kunert B, Saf R, Hofer F, Trimmel G (2013) Bismuth sulphide–polymer nanocomposites from a highly soluble bismuth xanthate precursor. J Mater Chem C 1:7825–7832
MacLachlan AJ, Rath T, Cappel UB, Dowland SA, Amenitsch H, Knall A-C, Buchmaier C, Trimmel G, Nelson J, Haque SA (2015) Polymer/nanocrystal hybrid solar cells: influence of molecular precursor design on film nanomorphology, charge generation and device performance. Adv Funct Mater 25:409–420
Rath T, MacLachlan AJ, Brown MD, Haque SA (2015) Structural, optical and charge generation properties of chalcostibite and tetrahedrite copper antimony sulfide thin films prepared from metal xanthates. J Mater Chem A 3:24155–24162
Fischereder A, Schenk A, Rath T, Haas W, Delbos S, Gougaud C, Naghavi N, Pateter A, Saf R, Schenk D, Edler M, Bohnemann K, Reichmann A, Chernev B, Hofer F, Trimmel G (2013) Solution-processed copper zinc tin sulfide thin films from metal xanthate precursors. Monatsh Chem 144:273–283
Tejos M, Rolón BG, del Río R, Cabello G (2008) CdS amorphous thin films photochemical synthesis and optical characterization. Mater Sci Semicond Process 11:94–99
Rath T, Padeste C, Vockenhuber M, Fradler C, Edler M, Reichmann A, Letofsky-Papst I, Hofer F, Ekinci Y, Griesser T (2013) Direct extreme UV-lithographic conversion of metal xanthates into nanostructured metal sulfide layers for hybrid photovoltaics. J Mater Chem A 1:11135–11140
Buchmaier C, Rath T, Pirolt F, Knall A-C, Kaschnitz P, Glatter O, Wewerka K, Hofer F, Kunert B, Krenn K, Trimmel G (2016) Room temperature synthesis of CuInS2 nanocrystals. RSC Adv 6:106120–106129
Al-Shakban M, Xie Z, Savjani N, Malik MA, O’Brien P (2016) A facile method for the production of SnS thin films from melt reactions. J Mater Sci 51:6166–6172. doi:10.1007/s10853-016-9906-7
Lewis EA, McNaughter PD, Yin Z, Chen Y, Brent JR, Saah SA, Raftery J, Awudza JAM, Malik MA, O’Brien P, Haigh SJ (2015) In situ synthesis of PbS nanocrystals in polymer thin films from lead(II) xanthate and dithiocarbamate complexes: evidence for size and morphology control. Chem Mater 27:2127–2136
MacLachlan AJ, O’Mahony FTF, Sudlow AL, Hill MS, Molloy KC, Nelson J, Haque SA (2014) Solution-processed mesoscopic Bi2S3:polymer photoactive layers. ChemPhysChem 15:1019–1023
Gottlieb HE, Kotlyar V, Nudelman A (1997) NMR chemical shifts of common laboratory solvents as trace impurities. J Org Chem 62:7512–7515
Blessing RH (1995) An empirical correction for absorption anisotropy. Acta Cryst A 51:33–38
Sheldrick GM (1990) Phase annealing in SHELX-90: direct methods for larger structures. Acta Cryst A 46:467–473
Sheldrick GM (2015) SHELXT- Integrated space-group and crystal-structure determination. Acta Cryst A 71:3–8
Spek AL (2003) Single-crystal structure validation with the program PLATON. J Appl Cryst 36:7–13
Müller P, Herbst-Irmer R, Spek A, Schneider T, Sawaya M (2006) Crystal structure refinement: a crystallographer’s guide to SHELXL. Oxford University Press, Oxford, p 56–91
Allen FH (2002) The Cambridge structural database: a quarter of a million crystal structures and rising. Acta Cryst B 58:380–388
Cox MJ, Tiekink ERT (1999) The crystal and molecular structures of some nickel(ll)bis(O-alkyldithiocarbonate)s and nickel(II)bis(N, N-dialkyldithiocarbamate)s: an evaluation of the coordination potential of 1,1-dithiolate ligands in their nickel(II) complexes. Z Kristallogr 214:242–250
Husain A, Nami SAA, Singh SP, Oves M, Siddiqi KS (2011) Anagostic interactions, revisiting the crystal structure of nickel dithiocarbamate complex and its antibacterial and antifungal studies. Polyhedron 30:33–40
Singh SK, Drew MGB, Singh N (2013) Self assembly of homoleptic Ni(II) dithiocarbamates and dithiocarbimates via Ni···H–C anagostic and C-H···π (chelate) interactions. CrystEngComm 15:10255–10265
Singh V, Chauhan R, Gupta AN, Kumar V, Drew MGB, Bahadur L, Singh N (2014) Photosensitizing activity of ferrocenyl bearing Ni(II) and Cu(II) dithiocarbamates in dye sensitized TiO2 solar cells. Dalton Trans 43:4752–4761
Gupta AN, Kumar V, Singh V, Manar KK, Drew MGB, Singh N (2014) Intermolecular anagostic interactions in group 10 metal dithiocarbamates. CrystEngComm 16:9299–9307
Kumar V, Singh V, Gupta AN, Singh SK, Drew MGB, Singh N (2015) Cooperative influence of ligand frameworks in sustaining supramolecular architectures of Ni(II)/Pd(II) heteroleptic dithio-dipyrrin complexes via non-covalent interactions. Polyhedron 89:304–312
Yadav MK, Rajput G, Prasad LB, Drew MGB, Singh N (2015) Rare intermolecular M···H–C anagostic interactions in homoleptic Ni(II)–Pd(II) dithiocarbamate complexes. New J Chem 39:5493–5499
Sathiyaraj E, Selvaganapathi P, Thirumaran S, Ciattini S (2016) Synthesis, spectral, structural and computational studies on NiS4 and NiS2NP chromophores: anagostic and C–H···π (chelate) interactions in [Ni(dtc)(PPh3)(NCS)] (dtc=N-(2-phenylethyl)-N-(4-methoxybenzyl)- dithiocarbamate and N-(2-phenylethyl)-N-(4-chlorobenzyl)dithiocarbamate). J Mol Struct 1119:385–395
Brookhart M, Green MLH, Parkin G (2007) Agostic interactions in transition metal compounds. Proc Natl Acad Sci 104:6908–6914
Huynh HV, Wong LR, Ng PS (2008) Anagostic interactions and catalytic activities of sterically bulky benzannulated N-heterocyclic carbene complexes of nickel(II). Organometallics 27:2231–2237
Okamura H, Naitoh J, Nanba T, Matoba M, Nishioka M, Anzai S, Shimoyama I, Fukui K, Miura H, Nakagawa H, Nakagawa K, Kinoshita T (1999) Optical study of the metal–nonmetal transition in Ni1-δS. Solid State Commun 112:91–95
Pein A, Baghbanzadeh M, Rath T, Haas W, Maier E, Amenitsch H, Hofer F, Kappe CO, Trimmel G (2011) Investigation of the formation of CuInS2 nanoparticles by the oleylamine route: comparison of microwave-assisted and conventional syntheses. Inorg Chem 50:193–200
Mourdikoudis S, Liz-Marzán LM (2013) Oleylamine in nanoparticle synthesis. Chem Mater 25:1465–1476
Acknowledgements
Open access funding provided by Graz University of Technology. The authors thank Josefine Hobisch and Verena Perner for experimental support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Buchmaier, C., Glänzer, M., Torvisco, A. et al. Nickel sulfide thin films and nanocrystals synthesized from nickel xanthate precursors. J Mater Sci 52, 10898–10914 (2017). https://doi.org/10.1007/s10853-017-1265-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-017-1265-5