
Abstract
Three new dinuclear triple helicates were synthesised using a ditopic semi-rigid pyridylylimine ligand L, separated by a diphenoxy-biphenol spacer providing considerable length to the backbone. L and the new large dinuclear triple helicate complexes [Fe2L3](BF4)4 (1), [Ni2L3](BF4)4 (2) and [Zn2L3](BF4)4 (3) have been characterised in solution and solid state. Single crystal X-ray diffraction was used to investigate overall complex ion shape as the coordination sphere was modulated by metal ion selection. Small differences in complex shape were seen to arise due to subtle distortions in coordination sphere environments. This study sheds light on how the length and twist of dinuclear triple helicates can be tuned by selection of coordinating metal ion.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Metal-directed self-assembly of complex supramolecular architectures remains an area of insatiable research interest [1,2,3,4,5]. Of particular interest is the dinuclear triple helicate, whose synthesis is made possible by design of suitable bis-bidentate ligand backbones [1,2,3,4,5,6,7,8,9,10,11,12,13,14]. These ligands must be tailored to provide suitable donor atoms to allow stable coordination environments with metal ions, while containing the delicate balance of required rigidity to undergo the conformational change necessary to achieve a helical assembly [1, 8,9,10,11, 15, 16]. The intrinsic nature in which these complexes assemble makes them good candidates to bind with DNA, so they are therefore interesting targets for molecular design [17,18,19]. Designable molecular assemblies that possess the ability to bind to DNA with recognition could give rise to a new class of therapeutic drugs [18, 20, 21].
As an extension to our previous work [6,7,8,9,10,11, 14] on dinuclear triple helicates we have designed three new dinuclear triple helicate assemblies. The ligand L (scheme 1) was achieved by incorporating the same long semi-rigid ligand backbone as our previous helicate [8], this time incorporating a pyridyl functional moiety that gives rise to a stronger ligand field. This study investigates the impact that coordination sphere size has on the shape and size of these assemblies, brought about by the changing of metal ions. The logic was to study the effect of three d-block metals with increasing coordination sphere size and distortion. For this, FeII, NiII and ZnII were selected, a d6, d8 and d10 electron configuration. With a strong-field ligand, these metals bear t2g6 occupancy with eg0, eg2 and eg4 configurations, respectively. By this method, we investigate the effect that d-orbital occupancy and coordination sphere size has on the shape and size of these assemblies. By investigating the effects that small changes have on the overall shape and size of these architectures, it may be possible to modulate the supramolecular contacts which they can form. This could result in interesting properties such as tuneable DNA binding affinity and varied crystal packing arrangements.

Synthesis of L from Schiff-base condensation reaction of 4,4′-(1,1′-Biphenyl-4,4′-diyldioxy)dianiline and 2-pyridinecarboxaldehyde in excess to ensure reaction completion
Here we describe three new [M2L3](BF4)4 helicates; [Fe2L3](BF4)4 (1), [Ni2L3](BF4)4 (2) and [Zn2L3](BF4)4 (3), have been characterised by SCXRD, HR ESI-MS, TGA-DSC, FT-IR and SEM-EDS. We also report structural characteristics for these new assemblies and relate them to the differences in their coordination sphere size. These structural characteristics were compared to those of our previously reported fully low spin (LS) FeII dinuclear triple helicate which employed 4-thiazolylimine chelating moieties. This work represents an important progression towards achieving new long dinuclear triple helicates, presenting a similar ligand bearing a stronger ligand field.
Experimental
Materials and instrumentation
All chemicals and reagents were purchased from commercial sources and used without further purification.
High-resolution electrospray ionisation–mass spectrometry (HR ESI-MS) data was obtained using a Waters Xevo QToF mass spectrometer, operating in positive ion mode. Samples were dissolved with acetonitrile (MeCN) and directly injected via syringe into the ESI source. Calibration of high-resolution masses was achieved using a Waters lock-spray system.
Fourier transform infrared (FT-IR) measurements were undertaken on a Bruker Vertex 70 with a diamond ATR crystal. Spectra were recorded over a 400–4000 cm− 1 range.
Thermogravimetric Analysis (TGA) and Differential Scanning Calorimetry (DSC) experiments were conducted using a NETZSCH STA449 Jupiter Simultaneous Thermal Analysis (STA) instrument. TGA measurements were conducted using Nitrogen for both the purge and protective gases; an aluminium crucible and a temperature range of 300–550 K at a rate of 10 K min− 1.
Scanning Electron Microscopy and Energy Dispersive X-ray Spectroscopy (SEM-EDS) analysis were carried out using an SEM JEOL JSM 6510LV with an attached silicon drift EDS detector and operated in low-vac with a chamber pressure of 30 Pa. The accelerating voltage was set to 25.0 kV. All samples were mounted to an aluminium stub with double-sided conductive carbon tape. Images were taken without surface coating.
Nuclear Magnetic Resonance (NMR) experiments were carried out on a Bruker Advance 400 MHz NMR Spectrometer.
Crystallographic data collection and refinement
Single Crystal X-ray Diffraction (SCXRD) experiments for complexes1–3 were carried out on a Bruker D8 Quest Single Crystal diffractometer with Photon II detector using a IµS 3.0 Microfocus source with Mo-Kα radiation (λ = 0.710723 Å) at the Mark Wainwright Analytical Centre at the University of New South Wales (UNSW). Data integration and reduction was undertaken using Bruker APEX 3 software. For all data, absorption corrections were applied using the multi scan method in SADABS [22]. The structures were solved by intrinsic phasing and the full-matrix least-squares refinements were carried out using a suite of SHELX programs [23, 24] via the OLEX2 graphical interface [25]. Non-hydrogen atoms were refined anisotropically except for some disordered anions and solvent molecules. Carbon-bound hydrogen atoms were included in idealised positions and refined using a riding model. In all structures, a solvent mask was applied to account for residual low intensity peaks, arising due to disordered solvent.
All crystallographic data in CIF format has been deposited at the Cambridge Crystallographic Data Centre with CCDC No: 2,329,598–2,329,600. It is available free of charge from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1 EZ, UK; fax: (+ 44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk. Specific refinement details and crystallographic data for each structure are present below in the crystallographic section.
Synthesis
Preparation of ligand (L)
To a stirring white suspension of 4,4′-(1,1′-Biphenyl-4,4′-diyldioxy)dianiline (1,000 mg, 2.71 mmol) in 25 mL of methanol (MeOH), 2-pyridinecarboxaldehyde (1,453 mg, 13.56 mmol) diluted in 10 mL of MeOH was added dropwise. Upon addition the suspension turned yellow. The reaction was allowed to reflux under nitrogen atmosphere overnight. The resultant creamy yellow precipitate was filtered while hot, the product was then washed with cold methanol and allowed to air dry, yielding the final product as a pale-yellow powder. Yield = 88%. HR ESI-MS, (positive ion detection, MeCN, m/z): calc. for [L + H]+, 547.2134; found 547.2081; FT-IR (ATR, νmax/cm− 1): 1580, 1567, 1496, 1433, 1268, 1252, 850, 826; 1H NMR (400 MHz, DMSO-d6) δ 8.72 (dd, J = 7.5, 1.6 Hz, 2 H, Ar-H), 8.65 (s, 2 H, N = C-H of Imine), 8.16 (dd, J = 7.5, 1.7 Hz, 2 H, Ar-H), 7.96 (td, J = 7.4, 1.5 Hz, 2 H, Ar-H), 7.70 (d, J = 8.5 Hz, 4 H, Ar-H), 7.53 (td, J = 7.4, 1.6 Hz, 2 H, Ar-H), 7.46 (d, J = 8.7 Hz, 4 H, Ar-H), 7.20–7.09 (m, 8 H, Ar-H).
Preparation of complexes 1–3
To a stirring suspension of L (200 mg, 0.36 mmol) in 20 mL of acetonitrile (MeCN), a solution of iron(II) tetrafluoroborate (82 mg, 0.24 mmol) in 5 mL of MeCN was added dropwise. The resulting deep purple solution was heated and reacted under a nitrogen atmosphere for 1–2 h. The resulting reaction mixture was filtered while hot, X-ray quality crystals were obtained by vapour diffusion of diethyl ether into the reaction mixture. Diffusion yielded large deep purple rod crystals, which were obtained by filtration, washed with diethyl ether, and dried in air. Yield = 44%. HR ESI-MS, (positive ion detection, MeCN, m/z): calc. for [Fe2L3]4+, 437.6229; found 437.5878. FT-IR (ATR, νmax/cm− 1): 1591, 1489, 1230, 1201, 1167, 1061.
To a stirring suspension of L (100 mg, 0.18 mmol) in 15 mL of MeCN, a solution of nickel(II) tetrafluoroborate (42 mg, 0.12 mmol) in 5 mL of MeCN was added dropwise. The resulting clear orange solution was heated and reacted under a nitrogen atmosphere for 1–2 h. The resulting reaction mixture was filtered while hot, X-ray quality crystals were obtained by vapour diffusion of diethyl ether into the reaction mixture. Diffusion yielded dark orange plate crystals, which were obtained by filtration, washed with diethyl ether, and dried in air. Crystals were not air stable. Yield = 29%. HR ESI-MS, (positive ion detection, MeCN, m/z): calc. for [Ni2L3]4+, 439.0992; found 439.1219; FT-IR (ATR, νmax/cm− 1): 1595, 1489, 1237, 1200, 1060.
To a stirring suspension of [L] (200 mg, 0.36 mmol) in 15 mL of MeCN, a solution of zinc(II) tetrafluoroborate (58 mg, 0.24 mmol) in 5 mL of MeCN was added dropwise. The resulting pale-yellow solution was heated and reacted under a nitrogen atmosphere for 1–2 h. The solution was filtered while hot, the remaining solvent was evaporated off under vacuum, to obtain a bright yellow precipitate. Yield = 64%. HR ESI-MS, (positive ion detection, MeCN, m/z): cald. for [Zn2L3]4+, 442.1187; found 442.0901; FT-IR (ATR, νmax/cm− 1): 1596, 1489, 1242, 1061. X-ray quality crystals were obtained by redissolving the crude product in MeCN, followed by vapour diffusion of diethyl ether into the solution, yielding clear yellow plate crystals. Crystals were not air stable.
Results and discussion
Synthesis and characterisation
The synthesis of L (Scheme 1) involves a Schiff-base condensation reaction using a 1:5 stoichiometric ratio of 4,4′-(1,1′-Biphenyl-4,4′-diyldioxy)dianiline and 2-pyridine carboxaldehyde in a methanol solution performed under inert conditions while the reaction mixture is refluxed overnight, adapted from previously reported methods [26]. Complexes 1–3 were all synthesised in a 3:2 stoichiometric ligand-to-metal (L:M) ratio. All reactions were performed under an inert atmosphere with MeCN as the solvent. Crystallisations were performed by slow diffusion of diethyl ether into the reaction mixture to yield X-ray quality crystals. Dark purple, dark orange, and clear yellow crystals of 1–3, respectively, were obtained in moderate-low yields.
High-resolution electrospray ionisation-mass spectrometry (HR ESI-MS) provided indication that the ligand L persists in solution (Fig. S1.1). The experimental m/z obtained for L, [L + H]+ = 547.2081 exp, is in excellent agreement with the calculated m/z = 549.2134 (inset Fig. S1.1). Fig. S1.2-1.4, show the quadruply charged complexes 1, 2 and 3, [Fe2L3]4+, [Ni2L3]4+, [Zn2L3]4+, respectively, confirming the complex persists in the solution state. The experimental isotopic distribution patterns for complexes 1–3 are in excellent agreement with the calculated patterns. Spectra confirm all complexes persist in a stoichiometric 2:3 ratio in the form [M2L3]4+ which also confers with crystal data.
FT-IR spectra for ligand L and complexes 1–3, [Fe2L3](BF4)4, [Ni2L3](BF4)4 and [Zn2L3](BF4) were all recorded at room temperature and are shown in Fig. S2.1-2.4, respectively. The FT-IR spectrum for L shows absorptions in the region 1580 cm− 1 that is attributable to imine (C = N) stretching modes (Fig. S2.1). Similarly, the absorptions in the region 1591–1595 cm− 1 for complexes 1–3 (Fig.S2.2-2.4) are all consistent with each other, exhibiting characteristic imine (C = N) stretching modes.
TGA-DSC experiments were performed on complex 1–3 after drying the samples at room temperature (Fig. S3.1-3.3). Heating these samples showed the complexes did not decompose by the experimental upper limit of 277 °C. A small amount of solvent loss is indicated in samples 1–3 of 3.43%, 1.79% and 3.01%, respectively.
Scanning electron microscopy (SEM) micrographs (Fig. S4.1-4.3) indicated that complexes 1 and 2 exist as large single crystals. Complex 3 analysis was performed on the crude powder product. Additionally, energy dispersive X-ray spectroscopy (EDS) analysis confirmed the presence of C, N, O, F and the intended metal Fe, Ni or Zn, respectively for complexes 1–3.
The 1H NMR spectrum obtained for L demonstrates the purity of the ligand L (S5).
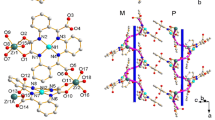
Crystal structures of compounds 1–3 were obtained by means of SCXRD experiments. All three lattices were isostructural, crystallising in the Monoclinic space group C2/c, with consistent lattice parameters as shown in Table S6. For all compounds, the asymmetric unit consisted of a half of a dinuclear triple helicate, with one crystallographically independent metal centre, one full ligand arm (ligand A) and one half-ligand arm (ligand B) (Fig. 1). Twofold symmetry generates a second instance of ligand A, as well as the second half of ligand B. Inversion and glide symmetry generates the opposite optical conformer, meaning that both the ΔΔ and ΛΛ helicate conformers are present in the structure. This means that each metal ion in the helicate assembly is chelated by three distinct groups, denoted Ai, Af and B (Fig. 1). Additionally, two BF4− anions and three (structures 1 and 2) or one (structure 3) acetonitrile molecules were found per asymmetric unit. In all cases, a solvent mask was applied to account for low intensity peaks, arising due to disordered solvent.

Asymmetric unit of dinuclear triple helicates from SCXRD experiments. One asymmetric unit is shown in bold colours: Metal ion (orange), ligand A (red) and ligand B (blue), and symmetry equivalents shown in lighter colours: Metal ion (pale yellow), ligand A (pink) and ligand B (light blue). The distinct chelate groups are highlighted by labels, each metal in the dinuclear triple helicate bears three distinct chelate groups, denoted Ai, Af and B. Figure was generated from the structure of 1 and is representative of structures 1–3
The crystal packing observed in 1–3 has been represented in Fig. 2. The crystal lattice is stabilised by intermolecular contacts between adjacent helicate units of opposite optical confirmation. When viewed along the [1,0,1] vector, 1D stacks of helicate units become apparent (Fig. 2b). These stacks feature both ΔΔ and ΛΛ conformers, interacting via π…π\(\pi \cdots \pi\)contacts in ligand B (Fig. 2c). Further, \(\pi \cdots \pi\) contacts between the ligand A arm of enantiomeric helicates interlinks 1D stacks together, manifesting the 3D network (Fig. 2d).

Crystal packing diagrams representing the presently reported compounds 1–3 (generated using 1). Hydrogen atoms have been omitted for clarity. The ΔΔ and ΛΛ conformers have been coloured red and blue, respectively. (a) The unit cell of 1–3 as viewed along the c-axis. Fragments at the cell boundaries have been completed to better represent the arrangement of helicate units, (b) Crystal packing viewed along the [1,0,1] direction, highlighting homochiral 1D helicate stacks (green box), interacting via ligand B, (c) Crystal packing as viewed along the c-axis. \(\pi \cdots \pi\) contacts between opposite conformers via ligand B are highlighted by green dashed lines, (d) Crystal packing diagram viewed along the b-axis. π…π\(\pi \cdots \pi\)contacts between opposite conformers via ligand A are highlighted by green dashed lines
Structural analysis and comparison of helicate shape
In the following section we compare the coordination environments of each of the presently reported helicate species 1–3, as well as the overall helical shape, as described by a few selected geometric parameters. Comparison will be made to the previously published [LS-LS] structure of the FeII thiazolylimine analogue, measured at 100 K [8]. We point out that the crystal packing differs, bearing an orthorhombic lattice in Pna21 symmetry. In this structure, one full dinuclear triple helicate unit is found per asymmetric unit.
Complex 1 exhibits octahedral distortion parameters consistent with the fully low spin conformation of the [LS-LS] FeII thiazolylimine helicate. The fully LS spin configuration of the presently reported FeII helicate is to be expected, considering the ligand field strength of 2-pyridylylimine coordinating moieties compared to those of 4-thiazolylimine coordinating moieties.
The coordination spheres, as described by the octahedral distortion parameters, bear a trend that might be expected with respect to the variation of population of the eg orbitals. As the population of the eg orbitals increases, the average coordinate bond length (D) increases (Table 1). The shortest D value was measured for the presently reported FeII helicate 1, while the longest was found in the ZnII analogue 3 (octahedron size). With the increase in eg orbital population and lengthening of coordinate bonds comes increased distortion of the octahedral cis angles, and projected twist angles, as demonstrated by Σ and Θ, respectively.
The change in coordination spheres brings an associated change to the overall shape of the helicate. This is qualitatively established by the measurement of the intramolecular intermetallic distance, as well as the helical twist, as demonstrated by the torsion angle between opposite chelating pyridyl nitrogen donors, averaged over the three different ligand arms Fig. S7.1.
Firstly, when comparing the presently reported compounds 1–3, a small difference in the intermetallic distance was measured, with the shortest intermetallic distance of 18.624 Å found in 1, and the longest distance of 18.801 Å observed for 3 (Table 2). With the increasing intermetallic distance, an associated decrease of the measured torsion angle was found, with this angle decreasing from 294.9° to 283.2° across the 1–3 series. This implies that the lengthening or contraction of these dinuclear triple helicate architectures is tied to the degree of ligand twist about the helical axis, with higher twist leading to shorter helical length, and vice versa. Further demonstrating this association, the previously reported LS FeII thiazolylimine structure [8], for which the measured intermetallic length and torsion angles were 19.322 Å and 273.7°, respectively, is the longest helicate among those compared herein with the lowest degree of twist.
Another key description regarding the shape of the helicate units can be made by quantifying the spread of the linking ligands. This may be achieved in various ways depending on the system. Here, we have opted to measure the average axis…O radius. We define this quantity as the average perpendicular distance from the intermetallic axis to the oxygen atoms in the ligand bridging sections (Fig. S8.1). The reported value gives some indication as to the spread of the helicate body, with respect to the bridging oxygen atoms.
In comparing these values, we see that in the presently reported series 1–3, there are only small variations when changing the identity of the coordinating metal ion. The shortest average radius of 4.246 Å is measured for 1, with the longest axis…O radius of 4.265 Å found for the NiII analogue 2, though the difference in average radius between NiII and ZnII analogues is negligible. A more significant difference is observed in our previously reported FeII thiazolylimine helicate, yielding an average axis…O radius of 4.197. The more compact spread of the body of this thiazolylimine helicate aligns well with the longer helical axis and the lesser degree of helical twist since the linker groups must approach the helical axis more closely in this case.
These generalised descriptions of the shape of each dinuclear triple helicate unit can be related back to subtle differences in the conformations of each coordination sphere. In an effort to convey this, we employ our previously described methodology for calculating the angles between chelate vectors and the intermetallic axis [6, 14]. For this analysis, we use the definition of the chelate vector as being the interval connecting the two coordinating atoms on one chelating moiety. This angle can be measured directly from the coordinate set of the crystal structure in three dimensions giving what we describe as the 3D chelate angle (Fig. S9.1). Additionally, the same angle can be measured in two orthogonal two-dimensional projections, yielding the values corresponding to the pitch and yaw of the chelate group (Fig. S9.2). These quantities respectively measure the back-and-forth rocking, and the side-to-side twisting of the chelate group, respectively. For full details see the Supporting Information S9 and our previously reported descriptions [6, 14].
Across the series of 1–3, the helicate units have been shown to lengthen, while the degree of twist in ligand components is seen to lessen. When probed in terms of the 3D chelate angle (Fig. 3a), it can be shown that these descriptions of the overall shape of helicates can be linked to realignment of the chelate vectors towards co-linearity with the intermetallic axis. As the pitch values across the series remain quite consistent (Fig. 3b), this trend arises primarily from the generally declining yaw values of the three distinct chelate groups (Fig. 3c).
As an interesting point of contrast, the long intermetallic length and low torsion value of our previously reported [LS-LS] FeII thiazolylimine helicate, which bore the longest intermetallic distance, can be rationalised in terms of the chelate vector alignments (see Fig. S10.1 for labelling of ligands in this structure). The 3D chelate angle was lower in two of the three chelate groups (designated ligand Ct and Bt in the associated .cif), with respect to 1, while a slightly higher value was measured in the two chelate groups of ligand At. This feature is mirrored by the yaw plot (Fig. 3c), though the associated pitch value is much lower in one of the chelate groups, bearing a value of ~ 2.8°, compared to the other measured values of 8°. One key contribution to the high intermetallic distance in the FeII thiazolylimine analogue would be the markedly lower pitch magnitude in several chelate groups, which would serve to push the metal ions away from the helicate centre.

Angles describing the orientations of chelate vectors with respect to the intermetallic axis. Colour scheme for 1–3: Ai - red solid squares, Af - red solid triangles, B - blue solid squares). Colour scheme for 4-thiazolylimine chelate rings (Fe1 Ct - red half filled squares, Fe2 Ct - red half filled triangles, Fe1 Bt - red hollow squares, Fe2 Bt - red hollow triangles, Fe1 At - blue half filled squares, Fe2 At - blue half filled triangles. a) Angle between the chelate vector and the intermetallic axis, measured in three dimensions, b) Pitch of chelate rings, c) Yaw of chelate rings. Areas marked with an asterisk (*) indicate multiple overlapping points. See S9 for full descriptions and calculated values
By this analysis we demonstrate that the distortion of the octahedral coordination sphere has a direct impact on the shape of the dinuclear triple helicate architecture. Subtle angular differences between the intermetallic axis and chelate vectors are correlated with slight changes to the shape of the overall complex ion (Fig. 4). Of course, larger coordination spheres contribute to the helicate length and twist, but these are not the only factor, as demonstrated by the analysis of the [LS-LS] FeII thiazolylimine helicate. There would be some contribution by the crystal packing on the helicate shape, which would act to balance the values of the chelate group angles. This is demonstrated by the relatively stable shape parameters in the presently reported series 1–3, as compared to the values established with the [LS-LS] FeII thiazolylimine dinuclear triple helicate.

Overlays of helicate units from SCXRD experiments. (a) The three structurally similar helicates of the presently reported series 1–3, coloured red (1), blue (2) and green (3), (b) The two structurally dissimilar Fe(II) helicates discussed in this study. 1 is shown in darker colours, while the 4-thaizolyl helicate is shown in lighter colours. Colour scheme for 1: Ligand A (red), symmetry equivalent ligand A (blue), ligand B (green). Colour scheme for 4-thiazolylimine analogue: Ligand C (pink), ligand B (light blue), ligand A (light green). Note that the disordered second position of ligand C has been omitted for clarity
Conclusion
We have presented three new species of dinuclear triple helicate, employing bis-bidentate pyridylylimine coordination moieties separated by a long aromatic spacer. Geometric analysis of crystal structures revealed that across the presently reported series of 1–3, higher eg orbital population led to lengthened intermetallic distances, along with decreased helical twist, as quantified by the average torsion angle. Angles describing the orientation of chelate groups with respect to the intermetallic axis were implemented, revealing that the increased length and lowered twist are associated with lowered yaw values. Comparison to our previously reported FeII thiazolylimine analogue showed that the longer helicate length in that instance was primarily contributed by low pitch angles in several chelate groups. This study has demonstrated how d orbital occupancy can be exploited to tune the shape of dinuclear triple helicates, which may allow for the targeted binding of helicates, or variation of solid state packing arrangements.
References
Albrecht, M., Blau, O., Fröhlich, R.: Size-selectivity in the template-directed assembly of dinuclear triple-stranded helicates. Proc. Natl. Acad. Sci. U S A. 99(8), 4867–4872 (2002). https://doi.org/10.1073/pnas.062600799
Miyake, H., Sugimoto, H., Tamiaki, H., Tsukube, H.: Dynamic helicity inversion in an octahedral cobalt(ii) complex system via solvato-diastereomerism. Chem. Comm. 34, 4291–4293 (2005). https://doi.org/10.1039/B506130J
Piguet, C., Bernardinelli, G., Hopfgartner, G.: Helicates as Versatile Supramolecular complexes. Chem. Rev. 97(6), 2005–2062 (1997). https://doi.org/10.1021/cr960053s
Glasson, C.R.K., Meehan, G.V., Motti, C.A., Clegg, J.K., Turner, P., Jensen, P., Lindoy, L.F.: Nickel(ii) and iron(ii) triple helicates assembled from expanded quaterpyridines incorporating flexible linkages. Dalton Trans. 40(45), 12153–12159 (2011). https://doi.org/10.1039/C1DT10820D
Chinnaraja, E., Arunachalam, R., Suresh, E., Sen, S.K., Natarajan, R., Subramanian, P.: Binuclear double-stranded helicates and their catalytic applications in desymmetrization of Mesodiols. Inorg. Chem. 58(7), 4465–4479 (2019). https://doi.org/10.1021/acs.inorgchem.8b03643
Wallis, M.J., Craze, A.R., Zenno, H., Tokunaga, R., Taira, T., Min, H., Bhadbhade, M.M., Bhattacharyya, S.K., Tian, R., Rich, A.M., Hayami, S., Clegg, J.K., Marjo, C.E., Lindoy, L.F., Li, F.: Unique spin crossover pathways differentiated by scan rate in a new dinuclear fe(ii) triple helicate: Mechanistic deductions enabled by synchrotron radiation studies. J. Mater. Chem. C. 11(26), 8908–8918 (2023). https://doi.org/10.1039/D2TC04998H
Craze, A.R., Bhadbhade, M.M., Komatsumaru, Y., Marjo, C.E., Hayami, S., Li, F.: A rare example of a complete, incomplete, and Non-occurring spin transition in a [Fe2L3]X4 Series Driven by a combination of Solvent-and halide-anion-mediated steric factors. Inorg. Chem. 59(2), 1274–1283 (2020). https://doi.org/10.1021/acs.inorgchem.9b02995
Howard-Smith, K.J., Craze, A.R., Zenno, H., Yagyu, J., Hayami, S., Li, F.: A large dinuclear fe(ii) triple helicate demonstrating a two-step spin crossover. Chem. Comm. 56(62), 8838–8841 (2020). https://doi.org/10.1039/D0CC03708G
Li, L., Craze, A.R., Akiyoshi, R., Tsukiashi, A., Hayami, S., Mustonen, O., Bhadbhade, M.M., Bhattacharyya, S., Marjo, C.E., Wang, Y., Lindoy, L.F., Aldrich-Wright, J.R., Li, F.: Direct monitoring of spin transitions in a dinuclear triple-stranded helicate iron(ii) complex through X-ray photoelectron spectroscopy. Dalton Trans. 47(8), 2543–2548 (2018). https://doi.org/10.1039/C7DT04190J
Craze, A.R., Sciortino, N.F., Badbhade, M.M., Kepert, C.J., Marjo, C.E., Li, F.: Investigation of the spin crossover properties of three Dinulear Fe(II) Triple Helicates by Variation of the Steric Nature of the ligand type. Inorganics. 5(4), 62 (2017). https://doi.org/10.3390/inorganics5040062
Craze, A.R., Zenno, H., Pfrunder, M.C., McMurtrie, J.C., Hayami, S., Clegg, J.K., Li, F.: Supramolecular modulation of spin crossover in an fe(II) Dinuclear Triple Helicate. Inorg. Chem. 60(9), 6731–6738 (2021). https://doi.org/10.1021/acs.inorgchem.1c00553
Hannon, M.J., Childs, L.J.: Helices and helicates: Beautiful supramolecular motifs with emerging applications. Supramol Chem. 16(1), 7–22 (2004). https://doi.org/10.1080/10610270310001632386
Gusev, A.N., Shul’gin, V.F., Riush, I.O., Lyssenko, K.A., Eremenko, I.L., Linert, W.: A new family of Co(II), ni(II), Fe(II) triple helicate systems on 5,5′-di(pyridin-2-yl)-3,3′-bi(1,2,4-triazole) basis: Synthesis, structure and magnetic studies. Inorganica Chim. Acta. 456, 136–141 (2017). https://doi.org/10.1016/j.ica.2016.10.040
Wallis, M.J., Min, H., Lindoy, L.F., Li, F.: Investigating the conformations of a family of [M(2)L(3)](4+) helicates using single crystal X-ray diffraction. Molecules. 28(3), 1404 (2023). https://doi.org/10.3390/molecules28031404
Schneider, H.-J., Yatsimirsky, A.: Principles and Methods in Supramolecular Chemistry. Wiley (2000)
Piguet, C., Bernardinelli, G., Bocquet, B., Quattropani, A., Williams, A.F.: Self-assembly of double and triple helices controlled by metal ion stereochemical preference. J. Am. Chem. Soc. 114(19), 7440–7451 (1992). https://doi.org/10.1021/ja00045a016
Uhlenheuer, D.A., Petkau, K., Brunsveld, L.: Combining supramolecular chemistry with biology. Chem. Soc. Rev. 39(8), 2817–2826 (2010). https://doi.org/10.1039/B820283B
McLaughlin, C.K., Hamblin, G.D., Sleiman, H.F.: Supramolecular DNA assembly. Chem. Soc. Rev. 40(12), 5647–5656 (2011). https://doi.org/10.1039/C1CS15253J
Pascu, G.I., Hotze, A.C.G., Sanchez-Cano, C., Kariuki, B.M., Hannon, M.J.: Dinuclear Ruthenium(II) Triple-stranded helicates: Luminescent supramolecular cylinders that bind and Coil DNA and exhibit activity against Cancer Cell lines. Angew Chem. Int. Ed. 46(23), 4374–4378 (2007). https://doi.org/10.1002/anie.200700656
Alessio, E., Mestroni, G., Bergamo, A., Sava, G.: Ruthenium antimetastatic agents. Curr. Top. Med. Chem. 4(15), 1525–1535 (2004). https://doi.org/10.2174/1568026043387421
Hartinger, C.G., Zorbas-Seifried, S., Jakupec, Jakupec, M.A., Kynast, B., Zorbas, H., Keppler, B.K.: From bench to bedside–preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)]. J. Inorg. Biochem. 100(5–6), 891–904 (2006). https://doi.org/10.1016/j.jinorgbio.2006.02.013
Bruker, Bruker: AXS Inc., Madison, Wisconsin, USA, (2016)
SHELX-2014: : Programs for Crystal Structure Analysis; University of Göttingen: Göttingen 2014
Sheldrick, G.: Crystal structure refinement with SHELXL. Acta. Crystallogr. Sect. C. 71(1), 3–8 (2015). https://doi.org/10.1107/S2053229614024218
Dolomanov, O.V., Bourhis, L.J., Gildea, R.J., Howard, J.A.K., Puschmann, H.: OLEX2: A complete structure solution, refinement and analysis program. J. App Cryst. 42(2), 339–341 (2009). https://doi.org/10.1107/S0021889808042726
Zhang, Y.B., Xiong, P., Cai, Cheng, G.Z.: Synthesis and properties of dinuclear ruthenium complexes with Schiff base bridging ligands. Chin. J. Inorg. Chem. 28, 2437–2443 (2012)
Acknowledgements
The authors would like to thank Western Sydney University (WSU) for research funding. We would like to thank the Advanced Materials Characterisation Facility at WSU for access to instrumentation. We thank the Biomedical Magnetic Resonance Facility at WSU for access to NMR instruments. Crystallographic data was collected at the Mark Wainwright Analytical Centre at UNSW. K.J.H-S. also wishes to thank WSU for funding and support of the PhD candidature and scholarship.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. The authors declare no external funding was received for this project.
Author information
Authors and Affiliations
Contributions
K.J.H-S. performed the synthetic work and majority of characterisation experiments. M.J.W. performed structural analysis. The manuscript was drafted by K.J.H-S. and M.J.W. J.P.F. and J.C.T collected and processed the NMR data. SCXRD data were collected by H.M., R.T., M.M.B., and C.E.M. All authors were involved in editing the final manuscript. F.L. directed the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare that there are no competing interests involved relating to the work presented in this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Howard-Smith, K., Wallis, M., Flood, J. et al. Conformational investigations on three large dinuclear triple helicates by single crystal X-ray diffraction. J Incl Phenom Macrocycl Chem 104, 199–207 (2024). https://doi.org/10.1007/s10847-024-01235-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10847-024-01235-3