
Abstract
Purpose
To evaluate the clinical validity of preimplantation genetic testing (PGT) to prevent hereditary hearing loss (HL) in Chinese population.
Methods
A PGT procedure combining multiple annealing and looping-based amplification cycles (MALBAC) and single-nucleotide polymorphisms (SNPs) linkage analyses with a single low-depth next-generation sequencing run was implemented. Forty-three couples carried pathogenic variants in autosomal recessive non-syndromic HL genes, GJB2 and SLC26A4, and four couples carried pathogenic variants in rare HL genes: KCNQ4, PTPN11, PAX3, and USH2A were enrolled.
Results
Fifty-four in vitro fertilization (IVF) cycles were implemented, 340 blastocysts were cultured, and 303 (89.1%) of these received a definite diagnosis of a disease-causing variant testing, linkage analysis and chromosome screening. A clinical pregnancy of 38 implanted was achieved, and 34 babies were born with normal hearing. The live birth rate was 61.1%.
Conclusions and relevance
In both the HL population and in hearing individuals at risk of giving birth to offspring with HL in China, there is a practical need for PGT. The whole genome amplification combined with NGS can simplify the PGT process, and the efficiency of PGT process can be improved by establishing a universal SNP bank of common disease-causing gene in particular regions and nationalities. This PGT procedure was demonstrated to be effective and lead to satisfactory clinical outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hearing loss (HL) is the third-largest cause of disability as reported by the Global Burden of Disease (2019). The report stated that 1.57 billion people had HL, which was defined as hearing loss in the better ear of over 20 decibels (dB) [1]. A population-based survey conducted in China in 2016 revealed 70 million individuals as having HL. The prevalence of disabling HL (loss in the better ear of more than 40 dB) was 0.85% in children from the provinces of Jilin, Gansu, Guangdong, and Shanxi, in the north, northwest, and southeast of China [2]. Disabling HL in childhood inhibits language development, educational achievement, and social-emotional development. Although hearing aids and cochlear implants are capable of restoring some auditory stimulation, these devices cannot restore natural hearing. Furthermore, the cost of hearing rehabilitation devices is a financial burden for many, and is not covered by insurance in developing countries.
The causes of HL include a variety of environmental factors, such as infection and ototoxic medicine; however, in developed countries, genetic factors account for 50–60% of childhood HL [3]. The World Health Organization (WHO) has recommended strategies for the prevention of HL due to maternal/infant infection and adverse events at the time of birth, and prompt initiation of habilitation for HL once diagnosed (https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss). However, there is no effective and easily employed prevention strategy for hereditary HL yet.
Hereditary HL, a typically monogenic disorder, is highly heterogeneous, involving 124 known genes (https://hereditaryhearingloss.org/) and exhibiting all four Mendelian modes of inheritance. In Asia, Europe, and America, 35~60% of HL patients were identified as having a genetic etiology through targeted genomic enrichment with massively parallel sequencing [4,5,6]. Genetic testing cannot only identify a precise molecular etiology but can also serve as the basis of prenatal diagnosis (PND) for patients with hereditary diseases to prevent recurrence. Preimplantation genetic diagnosis, more recently described as preimplantation genetic testing for monogenic disease (PGT-M) [7], first appeared in 1990 [8]. PGT-M is an effective means of preventing birth defects associated with hereditary disorders where the genetic etiology can be confirmed [7]. However, attitudes towards PND and PGT for non-lethal disorders such as HL vary greatly depending upon the culture, financial status, and religion of the at-risk individuals, as well as government policies. These factors, combined with the complex technical processes, have resulted in fewer PGT cases with HL being reported (Table S1) [9,10,11,12,13,14,15,16,17] than are in cases of another monogenic disease such as Huntington disease, cystic fibrosis, and neurofibromatosis.
In China, approximately 80% of HL cases diagnosed as hereditary HL have been shown to be autosomal recessive genetic, with both parents having normal hearing and carrying a single copy of the altered gene [18]. Thus, the prevention of hereditary HL is targeted not only at families with multi-generational histories of HL but also toward at-risk carrier families. The purpose of this study was to develop an efficient PGT strategy to prevent births with HL in at-risk families.
Materials and methods
Patients’ recruitment
Forty-seven couples were recruited from the Genetic Testing Center for Deafness, Chinese PLA General Hospital between October 2016 and July 2020. Thirty-seven normal hearing couples are carrier-carrier pattern; they have already had HL children. Eight couples are patient-carrier pattern. One carrier couple was detected by a proactive pre-pregnancy genetic testing. One carrier couple was detected by neonatal genetic screening; their first child was a HL gene carrier. All participants desired to have children with normal hearing and underwent face-to-face genetic counseling along with their immediate family members.
Variants interpretation
All the variants identified in the study participants were assessed in detail (Table 1). Variants were classified according to the American College of Medical Genetics (ACMG) guidelines [19], and variant information was retrieved from the ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and Deafness Variation Database (http://deafnessvariationdatabase.org/) websites.
Clinical PGT procedure
The PGT-M procedure combined multiple annealing and looping based amplification cycles (MALBAC) and single-nucleotide polymorphisms (SNPs) linkage analyses with a single low-depth next-generation sequencing run, which was developed base on MASALA [20] (mutated allele revealed by sequencing with aneuploidy and linkage analyses).
Embryo biopsy
Embryo biopsy procedures were performed at the Reproductive Medicine Center of Chinese PLA General Hospital as described in a previous article [21]. Intracytoplasmic sperm injection (ICSI) was performed to avoid surplus sperm contamination. All embryos were biopsied at the blastocyst stage; three to five trophectoderm cells were biopsied from the expanding blastocysts using a rubbing dissection method on day 5.
Whole-genome amplification by MALBAC and genetic analysis
Cell lysis and whole-genome amplification (WGA) were performed using a MALBAC single-cell WGA kit (Yikon Genomics Inc., catalog number: YK001A/B) according to the manufacturer’s protocol.
Variant site detection and linkage analysis
Ninety SNP markers linked to alleles with pathogenic variants were selected for each gene within 1 Mb upstream or downstream of the gene. The frequencies of the major alleles were greater than 0.1 in the Asian population. We performed multiplex PCR using the MALBAC amplification product as a template with 90 pairs of SNP-specific primers in each PCR. The variant sites were amplified in individual PCRs using specific primers. These PCR products were purified, subjected to library construction, and then sequenced on the Illumina HiSeq 2500 platform (1 M reads). The Genome Analysis Toolkit, version 3.5, Best Practices workflow was used to identify variants in the PCR-amplified regions. SNPs with less than 100× coverage were defined as “low depth”. Sanger sequencing was used to further confirm the detection of variants.
CNVs analysis
The MALBAC-amplified products were purified and sequenced on the Illumina HiSeq 2500 platform (1.5 M reads). Uniquely mapped reads were extracted from the alignment reads (bam file). The entire reference genome was divided into non-overlapping observation windows (bins), each 1 Mb in size. The read numbers and GC content were calculated for each bin. GC bias correction was applied for every 1% of GC content. R software (version 3.0.0) was used to graphically display the GC-corrected relative read numbers of each bin for the visualization of CNVs.
Transfer of embryos and pregnancy follow-up
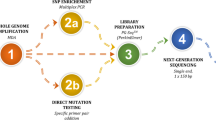
The first transfer was performed 2–3 months after ICSI via artificial or natural cycles. After the exclusion of aneuploid, affected, or undetermined embryos, one or two unaffected embryos were transferred. The human chorionic gonadotropin test was performed on day 10 following transfer. Pregnancy was confirmed by ultrasound at 8 weeks’ gestation. Clinical pregnancy was defined as the observation of fetal heartbeat. Prenatal diagnosis was performed by amniocentesis at 20 weeks gestation. Auditory brainstem response, otoacoustic emissions, and gene detection in cord blood were performed following birth. The clinical PGT procedure and testing workflow are shown in Fig. 1.

PGT clinical procedure and testing workflow. Trophectoderm cell lysis and WGA were performed using a MALBAC single-cell WGA kit. Enrichment PCR amplification of the WGA products was conducted using SNPs- and variant-specific primers. The PCR products were then sequenced by NGS; linkage analysis, chromosome aneuploidy analysis, and variant diagnosis were carried out simultaneously
The study was approved by the Research Ethics Committee of the Chinese PLA General Hospital (approval number S2019-203-01). Informed written consent was obtained from all patients or their guardians prior to genetic analysis and PGT.
Results
Forty-seven couples were enrolled in the PGT-M with PGT-A procedure. The ages of the female participants undergoing PGT-M ranged from 25 to 40 years, with a mean age of 32.3 years. Forty-three couples carried pathogenic variants in autosomal recessive non-syndromic HL genes, GJB2 and SLC26A4, and one case carried a variant in the autosomal dominant non-syndromic HL gene, KCNQ4. Three cases were diagnosed with syndromic HL, including one case of Noonan syndrome (PTPN11), one case of Waardenburg syndrome (PAX3), and one case of usher syndrome (USH2A).
The haplotypes were constructed using heterozygous SNPs from the couples and their existing children with HL. However, in the case of two couples (F15421, F1884), they had no child, and as such the haplotypes was constructed from the couples and their parents. Genotyping of 90 SNPs in the proximity (1 Mb upstream or downstream) of each gene included in the PGT procedure led to the identification of between 14 and 54 informative heterozygous SNPs for each family that were subsequently used in linkage analyses.
A total of 54 ICSI cycles were implemented on 47 couples, 40 (out of 47; 85.1%) of which underwent one cycle and the remaining 7 (14.9%) underwent two cycles. A total of 340 embryos were cultured and biopsied. One hundred and fourteen (33.5%) euploid embryos were found to be either without pathogenic variants or with a heterozygous pathogenic variant only involving autosomal recessive genes. Fifty-nine transfers were completed, and a clinical pregnancy was achieved in 38 cases, with a clinical pregnancy rate of 64.4%. Four miscarriages (6.8%) occurred in early pregnancy. We performed the chromosome analysis on the abortion tissues in 3 cases. A del (7) (q11.21q11.23) mosaicism was detected in one case, which was inconsistent with the embryo diagnosis result. We are not sure if it was a PGT-A error or a mosaic embryo. The other two cases were euploids, and we assumed the cause of the miscarriage was not due to embryonic factors. The abortion tissue analysis was not carried out in case F8241. One intrauterine death happened in the 38th week due to torsion of the umbilical cord. Thirty-four babies were born with normal hearing, including a set of twins, with a cumulative live birth rate of 61.1% (33 live births/54 cycles). Five couples still have embryos available and are waiting for transfer. Eight couples (17.0%) withdrew from the procedure due to unsuccessful pregnancy in one or two cycles. The auditory brainstem response and otoacoustic emission measurements were normal for all neonates. During the 6- to 52-month follow-up, no infant developed HL or any other developmental abnormalities. The details of each cycle are presented in Table 2.
In our study group, 44 couples were identified as carriers of autosomal recessive HL gene, GJB2, SLC26A4, and USH2A. One hundred and seven transferable embryos were screened out, including 40 (40/107, 37.4%) wild type and 67 (67/107, 62.6%) carrier embryos. Eighteen (18/44, 40.9%) couples did not get any wild type embryo, and they chose to transfer the carrier embryos, and 4 (4/44, 9.1%) couples chose to transfer carrier embryos after wild type embryos were used up. No couples refused to transfer a carrier embryo in our study. Details were described in Table S2.
Three hundred and three (303/340, 89.1%) embryos were given a definite diagnosis as a result of the disease-causing variant testing, linkage analysis, and preimplantation genetic testing for aneuploidy (PGT-A) (Fig. 2A–E). Any CNVs greater than 4 Mb were reported, and 130 (130/340, 38.2%) embryos were identified as aneuploid (Fig. 2B, C). The difference in the aneuploid ratio between cycles ranged from 0% to 100% (Median 42.9%). There was no significant association between the aneuploid ratio and the women age at implant (Mann–Whitney test U=207.5, p=0.59).

Preimplantation genetic testing for aneuploidy (PGT-A) result samples: A Chromosome euploid; B trisomy 16; C del1q21.1qter mosaicism; D multi-chromosome abnormality; E contamination
The diagnostic results were inconclusive in 37 (37/340, 10.9%) embryos. MALBAC failure was detected in three (3/340, 0.9%) embryo cell samples upon quality control testing. For 337 embryos, 501 rounds of PCR and Sanger sequencing were carried out successfully, and 14 ADOs were identified with an ADO rate of 2.8% (14/501). In 10 cases, the ADO was identified through linkage analysis. In the other 4 cases, the non-mutant allele dropped out, and these did not affect the diagnosis. For instance, in one case (F10284) embryo number 4, we detected a homozygotic mutation of SCL26A4 c.2168A>G, but the maternal and paternal mutations were SCL26A4 c.2168A>G and SCL26A4 c.697G>C. It was clear that the non-mutant allele had dropped out, and as such, when taken in combination with the result of the linkage analysis, the diagnosis was clear. The CNV detection results of 9 embryos showed multiple chromosomal abnormalities (Fig. 2D). In these cases, the biopsy material was assumed to have contained broken cells. For 4 embryos, much of the sequencing data did not match the reference genome due to loss or contamination of biopsy cells (Fig. 2E). Variant or SNP sequencing failures of unknown cause occurred in 11 cases, also leading to inconclusive diagnoses.
Discussion
HL is the most common neurosensory disorder worldwide [22]. With the development of next-generation sequencing (NGS) techniques, hereditary HL can easily be detected at the molecular level, heralding the arrival of the era of precision medicine. A recent etiological study in China showed that the use of next generation sequencing (NGS) to detect 129 HL genes led to 52.19% diagnostic rate in sporadic HL patients (n = 433), and 56.67% in HL pedigrees (n = 78) [18]. Meanwhile, with the implementation of concurrent newborn hearing and genetic screening [23, 24], large numbers of carriers in normal hearing population can be identified in advance. Our recent study reported that among 180,469 newborns in Beijing, China, the carrier rate of the nine major pathogenic variants across four genes (GJB2, GJB3, SLC26A4, and mtDNA 12S rRNA) was 4.5% [23]. With the context of genetic epidemiology, the three-level prevention of hereditary hearing loss has been conceived and implemented in China over the years. Briefly, the primary prevention includes genetic screening before pregnancy and PGT-M; Secondary prevention includes the detection of deafness genes in pregnant women and the prenatal diagnosis; The tertiary prevention includes the joint screening of hearing and genes in newborns, as well as the genetic diagnosis and hearing rehabilitation of deaf patients. Therefore, in addition to those families who have already had HL members, many at-risk families with carriers were emerging by pre-pregnancy genetic screening and neonatal genetic screening. Both kind of conditions were enrolled in our PGT-M procedure. Prospectively, PGT-M procedure with great efficiency should be applied to a wider population.
The gene and variant spectra of hereditary HL vary greatly according to race. In a mixed-race genetic study from the USA with 1119 cases [6], nearly 75% of the genetic diagnoses were attributable to 10 HL genes, with GJB2 being the most common and accounting for the etiology in 22% of the diagnosed patients. The next three most-frequently implicated genes were STRC (16%), SLC26A4 (7%), and TECTA (5%). GJB2 and STRC are the most common HL genes in Caucasians as well as in Hispanics. The Middle East has a high rate of consanguineous marriage, thereby increasing the risk of recurrent hereditary HL. GJB2, SLC26A4, MYO15A, and TMC1 are the most prevalent genes involved in non-syndromic HL in the Middle East (Iran, Turkey, and Pakistan) [25]. In sub-Saharan Africa, the etiologic genetic diagnosis rate is 4% for certain specific genes (MYO7A, MYO6, SLC26A4, SIX1, TRIPBP, and POU3F4) [26]. According to the previous study [18], GJB2 and SLC26A4 were the predominant etiologies of HL in the Chinese population with HL (43.5% of GJB2 and SLC26A4 vs. 8.7% of other rare HL genes). The pathogenicity of hotspot mutations in these two genes is well defined. Thus, a PGT-M strategy target to GJB2 and SLC26A4 should covered over 80% genetic diagnosed at-risk families.
Prenatal diagnosis plays an important role in preventing congenital disabilities caused by severe genetic diseases. A quantitative circulating single-molecule amplification and resequencing technology cSMART assay was developed for the non-invasive prenatal diagnosis (NIPD) of autosomal recessive non-syndromic HL caused by GJB2 and SLC26A4 pathogenic variants [27]. Zhang et al. [28] implemented a novel NIPD approach for de novo mutations in causative genes implicated in common dominant monogenic diseases including HL. Surveys in developed countries have shown that people often undergo PND to inform the use of a neonatal early hearing habilitation plan, and are unlikely to terminate the pregnancy [29, 30]. However, also in developing countries, hearing parents of children with HL expressed a desire to terminate a future pregnancy if the fetus were found to be affected [31]. Genetic counselors must be cautious in giving advice, providing detailed information and psychological support to advocate as wide a range of informed choices as possible. However, as a non-fatal monogenetic disease, the timing for HL prevention is more suited to a prospective technique such as PGT than either invasive or non-invasive prenatal testing methods.
PGT has been in development for almost 30 years, with the successful addition of techniques such as nested-PCR, multiple displacement amplification (MDA), MALBAC, array comparative genomic hybridization (CGH), SNP array and NGS [8, 32,33,34]. Routine PGT-M requires a complex process of preparatory work that incorporates linkage marker selection, primer design, development of amplification conditions, and single-cell validation [34]. These procedures are both time- and labor-intensive. Compare to STR marker, using SNP marker makes PGT-M procedure more efficient and simple. Our PGT procedure was developed base on MARSALA (mutated allele revealed by sequencing with aneuploidy and linkage analyses), which was first reported in 2015 [20] and had been proven to offer satisfied accuracy in linkage analysis [35]. We created a mini-SNP bank consisting of 90 SNPs within a 1 Mb region (upstream or downstream) of the GJB2 and SLC26A4 genes (Table S3 and Table S4). The frequencies of the major alleles in the SNPs were greater than 0.1 in the Asian population. This strategy allowed every case to provide sufficient heterozygous SNPs necessary for the linkage analysis. All families with GJB2 and SLC26A4 mutant genes were able to start the PGT process immediately, without any of the previously required preparatory work.
Whole-genome sequencing data provide euploid information simultaneously. Our results showed that 38.2% of the tested embryos exhibited aneuploidy, including monosomy, trisomy, or segmental CNVs, which may lead to implantation failure, miscarriage, or congenital disabilities. Thus, the simultaneous screening of CNVs, alongside the use of PGT-M, could increase clinical pregnancy rates to the level of 64.4% reported in this study, which is much higher than rates seen using PGT-M cycles without PGT-A reported by the ESHRE PGT consortium XIX-XX (24%) [36], similar to rates reported by groups in the USA and the UK (40.9–52.5%) [37, 38]. Previous studies indicated improvements in clinical outcome were obtained by trophectoderm biopsy, concurrent PGT-M, and PGT-A, and frozen embryo transfer. However, it remains to be seen whether different PGT-A methods (NGS, SNP array, or CGH array) can facilitate similar improvements in clinical outcomes.
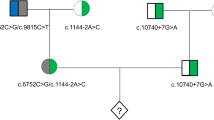
Hereditary HL is a typical monogenic disorder associated with high genetic heterogeneity, involving 124 genes that have been related to non-syndromic HL and more than 40 genes that have been related to syndromic HL (https://hereditaryhearingloss.org/). Identifying the pathogenic variants of rare HL genes is still the major challenge for clinical diagnosis and the clinical implementation of PGT. The present study enrolled 4 families with rare HL genes (PTPN11, KCNQ4, PAX3, and USH2A) into the cohort undergoing the PGT procedure. The variants were interpreted according to the ACMG guidelines and available databases (ClinVar and Deafness Variation Database). The pathogenicity of the variant KCNQ4 c.733 G>A (p.Gly245Arg) was classified as uncertain and that of PAX3 c.210 C>A (p.Cys70*) was classified as likely pathogenic. These classifications were upgraded to likely pathogenic and pathogenic through the co-segregation analyses of their pedigrees (Fig. 3). With the extensive application of multi-gene NGS panel and whole-genome sequencing, more HL patients will be offered access to an exact genetic diagnosis and PGT implementation for rare HL gene variants. Having said this, progress should be cautious, and a solid evaluation of the pathogenicity of variants is necessary.

Pedigree chart of the four families carried rare HL gene variants
Although the improvements made to the PGT procedure helped to reduce implementation difficulties and economical costs, more at-risk families still showed hesitance due to a lack of knowledge or fear of new technology, and/or worry about possible complications. As such, more cases need to be accumulated in order to objectively evaluate PGT and to streamline the process of introducing the possible intervention for at-risk families.
Conclusions
In both the HL population and in hearing individuals at risk of giving birth to offspring with HL in China, there is a practical need for a PGT procedure to prevent babies from being born with HL. Considering the genetic heterogeneity of HL, defining the mutational spectrum through a large-scale molecular etiology study, identifying the target variants and a high-density set of SNP markers with high heterozygosity are key aims in the crusade for the prevention of hereditary HL. The modified MARSALA PGT procedure has the advantages of a simplified process, a satisfactory live birth rate, better generalizability, and has been validated as an effective and precise approach for preventing common hereditary HL in at-risk families, offering an alternative option to PND or to giving birth to children with HL who will require lifelong habilitation.
Change history
24 April 2023
A Correction to this paper has been published: https://doi.org/10.1007/s10815-023-02807-x
References
Collaborators GBDHL. Hearing loss prevalence and years lived with disability, 1990-2019: findings from the Global Burden of Disease Study 2019. Lancet. 2021;397(10278):996–1009. https://doi.org/10.1016/S0140-6736(21)00516-X.
Hu XY, Zheng XY, Ma FR, et al. Prevalence of hearing disorders in China: a population-based survey in four provinces of China. Chin J Otorhino Head and Neck Surg. 2016;51(11):819–25. https://doi.org/10.3760/cma.j.issn.1673-0860.2016.11.004.
Morton CC, Nance WE. Newborn hearing screening--a silent revolution. N Engl J Med. 2006;354(20):2151–64. https://doi.org/10.1056/NEJMra050700.
Moteki H, Azaiez H, Booth KT, et al. Comprehensive genetic testing with ethnic-specific filtering by allele frequency in a Japanese hearing-loss population. Clin Genet. 2016;89(4):466–72. https://doi.org/10.1111/cge.12677.
Cabanillas R, Dineiro M, Cifuentes GA, et al. Comprehensive genomic diagnosis of non-syndromic and syndromic hereditary hearing loss in Spanish patients. BMC Med Genomics. 2018;11(1):58. https://doi.org/10.1186/s12920-018-0375-5.
Sloan-Heggen CM, Bierer AO, Shearer AE, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135(4):441–50. https://doi.org/10.1007/s00439-016-1648-8.
Group EP-MW, Carvalho F, Moutou C, et al. ESHRE PGT Consortium good practice recommendations for the detection of monogenic disorders. Human Reproduction Open. 2020;2020(3):hoaa018. https://doi.org/10.1093/hropen/hoaa018.
Handyside AH, Kontogianni EH, Hardy K, et al. Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature. 1990;344(6268):768–70. https://doi.org/10.1038/344768a0.
Chen HL, Lin PH, Chiang YT, et al. Preimplantation genetic diagnosis in hereditary hearing impairment. Diagnostics. 2021;11(12). https://doi.org/10.3390/diagnostics11122395.
Shi WH, Ye MJ, Chen SC, et al. Case report: preimplantation genetic testing and pregnancy outcomes in women with Alport syndrome. Front Genet. 2021;12:633003. https://doi.org/10.3389/fgene.2021.633003.
Luo H, Chen C, Yang Y, et al. Preimplantation genetic testing for a family with usher syndrome through targeted sequencing and haplotype analysis. BMC Med Genomics. 2019;12(1):157. https://doi.org/10.1186/s12920-019-0600-x.
Karimi Yazdi A, Davoudi-Dehaghani E, Rabbani Anari M, et al. The first successful application of preimplantation genetic diagnosis for hearing loss in Iran. Cell Mol Biol. 2018;64(9):1718.
Hao Y, Chen D, Zhang Z, et al. Successful preimplantation genetic diagnosis by targeted next-generation sequencing on an ion torrent personal genome machine platform. Oncol Lett. 2018;15(4):4296–302. https://doi.org/10.3892/ol.2018.7876.
Yahalom C, Macarov M, Lazer-Derbeko G, et al. Preimplantation genetic diagnosis as a strategy to prevent having a child born with an heritable eye disease. Ophthalmic Genet. 2018;39(4):450–6. https://doi.org/10.1080/13816810.2018.1474368.
Liss J, Mirecka A, Kitowska K, et al. Preimplantaion genetic diagnosis of hearing loss with 35delG mutation in GJB2 gene - preliminary report. Pol Otolaryn. 2011;65(6):443–6. https://doi.org/10.1016/S0030-6657(11)70738-7.
Wu CC, Lin SY, Su YN, et al. Preimplantation genetic diagnosis (embryo screening) for enlarged vestibular aqueduct due to SLC26A4 mutation. Audiol Neurootol. 2010;15(5):311–7. https://doi.org/10.1159/000284349.
Altarescu G, Eldar-Geva T, Brooks B, et al. Preimplantation genetic diagnosis (PGD) for nonsyndromic deafness by polar body and blastomere biopsy. J Assist Reprod Genet. 2009;26(7):391–7. https://doi.org/10.1007/s10815-009-9335-5.
Yuan Y, Li Q, Su Y, et al. Comprehensive genetic testing of Chinese SNHL patients and variants interpretation using ACMG guidelines and ethnically matched normal controls. EJHG. 2020;28(2):231–43. https://doi.org/10.1038/s41431-019-0510-6.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
Yan L, Huang L, Xu L, et al. Live births after simultaneous avoidance of monogenic diseases and chromosome abnormality by next-generation sequencing with linkage analyses. Proc Natl Acad Sci USA. 2015;112(52):15964–9. https://doi.org/10.1073/pnas.1523297113.
Lu Y, Peng H, Jin Z, et al. Preimplantation genetic diagnosis for a Chinese family with autosomal recessive Meckel-Gruber syndrome type 3 (MKS3). PloS One. 2013;8(9):e73245. https://doi.org/10.1371/journal.pone.0073245.
van Wieringen A, Boudewyns A, Sangen A, et al. Unilateral congenital hearing loss in children: challenges and potentials. Hear Res. 2019;372:29–41. https://doi.org/10.1016/j.heares.2018.01.010.
Dai P, Huang LH, Wang GJ, et al. Concurrent hearing and genetic screening of 180,469 neonates with follow-up in Beijing, China. Am J Hum Genet. 2019;105(4):803–12. https://doi.org/10.1016/j.ajhg.2019.09.003.
Shearer AE, Shen J, Amr S, et al. A proposal for comprehensive newborn hearing screening to improve identification of deaf and hard-of-hearing children. Genet Med. https://doi.org/10.1038/s41436-019-0563-5.
Najmabadi H, Kahrizi K. Genetics of non-syndromic hearing loss in the Middle East. Int J Pediatr Otorhinolaryngol. 2014;78(12):2026–36. https://doi.org/10.1016/j.ijporl.2014.08.036.
Rudman JR, Kabahuma RI, Bressler SE, et al. The genetic basis of deafness in populations of African descent. J Genet Genomics. 2017;44(6):285–94. https://doi.org/10.1016/j.jgg.2017.03.008.
Han M, Li Z, Wang W, et al. A quantitative cSMART assay for noninvasive prenatal screening of autosomal recessive nonsyndromic hearing loss caused by GJB2 and SLC26A4 mutations. Gen in Med. 2017;19(12):1309–16. https://doi.org/10.1038/gim.2017.54.
Zhang J, Li J, Saucier JB, et al. Non-invasive prenatal sequencing for multiple Mendelian monogenic disorders using circulating cell-free fetal DNA. Nat Med. 2019;25(3):439–47. https://doi.org/10.1038/s41591-018-0334-x.
Thorsen A, Devantier L, Ovesen T. Attitudes towards prenatal diagnosis of deafness among parents to children with cochlear implants. Ugeskr Laeger. 2009;171(17):1387–91.
Middleton A, Hewison J, Mueller R. Prenatal diagnosis for inherited deafness--what is the potential demand? J Genet Couns. 2001;10(2):121–31. https://doi.org/10.1023/a:1009439630457.
Nahar R, Puri RD, Saxena R, et al. Do parental perceptions and motivations towards genetic testing and prenatal diagnosis for deafness vary in different cultures? Am J Med Genet A. 2013;161A(1):76–81. https://doi.org/10.1002/ajmg.a.35692.
Rubio C, Rodrigo L, Mir P, et al. Use of array comparative genomic hybridization (array-CGH) for embryo assessment: clinical results. Fertil Steril. 2013;99(4):1044–8. https://doi.org/10.1016/j.fertnstert.2013.01.094.
Handyside AH, Robinson MD, Simpson RJ, et al. Isothermal whole genome amplification from single and small numbers of cells: a new era for preimplantation genetic diagnosis of inherited disease. Mol Hum Reprod. 2004;10(10):767–72. https://doi.org/10.1093/molehr/gah101.
Natesan SA, Bladon AJ, Coskun S, et al. Genome-wide karyomapping accurately identifies the inheritance of single-gene defects in human preimplantation embryos in vitro. Genet Med. 2014;16(11):838–45. https://doi.org/10.1038/gim.2014.45.
Xiong L, Huang L, Tian F, et al. Bayesian model for accurate MARSALA (mutated allele revealed by sequencing with aneuploidy and linkage analyses). J Assist Reprod Genet. 2019;36(6):1263–71. https://doi.org/10.1007/s10815-019-01451-8.
van Montfoort A, Carvalho F, Coonen E, et al. ESHRE PGT Consortium data collection XIX-XX: PGT analyses from 2016 to 2017(dagger). Hum Reprod Open. 2021;2021(3):hoab024. https://doi.org/10.1093/hropen/hoab024.
Shaulov T, Zhang L, Chung JT, et al. Outcomes of preimplantation genetic testing for single gene defects in a privately funded period and publicly funded period: a North-American single center experience. J Reprod Infertil. 2020;21(2):107–15.
Ben-Nagi J, Jones B, Naja R, et al. Live birth rate is associated with oocyte yield and number of biopsied and suitable blastocysts to transfer in preimplantation genetic testing (PGT) cycles for monogenic disorders and chromosomal structural rearrangements. Eur J Obstet Gynecol. 2019;4:100055. https://doi.org/10.1016/j.eurox.2019.100055.
Acknowledgements
We sincerely thank all subjects and family members for their participation and cooperation in this study.
Funding
This work is supported by the National Key Research and Development Program of China (NO.2022YFC2703602,2016YFC1000706,2018YFC1003100), National Natural Science Foundation of China (81870731, 81900953), Beijing Natural Science Foundation (7191011, 7192234) and Hainan Provincial Department of Science and Technology (819MS110).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bi, Q., Huang, S., Wang, H. et al. Preimplantation genetic testing for hereditary hearing loss in Chinese population. J Assist Reprod Genet 40, 1721–1732 (2023). https://doi.org/10.1007/s10815-023-02753-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-023-02753-8