
Abstract
Novel lignin-chitosan polyelectrolyte fibres were produced through a reactive electrospinning process. Polyelectrolyte formation between the anionic lignin and cationic chitosan was controlled through the pH of the solution. Through manipulating the polyelectrolyte complex formation, fibres could be effectively produced from two biopolymers, which are normally very difficult to electrospin on their own. Though minimal amounts of the petroleum-derived polyethylene oxide were introduced into the solution to enhance the spinnability of the polyelectrolyte solution, it could be easily removed from the fibres post spinning by washing with water. Thus, pure biopolymer fibres could be produced. The optimum composition of lignin to chitosan was identified through SEM, FTIR and TGA analysis of the electrospun fibres. Fluorescence spectra of the electrospun fibres reveal the homogeneous distribution of lignin and chitosan components throughout the fibre network.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Carbon fibres have been studied extensively for their scientific and technological importance and are finding application in many fields including catalysis [1], composites [2–4], filtration [5–7], and alternative energy technologies [8]. Carbon fibres are advantageous due to being a light weight material with a high specific modulus and strength, good thermal/electrical conductivity and being structurally stable against various environmental changes [2, 9]. In recent years, the demand for carbon fibres has steadily increased and it is expected to continue to rise in the following years [10]. The highest demands for carbon fibres are in the automobile and aerospace industries as their light weight allows for improved fuel efficiencies [11]. However, carbon fibres also have potential applications in other areas such as electronics, which do not require as high of a quality of the fibres as in the aerospace industry. A recent advancement in carbon fibre technology is their nanofabrication. Various functional properties of the carbon fibres can be improved by reducing their diameter down to nanometer scale dimensions. The main challenge with establishing wider carbon fibre technologies is their higher cost; traditional carbon fibres are sourced from petroleum resource-based feedstocks, which tend to continually increase in price. Currently, the main precursor material used in carbon fibre production is the synthetic/petroleum-based polymer polyacrylonitrile (PAN) [9]. Some carbon fibres are also produced from petroleum pitch and rayon [9]. Depending on the precursor material used, the properties of the carbon fibre can vary. For example, pitch-based carbon fibres have greater thermal conductivity than rayon-based carbon fibres, and PAN-based carbon fibres possess higher compressive and tensile strengths compared to pitch-based carbon fibres [5]. In addition to the precursor material, the processing method is also very important in fabricating carbon fibres with desired properties.
One method of producing carbon fibre precursors, with the potential of commercial applicability, is electrospinning. It has previously been demonstrated that electrospinning can successfully produce precursor fibres that can be converted into high quality carbon fibres with controlled fibre diameters and morphologies [12–19]. The majority of electrospun carbon fibre precursors reported in the literature are PAN-based. The high cost of PAN [18], depleting petroleum resources and the toxicity of its solvent, dimethylformamide (DMF) [20], has motivated research to look into alternative electrospinnable materials to produce cheaper and more environmentally friendly carbon fibres. The fact that petroleum-based carbon resources exhibit negative environmental impacts and are of limited availability further motivates research towards green carbon fibres [21].
Recently, a wide range of renewable resource-based materials have been investigated for the fabrication of carbon materials. Among them, lignin has been looked at as a very promising candidate as a precursor for carbon materials due to its (i) carbon-rich phenolic chemical structure, (ii) chemical compatibility and (iii) abundance in nature. Lignin is a constituent of most plant materials and along with cellulose and hemicellulose, forms their structural basis [22]. The structure of lignin is very complex and unlike most polymers, it is a three-dimensional branched structure [23]. Different species of plants will contain lignin composed of different ratios of its three basic monomeric units: p-coumaryl alcohol, coniferyl alchohol and sinapyl alcohol [24, 25]. The greatest variation in the structure of lignin occurs as a result of the process of extracting lignin from the plant material, which may even add different functionalities to the lignin [24].
In the papermaking and cellulosic ethanol industries, lignin must be removed from the raw materials in a process known as delignification [26]. Due to this, lignin is massively produced as a co-product of these two industries. It is estimated that from the papermaking industry alone, 70 million tons are produced annually [26]. For the most part, this lignin is used as a low-energy density fuel by the factories [27] and 1–2 % of it is used for speciality products [24]. Thus, lignin is abundantly available and cheap. To establish lignin as an acceptable precursor material, it must be readily accessible from different sources. However, due to the variations in the properties and spinnability of the lignins based on their source and extraction methods, it is important to demonstrate the spinnability of a wide range of lignins, which may have different characteristics such as solubility and charge.
One of the main challenges associated with the production of lignin fibres through the electrospinning process are its low viscoelastic properties. A variety of work has demonstrated the benefits of blending lignin with small amounts of synthetic polymers such as polyethylene oxide (PEO) to improve the spinnability [28–31]. Recently, some work has emerged on the successful electrospinning of a variety of lignin-based fibres by utilising PEO [32]. Lignin has also been used to supplement PAN for carbon fibre precursor fibre production [19]. Pure lignin electrospinning has also been demonstrated through the use of co-axial electrospinning in which two concentric capillaries are used; the inner capillary containing the lignin solution while ethanol flowed from the outer capillary to stabilize the lignin jet [33, 34]. Different types of lignin are soluble in different solvents such as DMF [19, 32], ethanol [33, 34] and water [32]. Water would be the most environmentally friendly solvent to use due to its abundance and non-toxicity. However, the majority of lignin is water insoluble; the most well known water-soluble lignins being sulphur lignin (SL) and lignosulfunate (LS) [32]. In our previous study [35], we have used a water-soluble and sulphur-free anionic sodium carbonate lignin (SCL), which is obtained through a soda pulping process. In this study, PEO was used in order to enhance the spinnability of sodium carbonate lignin. However, the strong anionic nature of the SCL hindered it from continuous electrospinning.
In this paper, we attempt to neutralise the inherent negative charge of the lignin by combining SCL with another cationic polymer. Chitosan (CS), an abundant biopolymer derived from the naturally occurring chitin, is a cationic polymer and thus may be able to neutralise the anionic charges on the SCL. In addition, CS is soluble in acidic aqueous solutions, has been found to produce small diameter fibres with low levels of beading defects [36, 37] and has been demonstrated to be carbonizable [38–40]. Therefore, it would maintain the green nature and the desired morphology of a carbon fibre precursor. CS itself has been shown to be a difficult material to electrospin due to difficulties in creating solutions with a high enough polymer concentration and chain entanglement while still having a low enough viscosity to electrospin [41, 42]. However, as with lignin, chitosan has been successfully electrospun through blending with other easily electrospun polymers such as PEO [36, 37, 43, 44].
When a polyanion and polycation interact, it is well known that a structure known as a polyelectrolyte complex (PEC) can form. PECs are polymeric structures in which the constituent polyions are bound together by ionic linkages [45]. In general, PECs formed between two strong polyions will have a 1:1 ionic stoichiometry ratio [46]. However, if the constituent polyions are mixed in a non 1:1 ionic stoichiometry, the resultant PECs can be strongly overcharged rather than neutrally charged [46]. Thus, in the present study, we take advantage of the PEC forming ability of SCL and CS in order to improve their electrospinnability while reducing the PEO content. SCL, CS and PEO are blended in aqueous solutions of acetic acid and subsequently electrospun.
Experimental
Materials
SCL was purchased from Northway Lignin Chemical as Polybind 300 (liquid). Before use, the SCL was dried in a 100 °C laboratory oven until dry. The dry weight of the lignin was ~50 % of the as-received wet lignin weight. Medium molecular weight CS (viscosity average Mw 190–310 kDa, 75–85 % deacetylation) and 5,000,000 Mw PEO were purchased from Sigma-Aldrich and used as-received without further purification. Acetic acid (≥99.5 % pure) was purchased from Acros Organics. Deionized water was used as the primary solvent.
Method
Solution preparation
Various electrospinning solutions were prepared from a blend of SCL, CS and PEO. All polymer concentrations are reported as w/v % (g/ml). The solutions were prepared with 0.6 % PEO, 1.5 % chitosan and 1.5, 2.0, 2.5, or 3.0 % SCL. Initially, CS and PEO were dissolved in acetic acid and deionized water, while SCL was dissolved in deionized water. The two solutions were then mixed under constant stirring conditions to achieve the desired polymer concentrations in a 40(v/v) % acetic acid solution. The solutions are labelled as 1.5, 2.0, 2.5 and 3.0L based on their respective lignin content.
Electrospinning
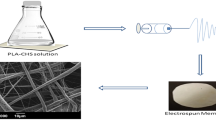
Electrospinning was performed in a NANON-01A electrospinning setup, MECC Co., Ltd. Japan. A custom-made plate collector, which allowed for a variation in the height of the collector (the collector, which came with the machine had a fixed height), was employed to achieve a greater working distance. Solutions were electrospun using a 24 gauge needle, 14 kV applied voltage, 0.1 ml/h flow rate and a 22.5 cm working distance in ambient conditions.
Characterization
Solution viscosities were measured using a Brookfield DV-II + Pro viscometer at room temperature with a #4 spindle at 100 rpm. Each measurement had an inherent 1 % error, and three independently prepared solutions were measured to calculate an average and standard deviation. Solution surface tensions were measured using the Du Nuoy ring method with the ring attached to a microbalance at room temperature where the ring was slowly pulled out of the solution, and the surface tension being recorded just before the ring broke contact with the solution surface. Each measurement had an inherent 5 % error, and three independently prepared solutions were measured to calculate an average and standard deviation. The resulting fibre morphologies were analysed using an FEI - Inspect S50 scanning electron microscope (SEM) operated at 15 kV. The fibres were sputter-coated with ~20 nm of gold to make them conductive. The obtained SEM images were further analysed using the ImageJ software to measure the fibre diameters using over 100 measurements for each formulation to obtain an average and standard deviation. A model TCS SP5 II laser scanning confocal microscope (LCM) (Leica Microsystems, Exton, PA) was used in the ‘xyλ’ mode to obtain fluorescence emission spectra of the resulting fibres and the constituent materials as well as to resolve the spatial distribution of the constituent materials within the fibres. Thermal characteristics of the fibres were measured using a TA instruments thermogravimetric analyser (TGA) Q500. TGA thermograms were obtained from 20 to 800 °C at a ramp rate of 10 °C/min. Heating took place in a nitrogen atmosphere injected at a flow rate of 15 ml/min. Structural characteristics of the fibres and constituent polymers were investigated using a Thermo Scientific Nicolet 6700 Fourier transform infrared (FTIR) spectrometer with a GladiATR single reflection ATR accessory operated between 400 and 4000 cm−1. The spectra were collected at a resolution of 4 cm−1 with 32 scans per sample at room temperature. Elemental analysis was performed on the fibres using a model S3400-N SEM (Hitachi Hitachi-Technologies, Pleasanton, CA) coupled to a Noran System 6 energy dispersive x-ray spectrometer (Thermo Electron Corp., Madison, WI). To confirm that the resultant fibres were composed of PECs, the fibres were soaked in water for at least a day and analysed through SEM, LCM, EDS, FTIR and TGA.
Result and discussion
Polyelectrolyte complex of sodium carbonate lignin and chitosan
Based on the polyionic nature of the SCL and CS, it was expected that they would form PECs with each other (Fig. 1). It was undesirable for them to form PECs in solution as it would cause clogging of the needle. It was observed that above an acetic acid concentration of 40 % in water, no precipitates formed in solution. This indicates that the pKa of the anionic groups on the SCL is about 4.3, below, which the carboxyl groups on the SCL are protonated and thus unable to form PECs. It was expected that during the electrospinning process, as solvent is evaporated from the solution, the pH rises above the pKa of the carboxyl groups on the SCL, thus allowing for PEC formation to occur.

Schematic of polyelectrolyte complexation between sodium carbonate lignin and chitosan. (Circled regions indicate the ionic bonds.)
Solution properties and their electrospinning behaviour
The properties of the various prepared solutions are shown in Fig. 2. It was found that the viscosity of the solutions dropped dramatically with increasing SCL content until a saturation point was reached. This is in agreement with our previous report [35] and is due to the low intrinsic viscosity of lignin [47]. Although the viscosity decreased dramatically, the high viscosity imparted by the chitosan still allowed for the solutions to be electrospinnable. The surface tension of the solutions was found to remain constant across the different formulations. This was expected based on the report by Geng et al. [48], which showed that in chitosan-based solutions, surface tension was greatly affected by the acetic acid concentration but not by polymer concentration.

Viscosity and surface tension properties of the various prepared solutions
All solutions were successfully electrospun using the applied electrospinning parameters. However, only the electrospinning jet of the 2.0L solution formed a stable region (region where the jet underwent no instabilities) whilst the 1.5, 2.5 and 3.0L solutions all had no stable region. In addition, of all the formulations, only the 2.0L solution deposited as a flat, dense fibre mat. The other formulations formed floating fibres (fibres that rise off from the plate collector towards the capillary caused by the deposited fibres having a high net charge density so that they repel each other and are attracted towards the applied voltage). Floating fibres are undesirable as they reduce the working distance available in which the electrospinning jet can elongate and solidify and also change the electric field. These effects cause the deposition of fibres with uneven fibre distributions, more beading defects and fibre bundling. The 1.5L and 2.5L fibres deposited as a loose, cotton-like mat. The morphology of the 1.5L mat was better than that of the 2.5L mat as the 2.5L mat formed some fibre bundles. The 3.0L mat was composed predominantly of fibre bundles. The 2.0L formulation (4:3 SCL:CS ratio) produced the best fibre mats and thus likely contained an almost stoichiometric charge ratio of SCL and CS functional groups. It may be expected that the use of CS with a higher degree of deacetylation would allow for optimised fibres with higher SCL:CS ratios to be produced due to the greater number of charged groups per CS molecule.
Characterization of electrospun fibres
In order to confirm the PEC nature of the electrospun fibres, the fibre mats were soaked in water for at least a day. This was to test if the fibre structure was retained after soaking as the predominant component, SCL, is water soluble. As PEO does not take place in PEC formation and is water soluble, it is also expected that PEO can be removed from the fibres through the water-soaking process. The fibres before and after being soaked in water were characterised for their morphological (SEM), chemical (EDS), thermal (TGA), spectroscopic (fluorescence) and structural (FTIR) properties.
SEM/EDS characterization
SEM micrographs of the four different fibre mats produced are shown in Fig. 3. The 2.0L fibres were found to possess the best fibre morphology out of the four samples. In the 2.0L fibres, beading was not observed while the 1.5L had some, the 2.5L more, and the 3.0L fibres were composed almost entirely of beads. The diameters of the 2.0L fibres were also much smaller and more uniform than the other fibres at 253 ± 77 nm. The 1.5L fibres possessed diameters of 1.92 ± 0.87 µm. The diameters of the 2.5L fibres were much too varied and the 3.0L fibres were too beaded to measure. The morphology of the 2.0L fibres was not perfect and showed some fusing between fibres indicating that the solvent did not completely evaporate upon deposition. However, for the desired application as carbon fibres, fused fibres may be beneficial as they provide greater electrical contact between the fibres and thus a greater electrical conductivity throughout the fibre mat. Thus, based on both the macroscale fibre mat morphologies and microscale fibre morphologies, the 2.0L formulation was optimal and was the focus of subsequent characterisations.

SEM images of a 1.5L, b 2.0L, c 2.5L and d 3.0L electrospun fibres
Figure 4a and b shows the SEM micrographs of the 2.0L fibres after being soaked in water (2.0L-W). The fibres can be observed to have retained their structure and be very similar in morphology to the as-spun 2.0L fibres with diameters of 248 ± 56 nm. Thus, the fibres appear stable in water; supporting their PEC composition. Figure 4c shows the EDS spectra of the 2.0L and 2.0L-W fibres. The N peak of chitosan did not show up in the EDS spectra due to the N x-ray fluorescence being quenched by O. It is clearly observed that while the samples contained sodium before being soaked in water, after soaking, there was no visible peak due to sodium (originating from the SCL). As is well established of PECs, upon forming, the couterions originally associated with the constituent polyions are excluded. Thus, through soaking the fibres in water, the free sodium ions could easily be washed out of the fibres. Removal of these sodium ions would lead to a more pure carbon fibre.

a,b SEM images of the 2.0L-W fibres. c EDS spectra of the 2.0L (solid line) and 2.0L-W (dotted line) fibres
Thermal properties
The TGA thermograms of the PEO, SCL and CS are shown in Fig. 5a and the 2.0L and 2.0L-W fibres are shown in Fig. 5b. The PEO and CS exhibit one-step degradations with the degradation of PEO being very rapid upon onset and leaving virtually no residue while the degradation of CS is more gradual and leaves some residue. SCL exhibits a three-step degradation over a wide temperature range, which is common of lignin due to differences in the thermal stability of oxygen-containing functional groups [49]. The first degradation is likely due to cleavage of the functional groups, while the second is likely due to rearrangement of the phenolic backbone [49]. The third degradation is not usually observed in lignins and may be due to volatilization of the sodium counterions present in SCL. The first degradation of SCL and CS are very close.

TGA thermograms of a PEO, SCL and CS b the 2.0L fibres and the 2.0L-W fibres
The 2.0L fibres, as well as the other fibres (not shown), exhibit a three-step degradation. The first degradation peak matches the first degradation peaks of SCL and CS. The second corresponds to that of PEO and the third to the third degradation of SCL. In comparing the degradation curve of the 2.0L-W fibres to the 2.0L fibres, it can be seen that the second degradation peak is greatly diminished while the third peak disappears. The diminishment of the second peak supports the removal of PEO from the fibres with water soaking. As the PEO used was of an extremely high molecular weight, it is expected that the smaller fractions diffused out of the fibres first and the larger fractions may require more time to diffuse out completely. The removal of PEO from the fibres is desirable as PEO has a low melting point (66–70 °C), which makes it an undesirable component in the thermal stabilization and carbonization process. The disappearance of the third peak supports that the peak was caused by the sodium ions as the EDS data revealed large amounts of sodium to be removed from the fibres upon water soaking. Though EDS only characterised the surface of the fibres, the disappearance of this TG peak indicates that the sodium was also removed from the bulk as well. The first peak also appears to have been slightly shifted to a higher onset of degradation indicating some enhanced thermal stability.
Fluorescence characterization
Lignin, due to its aromatic structure is well known to fluoresce; the fluorescence most likely arising from groups such as stilbene, biphenyl and phenylcoumarin [50]. In their pure forms, CS and PEO do not fluoresce. However, fluorescent impurities can exist in unpurified samples, which give them fluorescent peaks. The fluorescence emission spectra of PEO, SCL, CS, the 2.0L fibres and the 2.0L-W fibres are shown in Fig. 6a. Both CS and PEO share a peak emission at the wavelength of 544 nm; indicating the presence of a similar impurity. SCL has a broad peak emission between 583–598 nm, a shoulder at 558 nm and another slight shoulder at 647 nm. The 2.0L fibres before and after being soaked in water possess nearly identical emission spectra. The first peak occurs at 549 nm and is slightly shifted compared to the emission due to CS and PEO. The remaining emission peaks on the fibres are due to lignin. The peak at 563 nm matches one shoulder from SCL and is quite prominent. A peak and a shoulder appear at 583 nm and 598 nm, respectively, and arise from the extremes of the broad peak on SCL. The final shoulder occurs at 661 nm and is greatly shifted from the slight peak on lignin at 647 nm.

a Fluorescence spectra of PEO, CS, SCL, the 2.0L fibres and the 2.0L-W fibres. Topographical fluorescence microscopy images of the 2.0L fibre mat imaged between b 570–620 nm and c 520–560 nm. d Cross-sectional fluorescence microscopy image of the 2.0L fibre mat
The slight and large red-shift in the peaks at 549 nm and 647 nm, respectively, in the spectra of the 2.0L fibres compared to the spectra of the constituent materials is indicative of some conformational changes occurring in the polymers upon PEC formation and electrospinning. With a conformational change, the degree of π–π interactions between aromatic rings on the SCL can change [51]—a red-shift indicating closer packing of the polymers [52]. Thus, upon PEC formation between SCL and CS during the electrospinning process, the constituent polymers could be thought to pack much closer together than in their raw states. The linear structures of PEO and CS prevent much changes to their packing, and thus no shifts are visible in peaks corresponding to the fluorescence of either PEO or CS. SCL on the other hand is branched and through PEC formation, it would be expected to align somewhat with the linear CS chains, which is supported by the the large shift in one of the SCL peaks.
The fibres were imaged through confocal fluorescent microscopy in order to investigate the distribution of the component polymers throughout the fibres (Fig. 6b–d). Two fluorescence channels were used: one channel operated between 520–560 nm (coloured red) and another channel operated between 570–620 nm (coloured green) corresponding to the peak fluorescence emissions of CS and SCL, respectively. Topographical images of the fibre imaged using the SCL and CS peak emissions are shown in Fig. 6b and c, respectively. From these images, it can be observed that the two components are evenly distributed throughout the fibres. However, a cross- sectional image of a fibre mat (Fig. 6d) reveals some inhomogeneity in the polymer distribution through the fibre mat. The bottom of the fibre mat contains more CS while the top more SCL. This inhomogeneity may arise from the charges applied during the electrospinning process. Initially, the collector is negatively charged with respect to the needle, causing the positively charged CS to be more attracted to it then SCL, resulting in the deposition of PECs with a higher CS to SCL ratio. As the fibres continue to be deposited, an insulating layer develops over the collector; reducing the voltage bias and allowing more SCL to compose the deposited PEC fibres. Thus, during the deposition of the fibres, the PEC composition may vary slightly.
FTIR characterization
The FTIR spectra of PEO, CS, SCL and the 2.0L and 2.0L-W fibres are shown in Fig. 7. It can be observed that the spectrum of the 2.0L fibres is quite close to that of the SCL. No noticeable variation was observed between the FTIR spectra of the electrospun fibre mats produced from the various solutions (data not shown); as would be expected from simple variations in polymer composition. The effects of the PEO signal are minimal in the fibre spectrum as PEO is the smallest constituent of the fibre. The CS curve is also similar to and not as intense as the SCL signals and so are not very distinct in the 2.0L spectrum. Comparing the SCL curve to the 2.0L curve, some slight shifting is observed from 1570 and 1410 cm−1 in SCL to 1540 cm−1 and 1400 cm−1 in the fibres, respectively (C = O stretching of carbonyl groups, circled in Fig. 7). Thus, there is some change in the structure of SCL during PEC formation as expected.

FTIR spectra of a PEO, b CS, c SCL, d 2.0L fibres and e 2.0L-W fibres
A surprising result was the dramatic change in the spectra between the 2.0L and 2.0L-W fibres. The two peaks due to the C = O stretching of carbonyl groups disappear at 1540 and 1400 cm−1 and a new peak, unobserved in any of the constituent materials, appears at 1620 cm−1. Further studies will be required to elucidate the exact mechanism of this change but it can be speculated that the removal of the associated counterions and PEO from the fibres upon water soaking allowed the ionic PEC bonds to become much stronger. Thus, the stretching of the carbonyl groups could be largely extinguished, while the stretching of the aromatic diene bonds become much more prominent.
Thus, based on the increased thermal stability as seen from the TGA studies and the significant structural changes as seen from FTIR studies, it was revealed that soaking the PEC fibres in water significantly enhanced the stability of the SCL-CS-PEC structures.
Conclusion
Polyelectrolyte complex (PEC) fibres of SCL and CS were successfully produced by manipulating the rapid solvent evaporation that takes place during the electrospinning process to change the solution pH and induce PEC formation as the fibres are being formed. SCL-CS-PEO blend solutions with varying SCL contents were electrospun. The solution containing a 4:3 SCL:CS ratio (2.0L solution) produced the best fibre morphologies due to the charges on SCL and CS being stoichiometrically balanced. Through soaking the fibres in water, the PEC composition of the fibres was confirmed and the ability to remove PEO and sodium ions present in the fibres was demonstrated. Further, it was revealed that soaking the fibres in water increased the strength of the PEC bonds leading to an enhanced thermal stability. Thus, a novel method of producing pure-biopolymer electrospun fibres with green solvents was demonstrated. Future works are required to explore the thermal stabilization and carbonization of these PEC fibres.
References
Serp P (2003) Carbon nanotubes and nanofibers in catalysis. Appl Catal A 253:337
Chand S (2000) Carbon fibres for composites. J Mater Sci 35:1303
Park SB (1991) Experimental study on the engineering properties of carbon fiber reinforced cement composites. Cem Concr Res 21:589
Matsuo T (2008) Fibre materials for advanced technical textiles. Text Prog 40:87
Thiruvenkatachari R, Su S, An H, Yu XX (2009) Post combustion CO2 capture by carbon fibre monolithic adsorbents. Prog Energy Combust Sci 35:438
Heijman SGJ, Hopman R (1999) Activated carbon filtration in drinking water production: Model prediction and new concepts. Colloids Surf A 151:303
Martín-Gullón I, Font R (2001) Dynamic pesticide removal with activated carbon fibers. Water Res 35:516
Suarez-Garcia F, Vilaplana-Ortegoa E, Kunowskya M, Kimurab M, Oyac A, Linares- Solano A (2009) Activation of polymer blend carbon nanofibres by alkaline hydroxides and their hydrogen storage performances. Int J Hydrogen Energy 34:9141
Huang X (2009) Fabrication and properties of carbon fibers. Materials 2:2369
Roberts T (2006) The carbon fibre industry: Global strategic market evaluation 2006-2010. Materials Technology Publication
ORNL (2000) ORNL Review: Carbon-fiber composites for cars, vol 33. National Laboratory Review, Oak Ridge
Liu CK, Lai K, Liu W, Yao M, Sun RJ (2009) Preparation of carbon nanofibres through electrospinning and thermal treatment. Polym Int 58:1341
Pashaloo F, Bazgir S, Tamizifar M, Faghihisani M, Zakerifar S (2009) Preparation and characterization of carbon nanofibers via electrospun PAN nanofibers. Textile Science and Technology Journal 3:1
Moon S, Farris RJ (2009) Strong electrospun nanometer-diameter polyacrylonitrile carbon fiber yarns. Carbon 47:2829
Zhou Z, Lai C, Zhang L, Qian Y, Hou H, Reneker DH, Fong H (2009) Development of carbon nanofibers from aligned electrospun polyacrylonitrile nanofiber bundles and characterization of their microstructural, electrical, and mechanical properties. Polymer 50:2999
Zhang L, Hsieh YL (2009) Carbon nanofibers with nanoporosity and hollow channels from binary polyacrylonitrile systems. Eur Polym J 45:47
Kim C, Jeong YI, Ngoc BTN, Yang KS, Kojima M, Kim YA, Endo M, Lee JW (2007) Synthesis and characterization of porous carbon nanofibers with hollow cores through the thermal treatment of electrospun copolymeric nanofiber webs. Small 3:91
Inagaki M, Yang Y, Kang F (2012) Carbon nanofibers prepared via electrospinning. Adv Mater 24:2547
Seo DK, Jeun JP, Bin Kim H, Kang PH (2011) Preparation and characterization of the carbon nanofiber mat produced from electrospun pan/lignin precursors by electron beam irradiation. Rev Adv Mater Sci 28:31
Kennedy GJ, Sherman H (1986) Acute and subchronic toxicity of dimethylformamide and dimethylacetamide following various routes of administration. Drug Chem Toxicol 9:147
Hill J, Nelson E, Tilman D, Polasky S, Tiffany D (2006) Environmental, economic, and energetic costs and benefits of biodiesel and ethanol biofuels. PNAS 103:11206
Pouteau C, Baumberger S, Cathala B, Dole P (2004) Lignin–polymer blends: evaluation of compatibility by image analysis. CR Biol 327:935
Faulon JL, Hatcher PG (1994) Is there any order in the structure of lignin? Energy Fuels 8:402
Lora JH, Glasser WG (2002) Recent industrial applications of lignin: a sustainable alternative to nonrenewable materials. J Polym Environ 10:39
Whetten RW, MacKay JJ, Sederoff RR (1998) Recent advances in understanding lignin biosynthesis. Annu Rev Plant Physiol Plant Mol Biol 49:585
Satheesh Kumar MN, Mohanty AK, Erickson L, Misra M (2009) Lignin and its applications with polymers. J Biobased Mat Bioenergy 3:1
Luo J (2010) Lignin-based carbon fiber. MSc thesis. The University of Maine
Kubo S, Kadla JF (2004) Poly(ethylene oxide)/organosolv lignin blends: Relationship between thermal properties, chemical structure, and blend behavior. Macromolecules 37:6904
Kubo S, Kadla JF (2005) Kraft lignin/poly(ethylene oxide) blends: effect of lignin structure on miscibility and hydrogen bonding. J Appl Polym Sci 98:1437
Kubo S, Kadla JF (2006) Effect of poly(ethylene oxide) molecular mass on miscibility and hydrogen bonding with lignin. Holzforschung 60:245
Kadla JF, Kubo S (2004) Lignin-based polymer blends: analysis of intermolecular interactions in lignin–synthetic polymer blends. Compos A 35:395
Dallmeyer I, Ko F, Kadla JF (2010) Electrospinning of technical lignins for the production of fibrous networks. Wood Chem Technol 30:315
Ruiz-Rosas R, Bedia J, Lallave M, Loscertalesc IG, Barrero A, Rodríguez-Mirasol J, Cordero T (2010) The production of submicron diameter carbon fibers by the electrospinning of lignin. Carbon 48:696
Lallave M, Bedia J, Ruiz-Rosas R, Rodríguez-Mirasol J, Cordero T, Otero JC, Marquez M, Barrero A, Loscertales IG (2007) Filled and hollow carbon nanofibers by coaxial electrospinning of alcell lignin without binder polymers. Adv Mater 19:4292
Schreiber M, Vivekanandhan S, Mohanty AK, Misra M (2012) A study on the electrospinning behaviour and nanofibre morphology of anionically charged lignin. Adv Mat Lett 3:476
Zhang YZ, Su B, Ramakrishna S, Lim CT (2008) Chitosan nanofibers from an easily electrospinnable UHMWPEO-doped chitosan solution system. Biomacromolecules 9:136
Chang W, Ma G, Yang D, Su D, Song G, Nie J (2010) Electrospun ultrafine composite fibers from organic-soluble chitosan and poly(ethylene oxide). J Appl Polym Sci 117:2113
Bengisu M, Yilmaz E (2002) Oxidation and pyrolysis of chitosan as a route for carbon fiber derivation. Carbohydr Polym 50:165
Zawadzki J, Kaczmarek H (2010) Thermal treatment of chitosan in various conditions. Carbohydr Polym 80:394
Kaczmarek H, Zawadzki J (2010) Chitosan pyrolysis and adsorption properties of chitosan and its carbonizate. Carbohydr Res 345:941
Kriegel C, Arrechi A, Kit K, McClements DJ, Weiss J (2008) Fabrication, functionalization, and application of electrospun biopolymer nanofibers. Crit Rev Food Sci Nutr 48:775
Desai K, Kit K, Li J, Zivanovic S (2008) Morphological and surface properties of electrospun chitosan nanofibers. Biomacromolecules 9:1000
Kriegel C, Kit KM, McClements DJ, Weiss J (2009) Electrospinning of chitosan- poly(ethylene oxide) blend nanofibers in the presence of micellar surfactant solutions. Polymer 50:189
Ojha SS, Stevens DR, Hoffman TJ, Stano K, Klossner R, Scott MC, Krause W, Clarke LI, Gorga RE (2008) Fabrication and Characterization of Electrospun Chitosan Nanofibers Formed via Templating with Polyethylene Oxide. Biomacromolecules 9:2523
Michaels AS (1965) Polyelectrolyte complexes. Ind Eng Chem 57:32
Thünemann AF, Müller M, Dautzenberg H, Joanny JF, Löwen H (2004) Polyelectrolyte complexes. Adv Polym Sci 166:113
Goring D (1962) The physical chemistry of lignin. Pure Appl Chem 5:233
Geng X, Kwon O, Jang J (2005) Electrospinning of chitosan dissolved in concentrated acetic acid solution. Biomaterials 26:5427
Brebu M, Vasile C (2009) Thermal degradation of lignin – A review. Cellul Chem Technol 9:353
Machado AEH, Nicodem DE, Ruggiero R, da Perez SD, Castellan A (2001) The use of fluorescent probes in the characterization of lignin: the distribution, by energy, of fluorophores in Eucalyptus grandis lignin. J Photochem Photobiol A 138:253
Zhu Z, Zhang L, Smith S, Fong H, Sun Y, Gosztola D (2009) Fluorescence studies of electrospun MEH-PPV/PEO nanofibers. Synth Met 159:1454
Kong F, Wu XL, Yuan RK, Yang CZ, Siu GG, Chu PK (2006) Optical emission from the aggregated state in poly [2-methoxy-5-(2′-ethyl-hexyloxy)-p-phenylene vinylene]. J Vac Sci Technol, A 24:202
Acknowledgments
The financial support from the Natural Sciences and Engineering Research Council (NSERC), Canada for the Discovery grant individual to Manjusri Misra; NSERC NCE AUTO21; the Ontario Ministry of Agriculture, Food, and Rural Affairs (OMAFRA) New Directions and Alternative Renewable Fuels research program; and the Ontario Research Fund (ORF) Research Excellence (RE) Round-4 from the Ontario Ministry of Economic Development and Innovation (MEDI) to carry out this research is gratefully acknowledged. The Department of Physics is acknowledged for use of its SEM facilities. Some of the microscopic imaging was done with equipment supported by the NSF through an MRI Award # 0959817.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Schreiber, M., Vivekanandhan, S., Cooke, P. et al. Electrospun green fibres from lignin and chitosan: a novel polycomplexation process for the production of lignin-based fibres. J Mater Sci 49, 7949–7958 (2014). https://doi.org/10.1007/s10853-014-8481-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-014-8481-z