
Abstract
Purpose
Is it possible to eliminate metastasised chronic myeloid leukaemia (CML) and acute myeloid leukaemia (AML) cells from ovarian cortex fragments by inhibition of Aurora B/C kinases (AURKB/C) without compromising ovarian tissue or follicles?
Methods
Human ovarian cortex tissue with experimentally induced tumour foci of CML, AML and primary cells of AML patients were exposed to a 24h treatment with 1 μM GSK1070916, an AURKB/C inhibitor, to eliminate malignant cells by invoking mitotic catastrophe. After treatment, the inhibitor was removed, followed by an additional culture period of 6 days to allow any remaining tumour cells to form new foci. Ovarian tissue integrity after treatment was analysed by four different assays. Appropriate controls were included in all experiments.
Results
Foci of metastasised CML and AML cells in ovarian cortex tissue were severely affected by a 24h ex vivo treatment with an AURKB/C inhibitor, leading to the formation of multi-nuclear syncytia and large-scale apoptosis. Ovarian tissue morphology and viability was not compromised by the treatment, as no significant difference was observed regarding the percentage of morphologically normal follicles, follicular viability, glucose uptake or in vitro growth of small follicles between ovarian cortex treated with 1 μM GSK1070916 and the control.
Conclusion
Purging of CML/AML metastases in ovarian cortex is possible by targeting the Mitotic Catastrophe Signalling Pathway using GSK1070916 without affecting the ovarian tissue. This provides a therapeutic strategy to prevent reintroduction of leukaemia and enhances safety of autotransplantation in leukaemia patients currently considered at high risk for ovarian involvement.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
With the increase in overall 5-year survival rates in cancer [8, 70] due to advances in early detection and treatment, the effect on fertility of anti-cancer treatment has been gaining growing attention. As both radiotherapy and chemotherapy regimens can lead to subfertility or even infertility [31, 67, 73], the options for fertility preservation should be discussed before start of therapy.
Nowadays, multiple options are available for fertility preservation in women, however, for prepubertal girls and women who cannot delay start of their anti-cancer treatment ovarian tissue cryopreservation (OTC) is currently the sole option [13]. Ovarian cortex fragments are generally cryopreserved before start of anti-cancer therapy or during the first remission phase and used for restoration of fertility [69]. After autotransplantation, the ovarian follicles in the graft resume development and ovarian activity are restored in more than 93% of the cases allowing these women to conceive naturally or through assisted reproductive technology [18, 20, 21]. The first child was born in 2004 with the aid of OTC and the number of live births has been steadily rising and currently exceeds 130 [19, 29]. Live birth rates per autotransplantation are ranging from 23% up to 69% [20, 71].
Despite these excellent results, there are serious concerns regarding safety of autotransplantation of frozen-thawed ovarian tissue in former cancer patients. As the ovarian cortex tissue is generally obtained prior to start of the therapy, possible presence of metastasised malignant cells in the tissue may lead to reintroduction of the malignancy after autotransplantation. Many cancer types are known to metastasise to the ovary and especially hematologic malignancies are considered high risk [3, 16, 17, 66]. The presence of leukaemic cells in ovarian tissue from patients has been demonstrated by PCR analysis, multicolor flow cytometry and xenografting to immunodeficient mice [1, 9, 57, 72, 83].
Several strategies to circumvent the possibility of reintroducing malignant cells with the graft and increase the safety of OTC are currently pursued. These are based on separating the follicles from the possibly contaminated stromal cell compartment, and include in vitro maturation of primordial follicles [54, 78] and in vivo grafting of isolated pre-antral stage follicles in an artificial ovary, a decellularised ovarian matrix or by xenografting [2, 12, 48, 63].
In addition to these strategies, we have recently shown ex vivo purging of ovarian cortex tissue is a promising technique to prevent reintroduction of cancer by eradicating contaminating malignant cells, without affecting the ovarian tissue and follicles [59]. Foci of rhabdomyosarcoma cells were completely eliminated from ovarian cortex fragments by suppression of the Hippo signalling pathway using Verteporfin as an inhibitor of YAP/TAZ oncoproteins. However, tissue contaminated with myeloid leukaemia was not completely purged by treatment with Verteporfin.
Purging was also recently applied on ovarian cortex tissue from patients with acute lymphocytic leukaemia (ALL) using dexamethasone, known for its anti-leukaemic activity, but was found to be ineffective in purging ovarian cortex from contaminating malignant cells [9]. This, together with our previous findings, clearly indicates that different malignancies require different purging regimes [59].
Uncontrolled proliferation is a universal property of malignant cells, while ovarian cortex tissue, including the primordial follicles, is essentially mitotically silent [34]. Oncosuppression by targeting the mitotic catastrophe signalling pathway leads a cell to an irreversible antiproliferative fate of death or senescence and could therefore be a promising purging strategy [52, 80]. Although this pathway can be activated by several mechanisms, the inhibition of specific kinases such as the Aurora protein kinases has been shown to be very effective in disrupting mitotic progression. Aurora kinases are a family of serine/threonine kinases that are indispensable to cell division [7]. Dysregulation of Aurora kinases leads to mitotic abnormalities, failure of cytokinesis leading to formation of syncytia and ultimately to apoptosis [24, 27, 35]. Human malignancies frequently overexpress Aurora kinases, making them attractive and logical targets for purging [58].
In this paper, we have investigated the use of GSK1070916, a reversible and ATP competitive inhibitor of Aurora B and Aurora C kinases (AURKB/C), to purge ovarian cortex tissue fragments contaminated with foci of chronic myeloid leukaemia (CML) or acute myeloid leukaemia (AML) from cell lines and of primary cancer cells from patients with AML. We provide evidence that GSK1070916 is effective in inducing mitotic catastrophe in malignant cells of both CML and AML origin without compromising the integrity of the ovarian cells, including the follicles. Preventing reintroduction of malignancies before autotransplantation of ovarian cortex tissue by the pharmacological inhibition of Aurora kinases could therefore provide an effective therapeutic strategy for enhancing the safety of OTC.
Materials and methods
Selection of tumour cell lines
The cell lines used for this study consisted of CML: JURL-MK1, K562 and MEG-01, and AML: KG-1a, NB4 and NOMO-1 (Table 1). These cell lines represent various types of myeloid leukaemia and have been widely used for in vitro and in vivo studies [23, 43, 53, 82]. Cell lines were obtained from ATCC (Manassas, VA, USA) and DSMZ (Braunschweig, Germany). All cell lines were cultured in RPMI (BioWhittaker, Lonza, Basel, Switzerland) supplemented with 10% Fetal Bovine Serum (FCS, Gibco, Life Technologies, Carlsbad, CA, USA) and 40 μg/mL Gentamycin (Centrafarm, Etten-Leur, The Netherlands) and kept in log phase.
Primary cells
Cryopreserved primary AML cells were obtained from four AML patients (aged 64–80). Cells were thawed in FCS containing DNAseI (67 μg/mL, Sigma-Aldrich, Saint Louis, MO, USA), Heparin (17 U/mL, pharmacy Radboudumc, Nijmegen, The Netherlands) and MgSO4 (7 nM, Boom, Meppel, The Netherlands), incubated for 10 min at room temperature and washed twice with Phosphate Buffered Saline (Sigma-Aldrich, Saint Louis, MO, USA) containing 1% FCS. Cells were resuspended in Dulbecco’s Modified Eagle’s Medium (DMEM, BioWhittaker, Lonza, Basel, Switzerland) with 10% FSC.
Collection of human tissue
Intact ovaries were obtained after informed consent from transgender men undergoing oophorectomy as part of their gender-affirming surgery (aged 18–24). Immediately after surgery, the ovaries were submerged in cold L15 medium (Lonza, Basel, Switzerland) and transported on ice to the laboratory. Cortical fragments of 10 × 10 × 1 mm were prepared and cryopreserved within 4 h after surgery according to clinical standards using previously described methods [61]. Testicular tissue was obtained from a patient undergoing a testicular sperm extraction and after the collection of spermatozoa was completed.
Western blot
Expression of AURKB and AURKC in myeloid leukaemic cells grown in suspension and enzymatically digested human ovarian cortex tissue was examined by SDS-PAGE and Western blot analyses. Human testicular tissue, known for high level expression of AURKC [10, 42], was used as a control. In brief, cells were harvested, counted and resuspended in SDS-PAGE sample buffer containing 100 mM Dithiothreitol. Approximately 50 μg of total cell protein was subjected to standard 10% SDS-PAGE. After electrophoresis, samples were transferred to nitrocellulose membranes (GE Healthcare, Chicago, IL, USA) and probed with primary antibodies directed against AURKB (1:2000, Invitrogen, Carlsbad, CA, USA) or AURKC (1:500, Bioss Antibodies, Woburn, MA, USA), followed by incubation with a labelled secondary antibody (Cell Signalling Technology, Danvers, MA, USA). Proteins were visualised by chemiluminescence using Odyssey CLx Imaging system (LI-COR Biosciences, Lincoln, NE, USA). As a control, an antibody against Glyceradehyde 3-phosphate dehydrogenase (GAPDH, Abcam, Cambridge, UK) was used.
Tumour induction
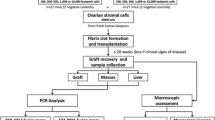
After thawing according to clinical standards [61], ovarian cortex fragments were micro-injected with exponentially growing CML cells (JURL-MK1, K562, MEG-01), AML cells (KG-1A, NB4, NOMO-1) or primary AML cells to induce micro metastases. The procedure was performed as previously described [61] using forceps and a 30-gauge needle with 0.3 mm outer diameter (Henke-Sass, Wolf GmbH, Nörten-Hardenberg, Germany). After tumour cell injection, the ovarium cortex fragments were cultured for 3 days to allow formation of small tumour foci.
Ex vivo purging
To select the optimal concentration for GSK1070916 (GlaxoSmithKline, Brentford, UK) in relation to tumoricidal activity, cancer cell lines were cultured in suspension in a 24-well plate for 24 h with increasing concentration (100 nM, 1 μM and 10 μM) of GSK1070916 or an equivalent concentration of solvent (dimethylsulfoxide, DMSO; WAK-Chemie Medical GmbH, Steinbach, Germany) and examined by light microscopy. In all subsequent purging experiments, ovarian cortex tissue was exposed to 1 μM GSK1070916916 or an equivalent concentration of solvent. After tumour formation, the cortex fragments were cut in half. One half was used for purging by 24 h incubation in 5 mL of culture medium consisting of DMEM supplemented with 10% FCS and 40 μg/mL Gentamycin containing 1 μM GSK1070916 dissolved in DMSO. The other half was used as a control and exposed to an equivalent concentration of solvent. After 24 h of treatment, the fragments were transferred to a 6-well plate and were subjected to four subsequent 10 min washes in 5 mL of fresh DMEM to remove the inhibitor and solvent. During washing, the fragments were placed on a shaking table at 37 °C for optimal diffusion. Next, fragments were cultured for an additional 6 days in 5 mL of culture medium to allow any remaining tumour cells to form new foci. The purging protocol was performed with ovarian cortex tissue of three subjects for all CML and AML cell lines. To confirm the effectiveness of the purging protocol on primary cells, the experiment was repeated with cells from four AML patients.
Histological examination
Fragments were fixed in Bouin’s fluid (Klinipath, Amsterdam, The Netherlands) for 1 h, washed with tap water and stored in 4% formaldehyde solution (Klinipath, Amsterdam, The Netherlands) before embedding in paraffin. The fragment was sectioned into 4 μm thick sections. Every 7th section was analysed for the presence of leukaemic cells after haematoxylin and eosin (HE) staining (Tissue Tek® Prisma™, Sakura, Alphen aan den Rijn, the Netherlands). Per cancer cell line, the entire fragment was sectioned and a total of 105 to 173 sections (representing a tissue surface of 9.3 to 17.4 cm2) of GSK1070916 treated tissue were examined by light microscopy for the presence of cancer cell foci by two independent observers.
Isolated tumour cells in suspension were fixated on a slide for immunohistochemical staining using acetic acid and methanol (1:3).
Proliferating cells were identified by immunohistochemical staining with Ki-67 antibody (1:100, DAKO, Agilent Technologies, Santa Clara, CA, USA) while apoptotic cells were highlighted by staining for active caspase-3 (1:200, AC3, BD Biosciences, San Jose, CA, USA). Immunohistochemical staining with polyclonal anti-Aurora B antibody (1:200, Invitrogen, Carlsbad, CA, USA) or polyclonal anti-Aurora C (1:200, Bioss Antibodies, Woburn, MA, USA) was performed to examine AURKB and AURKC expression in ovarian tissue and CML/AML cells. Immunostaining of cytokeratin AE1/AE3 (1:200, DAKO, Agilent Technologies, Santa Clara, CA, USA), a marker for unilaminar follicles [74], was used as a control.
Antigens were retrieved with citrate buffer (ScyTEK Laboratories, Logan, UT, USA). Next, sections were incubated with Vectastain Avidine/Biotine Blocking Kit followed by incubation with normal horse or goat serum (all from Vector Laboratories, Burlingame, CA, USA) depending on the secondary antibody. Subsequently sections were incubated with the primary antibodies followed by incubation with Biotinylated anti-Mouse or anti-Rabbit (both from Vector Laboratories, Burlingame, CA, USA). To visualise the target-antibody interaction Vectastain AB-complex and DAB (both from Vector Laboratories, Burlingame, CA, USA) incubation was used. After counterstaining with haematoxylin (Vector Laboratories, Burlingame, CA, USA), slides were evaluated by light microscopy and photographed using Visiontek® Live Digital Microscope (Sakura, Alphen aan den Rijn, The Netherlands).
Viability assays of ovarian cortex tissue and follicles
The viability of the ovarian cortex tissue and follicles before and after purging was determined by four different assays: conventional (immuno)histochemistry, a glucose uptake assay [30], Neutral Red staining [47] and in vitro growth (IVG) of small follicles [54, 55]. After thawing, ovarian cortex fragments were treated for 24 h with 1 μM GSK1070916 or solvent and washed. Cortex fragments were immediately used for analyses for glucose uptake assay and IVG assay. For histology and Neutral Red uptake, fragments were incubated for an additional 24 h culture, to allow possible damage to the tissue and follicles to become apparent before analysis. All assays were performed using ovarian cortex tissue from three subjects, except for the IVG assay which was performed in tissue from two subjects.
Glucose uptake assay
The glucose uptake assay was performed as described previously [30, 59, 62] and used as a measure of ovarian cortex tissue viability. In brief, ovarian cortex tissue fragments (approximately 1 × 1 × 1 mm) were cultured in DMEM/10% FCS/40 μg/mL Gentamycin. After 4 days, the glucose level in the spent culture medium was measured to determine glucose uptake per hour per mg of ovarian tissue. The glucose uptake assay was performed in four replicates per subject and condition.
Morphology of follicles
Follicle damage was assessed by HE staining of 4 μm sections as described previously [33, 40, 59]. Follicles were considered damaged if they had a pyknotic oocyte or granulosa cells, intensely eosinophilic cytoplasm or shrinkage of follicular cells. At least 100 primordial follicles per subject per condition were examined.
Neutral Red staining
Viability of follicles was analysed by Neutral Red uptake, a proven and reliable method to quantify follicle survival [4, 46, 47]. In brief, after treatment, ovarian cortex tissue was chopped to small fragments (approximately 0.5 × 0.5 × 0.5 mm) using tweezers and scalpel after which the fragments were incubated in 5 mL Ultraculture (Lonza, Basel, Switzerland) supplemented with Collagenase type IA (1 mg/mL, Sigma-Aldrich, Saint Louis, MO, USA) at 37 °C for 1 h to digest the tissue. The tissue was pelleted and resuspended with McCoy’s 5A medium supplemented with Neutral Red (to a final concentration of 50 mg/mL; Sigma-Aldrich, Saint Louis, MO, USA) for 90 min at 37 °C. A squash preparation was made with the partly dissolved tissue fragments and examined by light microscopy. The ratio between viable (red) and non-viable (colourless) follicles was determined by analysing 100 follicles.
In vitro growth assay
An IVG assay was performed to determine capacity of primordial follicles to grow after purging using a modified procedure described as culture step 1 of an in vitro maturation protocol [54]. Excess stromal tissue was removed from ovarian cortex tissue fragments purged with GSK1070916 and control tissue. Next, the tissue was dissected into approximately 1 × 1 × 0.5 mm cubes by using a scalpel and transferred to a 24-well plate containing 2 mL of IVG culture medium per well, consisting of McCoy’s 5a medium, glutamine (3 mM), penicillin G (0.1 mg/mL), streptomycin (0.1 mg/mL), transferrin (2.5 μg/mL), sodium selenite (4 ng/mL), human insulin (10 ng/mL), recombinant human FSH (rhFSH, 1 ng/mL), ascorbic acid (50 μg/mL) (all obtained from Sigma-Aldrich, Saint Louis, MO, USA) and human serum albumin (1 mg/mL, Sanquin Plasma Products, Amsterdam, The Netherlands). The tissue fragments were subsequently cultured for 8 days at 37 °C in humidified air with 5% CO2. Half the medium was replaced with fresh medium every other day. After 8 days of culture, the fragments were fixed and processed for (immuno)histochemistry. Non-cultured cortex fragments of the same patient were used as controls. HE-stained sections (4 μm) were evaluated and follicles were categorised according to their stage of maturation as previously described [55, 75].
Statistical analysis
Graphpad Prism®, version 5.03 for Windows (Graphpad Software Inc., San Diego, CA, USA), was used for statistical analyses. A paired parametric Student’s t test was applied and a P value <0.05 was considered statistically significant. Data from histology and viability were grouped for statistical analyses.
Ethical approval
This study was approved by the ethics committee of the Radboud university medical centre, Nijmegen, The Netherlands. Written informed consent was obtained from all patients.
Results
Expression of Aurora B and C kinases in CML/AML cells and ovarian cortex tissue
To assess the potential effect of the AURKB/C inhibitor GSK1070916 on myeloid leukaemia metastases in human ovarian tissue, we first used Western blot analysis to examine AURKB and AURKC expression levels in a panel of six myeloid leukaemia cell lines and in human ovarian cortex.
In suspension culture, all CML/AML cell lines tested expressed similar levels of AURKB. In the AML lines NB4 and NOMO-1, a single immunoreactive protein was detected whereas the other cell lines showed expression of an additional, slightly smaller AURKB protein. AURKC was detected at different expression levels in tumour cell lines JURL-MK1, K562, MEG-01 and KG-1a, but was absent in the AML cell lines NB4 and NOMO-1. AURKB expression was not detected in ovarian cortex tissue or testicular tissue, while AURKC was abundantly expressed in testicular tissue and at low levels in ovarian tissue (Fig. 1).

Expression of AURKB and AURKC by Western blot. Expression of AURKB and AURKC examined by Western blot analysis of CML (JURL-MK1, K562 and MEG-01) and AML (KG-1a, NB4 and NOMO-1) cell lines cultured in suspension and human gonadal tissues (ovarian cortex and testicular tissue). GAPDH was used as a loading control. AURKB was detectable in all CML/AML cell lines with as a single protein band of approximately 35 kDa (NB4 and NOMO-1), or in combination with a slightly smaller protein band (JURL-MK1, K562, MEG-01 an KG-1a). AURKB was undetectable in ovarian cortex tissue or testicular tissue. AURKC was detected in JURL-MK1, K562, MEG-01, KG-1a, ovarian cortex tissue and testicular tissue at different expression levels but absent in NB4 and NOMO-1
Immunohistochemical staining for AURKB and AURKC was performed to confirm the expression profile obtained by Western blot and to further delineate which cell type expressed AURKC in the ovarian cortex tissue (Fig. 2).

Expression of AURKB and AURKC by immunohistochemistry. Expression of AURKB was examined for all CML and AML cells in suspension and for ovarian cortex tissue by immunohistochemistry with cytokeratin AE1/AE3 as a control for small follicles (panel a). AURKB expression was not detectable in the oocytes or granulosa cells of either primordial, primary or secondary follicles (panel b and c). AURKB expression was detectable in the majority of cells of the CML/AML cell lines in suspension (only shown for the AML cell line KG-1a, panel d). Expression of AURKC was examined for CML and AML cells in suspension and ovarian cortex tissue separately. No AURKC expression was detectable in the oocytes or granulosa cell of primordial or primary follicles (panel e), but AURKC was present in oocytes of secondary follicles (panel f). AURKC expression was clearly detectable in the majority of CML/AML cell lines in suspension (only shown for the AML cell line KG-1a, panel g). Scale bars represent 20 μm
The polyclonal antibodies against AURKB and AURKC gave high background signals of extra cellular matrix components between cells, while leaving small follicles unstained (Fig. 2b, c, e and f). AURKB could not be detected in oocytes, granulosa cells of primordial, primary or secondary follicles or ovarian stromal cells. In contrast to AURKB, the oocytes of secondary follicles were positive for AURKC (compare Fig. 2c and f). Expression levels of AURKB and AURKC proteins fluctuate throughout the various stages of mitosis, which is reflected by the difference in immunohistochemical staining intensity of these proteins between individual cells (Fig. 2d and g) [6, 7].
Purging ovarian cortex tissue by inducing mitotic catastrophe in CML and AML tumour foci
Tissue sections derived from cultured human ovarian cortex fragments micro-injected with either CML or AML cells all showed efficient formation of numerous small tumour masses throughout the cortex fragment (Fig. 3a–f). Identification of CML/AML cells in ovarian cortex tissue was relatively straightforward based on the large size of the leukaemic cells, shape and HE-staining intensity. The size of tumour foci and the degree of cell dissemination varied considerably between cell lines. JURL-MK1, K562 and KG-1a all formed both small and large tumour foci of tightly packed cells whereas MEG-01, NB4 and NOMO-1 only formed small foci. K562, MEG-01, NB4 and NOMO-1 tumours showed extensive dissemination in the surrounding tissue (Table 2).

CML and AML tumour foci in ovarian cortex are sensitive to GSK1070916. HE staining showing that CML tumour foci (JURL-MK, K562 and MEG-01) and AML tumour foci (KG-1a, NB4 and NOMO-1) were abundantly present in control human ovarian cortex treated with solvent-only (panel a–f, white arrowheads indicate tumour foci, the white arrow points at a primordial follicle). After treatment for 24 h with 1 μM GSK1070916 followed by an additional culture for 6 days, tumour foci of JURL-MK1, K562, KG-1a, NB4 and NOMO-1 cells could no longer be detected in ovarian cortex tissue. Large syncytia with sometimes large vacuoles (panel g–r, black arrowheads indicate syncytia) were present in cortex tissue harbouring JURL-MK1, K562 and KG1-a cells after treatment with 1 μm GSK1070916. Apoptotic bodies (grey arrow heads) were present in all cell lines in ovarian cortex tissue after purging except for MEG-01, whereas NB4 and NOMO-1 showed almost exclusively apoptotic bodies with few small syncytia (panel q–r). In contrast to the other five cell lines, MEG-01 showed mostly tumour foci harbouring morphologically normal tumour cells (white arrowheads in panel i and o) next to few small syncytia with up to four nuclei after treatment (black arrowhead in panel o). Scale bars represent 20 μm in panel a–l and 10 μm in panel m-r
We investigated the purging capacity of a 24 h exposure to the AURKB/C inhibitor GSK1070916 of ovarian cortex tissue fragments harbouring CML or AML tumour foci. In the control tissue treated for 24 h with solvent-only all tumour cell lines displayed abundant numbers of tumour foci throughout the tissue fragment (Fig. 3a–f). The tumoricidal effect of GSK1070916 was determined by exposing ovarian cortex fragments with CML or AML tumour foci to 1 μM GSK1070916 for 24 h, followed by extensive washing to remove the inhibitor. The treated fragments were cultured for an additional 6 days before analysis to allow any remaining cancer cells to form new foci. To confirm the absence of morphologically normal tumour cells, serial sections of GSK1070916 treated tissue of all cell lines all in three different subjects was examined histologically. For each cell line, a total of 105 to 173 tissue sections, representing 9.3 to 17.4 cm2 of ovarian cortex tissue surface, was examined.
All cell lines reacted differently to GSK1070916 exposure. Whereas JURL-MK1 showed many syncytia with a diameter up to 40 μm containing up to 11 nuclei (Fig. 3g and m), K562 and KG-1a displayed even larger syncytia (≤ 80 μm) with up to 30 nuclei (Fig. 3h, j, n and p). Tumour foci of AML cell lines NB4 and NOMO-1 were almost exclusively reduced to apoptotic bodies with only few small syncytia (Fig. 3k, l, q and r). The megakaryoblast leukaemia cell line MEG-01 derived tumour foci appeared to be less sensitive to GSK1070916 treatment since only very few small syncytia could be detected among large numbers of morphologically normal MEG-01 cells (Fig. 3i and o).
Next, we investigated the presence of viable and apoptotic tumour cells in GSK1070916 treated tissue by immunohistochemical staining for the nuclear proliferation marker Ki-67 and the apoptosis marker AC3. Most syncytia were positive for Ki-67, suggesting that in these aberrant multinuclear structures part of the processes associated with cell proliferation are still active (Fig. 4g, h, j–l). Interestingly, syncytia harbouring Ki-67-positive nuclei do not show apoptosis as indicated by the absence of AC3 staining (compare Fig. 4h, n, j and p). Most of the smaller structures directly adjacent to the intact syncytia were positive for AC3, indicating large scale apoptosis.

Immunohistochemical analysis of ovarian cortex tissue harbouring CML and AML tumour foci after purging for 24 h with 1 μM GSK1070916. Large-scale formation of syncytia and apoptotic bodies in human ovarian tissue containing CML (JURL-MK1 and K562) and AML (KG-1a, NB4 and NOMO-1) tumour foci after 24-h treatment with 1 μM GSK1070916 followed by 6 days of culture. Nuclei of intact syncytia were positive for proliferation marker Ki-67 (panel g–j) whereas the degraded syncytia and surrounding apoptotic bodies were positive for the apoptotic marker anti-active caspase 3 (AC3) (panel m–r). Note that MEG-01 cells did not show extensive formation of syncytia or apoptotic bodies (panel c) with cells being positive for Ki-67 (panel i) but negative for AC3 (panel o). Scale bars represent 10 μm except in panel b, h and n where it represents 25 μm
The MEG-01 tumour foci reacted very differently to GSK1070916 treatment compared to the other cell lines as judged by the presence of histologically normal and mitotically active cells without any evidence for apoptosis (Fig. 4c, i and o).
Purging ovarian cortex tissue by inducing mitotic catastrophe in primary AML cells
To confirm the tumoricidal effect on primary AML cells, the purging experiment was repeated with primary cells of four AML patients. Similar to the cell lines, the primary AML cells formed both small and large tumour foci of tightly packed cells with dissemination in the surrounding tissue when treated for 24 h with solvent-only (Fig. 5a and c). In these tumour foci, a large number of cells were positive for the proliferation marker Ki67 (Fig. 5e and g), while only few apoptotic cells were observed (Fig. 5i and k). After the exposure to 1 μM GSK1070916 for 24 h, the primary AML cell foci of all four patients showed formation of both large and small syncytia (up to 28 nuclei, Fig. 5b and d) with signs of extensive apoptosis in some syncytia (Fig. 5j and l), while other syncytia contained numerous nuclei that were positive for Ki67 (Fig. 5f, note that the histology in two patients is shown. See supplementary Fig. 1 for the histology in the other two patients). To confirm the absence of morphologically normal tumour cells, serial sections of GSK1070916 treated tissue were examined histologically and no foci of morphologically normal tumour cells could be detected.

Primary AML tumour foci in ovarian cortex are sensitive to GSK1070916. Experimentally induced tumour foci of primary AML cells in human ovarian cortex tissue treated with solvent only showed normal cell morphology (panel a and c; white arrowheads indicate tumour foci) with a substantial part of the AML cells positive for the proliferation marker Ki67 (panel e and g) and very few cells positive for the apoptotic marker active caspase-3 (AC3, panel i and k). After treatment for 24 h with 1 μM GSK1070916 foci of normal tumour cells could no longer be detected and AML cells gave rise to large syncytia containing multiple nuclei and sometimes large vacuoles (panel b and d, black arrowheads indicate syncytia). Most nuclei of intact syncytia were positive for the proliferation marker Ki-67 (panel f and h) whereas degrading syncytia and apoptotic bodies were positive for the apoptotic marker active caspase-3 (panel j and l). Scale bars represent 30 μm. In supplemental Fig. 1, the results of purging primary cells from ovarian cortex tissue of two additional AML patients are shown
Inhibition of AURKB/C has no effect on ovarian tissue viability or follicular integrity
The effect of the 24 h purging protocol with 1 μM GSK1070916 on viability and follicular integrity of ovarian cortex tissue was examined using four different assays.
Glucose uptake assay
Metabolic activity of ovarian cortex tissue from two out of three subjects was not affected by treatment with GSK1070916 as glucose uptake was statistically not different from tissue exposed to solvent only. In the tissue from the third subject, a slight but statistically significant difference in glucose uptake in favour of the GSK1070916 treated tissue was observed (Table 3). In line with previous reports [30, 59, 68], glucose uptake by ovarian cortex tissue in vitro is variable between subjects ranging from 5.675 to 9.050 nmol/mg tissue/h.
Morphology of follicles
Morphological examination of ovarian follicles with standard HE stained sections from ovarian cortex treated with 1 μM GSK1070916 showed no statistically significant difference regarding the percentage of normal follicles compared with the control (P > 0.05, Fig. 6a). The majority of follicles were in the primordial stage.

Morphology and viability of follicles. Ovarian cortex tissue was exposed to a 24-h ex vivo treatment of solvent-only or 1 μM GSK1070916, washed and cultured for an additional 24 h, to allow tissue damage to become apparent. At least 100 follicles were evaluated per patient per condition. a Graphical depiction of the percentage of intact and damaged follicles after treatment. Morphology was examined by HE staining. No statistically significant difference (P = 0.83) was observed between follicles from the control or the tissue treated with the inhibitor. b Graphical depiction of the percentage of viable Neutral red positive follicles and non-viable Neutral red negative follicles. Ovarian cortex tissue was examined by Neutral red staining. No statistically significant difference (P = 0.92) was observed between follicles from the control or the tissue treated with the inhibitor
Neutral red uptake
In addition to follicular morphology, we examined the effect of GSK1070916 on the viability of preantral follicles in ovarian cortex tissue by Neutral red staining (Fig. 6b). The percentage of viable Neutral red positive follicles was not different between the fragments treated with 1 μM GSK1070916 or solvent (P > 0.05).
In vitro growth
Ovarian cortex tissue of two subjects was exposed to a 24h ex vivo treatment of 1 μM GSK1070916 or solvent-only and subsequently cultured for 8 days with IVG culture medium. Follicles were evaluated by standard HE staining and by immunohistochemistry for AC3 and Ki-67. Compared to the uncultured fragments (Fig. 7a and e), follicular growth could be clearly observed in fragments treated with solvent and in the GSK1070916 treated fragments, as illustrated by a shift from primordial to primary and secondary follicles after 8 days of IVG (Fig. 7b, f and i). GSK1070916-treated fragments showed no signs of apoptosis as illustrated by lack of AC3 expression in oocytes and granulosa cells after culture (Fig. 7c and g). Ki-67-positive granulosa cells could be detected in follicles of GSK1070916 treated tissue fragments indicating granulosa cell proliferation was still ongoing at day 8 of IVG (Fig. 7d and h).

In vitro growth of follicles after GSK1090716 exposure. Ovarian cortex tissue of two subjects was exposed to 1 μM GSK1070916 or solvent-only for 24 h, washed and subsequently cultured in IVG culture medium for 8 days. a Graphical depiction of percentage of primordial, primary and secondary follicles in ovarian cortex tissue of two subjects in uncultured tissue (control) and tissue subjected to IVG for 8 days after a 24 h exposure to 1 μM GSK1070916 or solvent only. At least 100 follicles were evaluated per sample. Standard HE staining showed mainly primordial follicles in uncultured ovarian cortex tissue (panel b and f) and a substantial increase in secondary follicles after IVG for 8 days irrespective of previous GSK1070916 treatment (panel c and g). Immunohistochemistry with AC3 indicated apoptosis was absent in oocytes or granulosa cells in GSK1070916 treated tissue (panel d and h) whereas Ki-67 positive granulosa cells showed ongoing folliculogenesis in secondary follicles (panel e and i, black arrowheads point to Ki-67 positive granulosa cells). Scale bars represent 20 μm
Discussion
Autotransplantation of cryopreserved ovarian cortex tissue remains a procedure that harbours the risk of reintroducing the cancer to the cured patient through malignant cells that may be present in the graft. Standard screening methods of ovarian cortex tissue prior to autotransplantation involve immuno-histological analysis and tumour-specific PCR examination, sometimes combined with xenotransplantation to immunodeficient mice, multicolor flow cytometry or next generation sequencing [15, 64, 72, 83]. Since the analysed tissue fragment is no longer available for autotransplantation, these screening methods do not assure that the remaining fragments are devoid of malignant cells. Tissue fragments of the same ovary were shown to give different results when tested for the presence of malignant cells by tumour-specific PCR leading to sampling bias [5, 9, 65]. To circumvent this problem, several strategies aimed at separating the ovarian follicles from the potentially contaminated stromal compartment before transplantation are currently being pursued [2, 48, 54, 63, 78]. However, these methods are not yet clinically available and rely on dissociation of the tissue by separating the follicles from their naturally surrounding stromal cells. Folliculogenesis depends heavily on the interaction of follicles with the surrounding tissue and dissociation may have an impact on follicle maturation after transplantation. To avoid problems caused by sampling bias or tissue dissociation, we recently developed a protocol based on pharmacological treatment to purge human ovarian cortex tissue of malignant cells without compromising ovarian tissue or follicular integrity. Rhabdomyosarcoma tumour foci were completely eliminated from ovarian tissue by a 24h ex vivo treatment using Verteporfin, an inhibitor of YAP/TAZ oncoproteins [36, 49, 59, 77]. Purging of ovarian cortex tissue therefore offers a potentially safe strategy to eliminate malignant cells from all tissue fragments prior to autotransplantation without affecting tissue viability.
In the current study, we have extended our efforts by developing efficient purging protocols for other clinically relevant malignancies. We investigated the use of GSK1070916, an onco-suppressive agent that induces mitotic catastrophe by driving cells to an irreversible antiproliferative fate of death [35, 52, 58, 80]. GSK1070916 exerts this effect by inhibiting AURKB/C and inhibits malignant cell proliferation in a wide range of tumour cell lines in vitro and in xenograft models of human tumours [35]. Aurora kinase inhibitors have not been approved for systemic cancer treatment in humans and only a Phase I clinical trial has been conducted with GSK1070916 [7, 56]. Neutropenia caused by Aurora B toxicity was found to be the limiting factor in achieving an effective systemic treatment in a clinical setting [44, 45, 56]. Obviously, this is of no concern to the current study in which ovarian cortex tissue is purged from malignant cells ex vivo, in the absence of perfusion.
CML and AML cell lines were selected to represent malignancies that are clinically relevant and an indication for OTC [14]. AML is one of the most common forms of haematological malignancies that occurs in girls and young women and requires immediate start of anti-cancer treatment with fertility threatening alkylating agents, sometimes followed by haematopoietic stem cell transplantation which further increases the risk of infertility [67]. Ovarian tissue fragments from patients with CML and AML have a high chance of being contaminated with malignant cells and ovarian cortex from a CML patient has actually been shown to transfer the disease upon xenotransplantation to immunodeficient mice [1, 3, 17]. Although ovarian cortex fragments of leukaemia patients are the most suitable for our studies, they are not available in the quantity required for purging experiments. Furthermore, because of sampling bias, it is impossible to predict which cortex fragments will actually contain metastases without a prior analysis that will render the fragment unsuitable for ex vivo purging. Moreover, cortex fragments of the same ovary may differ with respect to the presence of malignant cells, with some fragments being positive while the others were not [9, 65]. Cryopreserved ovarian cortex fragments from cancer patients might therefore be less suitable for purging experiments because it is not possible to determine the efficiency of purging when we are unable to establish the presence of malignant cells in the tissue before treatment.
To analyse the effect of GSK1070916 on metastasised CML and AML cells, we therefore used an established tumour model based on the micro-injection of cancer cells (both AML and CML cell lines and primary AML cells) into human ovarian cortex tissue followed by several days of culture to allow malignant cells to form small metastases [59, 61]. We first opted for cell lines since the cancer-type specific mutations are present in these cells and their long-term proliferation potential in vitro was required for our experiments. Furthermore, cell lines are widely and successfully used in cancer research and anti-cancer drug discovery [51]. To confirm our findings with the cell lines, we expanded our experiments using primary cancer cells which may represent the in vivo situation more closely.
The sensitivity of cancer cells to pharmacological inhibition depends on the rigidity and specific extra-cellular components of their microenvironment [25, 50, 59]. Our approach allowed us to test the inhibitory effect of GSK1070916 on leukaemic cells in the appropriate microenvironment. The ex vivo treatment was limited to 24 h in order to minimise culture induced activation of primordial follicles and subsequent follicular burn-out after autotransplantation [28, 55]. In vitro activation of small follicles has been shown to require special culturing conditions including ovarian fragmentation and Akt pathway stimulation [37, 38]. In addition, a 48-h culture period of ovarian cortex tissue prior to transplantation has resulted in live birth [37], suggesting that the short ex vivo culture used in the current study is unlikely to lead to extensive activation of follicles. Moreover, follicles in control tissue cultured for up to 10 days did not show any obvious signs of activation.
After purging, the entire cortex fragment was sectioned and analysed for the presence of morphologically normal tumour cells. Our extensive histological analysis revealed that tumour foci of both CML (JURL-MK1, K562) cells, AML (KG-1a, NB4 and NOMO-1) cells and primary AML cells were completely eliminated by a 24h treatment with 1 μM of GSK1070916, leaving only large multinuclear syncytia and apoptotic bodies. Inhibition of Aurora B is known to cause defective mitosis and cytokinesis failure leading to large polyploid syncytia and ultimately to apoptosis [35, 52, 58, 80].
This is in line with our immunohistochemical staining results for the proliferation marker Ki-67 and apoptosis marker AC3. Syncytia from both the cell lines and the primary cells were negative for AC3 but still expressed Ki-67, indicating that these cells were not arrested in mitosis but failed to divide. The Ki-67 negative/AC-3 positive small cellular bodies next to the syncytia with eosinophilic cytoplasm and condensed chromatin are most probably syncytia in the process of apoptosis. Strikingly, the number of nuclei (up to 30 nuclei per syncytia in the cell lines and 28 nuclei in the primary cells) observed in histological sections of syncytia was frequently higher than expected considering a 6-day culture period after purging and the 30–50 h doubling time of the CML and AML cells in suspension culture. This suggests that mitosis, in the absence of cytokinesis, of these cells is accelerated after treatment with GSK1070916, leading to the formation of more nuclei than under normal growth conditions.
The acute megakaryocytic leukaemia cell line MEG-01 was largely unresponsive to purging with GSK1070916. Megakaryocytes undergo polyploidization prior to the formation of a large number of platelets and subsequent release into the bloodstream. Downregulation of Aurora B during late anaphase of the endomitotic cycle has been implicated in polyploid formation [39, 81] and is therefore most likely the reason that the MEG-01 cells did not show extensive formation of syncytia or apoptosis after GSK1070916 treatment [32, 76]. Furthermore, cell lines with a polyploid phenotype and high chromosome number are associated with resistance to GSK1070916 [58]. Stratification by phenotyping and karyotyping malignant cells of patients could therefore serve as resistance-associated marker for prediction of response to purging with GSK1070916.
Western blot analysis of AURKB/C expression in CML/AML cell lines revealed AURKC and two AURKB proteins, with a slight difference in molecular weight, for JURL-MK1, K562, MEG-01 and KG-1a. The AML cell lines NB4 and NOMO-1 only expressed the larger AURKB protein but no AURKC. Aurora kinases are subjected to alternative splicing and different functions have been ascribed to the splice variants [26, 79]. Possibly, differences in the AURKB/C protein expression profile are related to their reaction to purging with GSK1070916 as NB4 and NOMO-1 showed mostly apoptosis and did not show the large scale formation of syncytia as observed in JURL-MK1, K562 and KG-1a cell lines.
In this study, we examined purging for 24 h with 1 μM GSK1070916 on CML/AML metastases of cell lines and primary cells in ovarian cortex by extensive histological examination after serial sectioning the entire treated tissue fragments to ensure no normal tumour cells were present. This clearly exceeds the standard clinical pathological examination involving only a limited number of sections of ovarian cortex prior to autotransplantation. Ideally, we would have confirmed the absence of CML/AML tumour cells by sensitive molecular techniques. We have shown previously effective ex vivo purging of ovarian cortex tissue from contaminating alveolar rhabdomyosarcoma with Verteporfin could be confirmed by an RT-PCR for the presence of the PAX3-FOXO1 transcript [59]. However, in contrast to Verteporfin, purging with GSK1070916 not only leads to large scale apoptosis but initially also leads to the formation of syncytia. Although these large multinuclear cells will ultimately go into apoptosis [35, 52, 58, 80], they are metabolically still active at the end of the culture period and therefore probably contain detectable tumour-specific transcripts. Long-term culture of the purged ovarian cortex fragments beyond day 6 to allow any remaining syncytia to be eliminated through apoptosis has been shown to result in significant loss of viability of ovarian tissue and is therefore not reliable [61]. Xenografting to immunodeficient mice could further substantiate the absence of viable tumour cells in purged ovarian cortex tissue.
In addition to its oncosuppresive properties, GSK1070916 should obviously have no compromising effects on the ovarian cortex tissue to maximise the chances of a successful pregnancy after autotransplantation. Our purging protocol with GSK1070916 did not affect overall tissue viability, follicle morphology, follicle viability or the capacity of small unilaminar follicles to develop into secondary follicles. This was not unexpected, as isolated ovarian cortex tissue is essentially mitotically and meiotically silent and not dependent on AURKB/C function.
To determine the capacity of primordial follicles to grow after purging, we used the first culture step of a recently published multi-step in vitro maturation protocol [54]. Culturing the tissue beyond the first step was not performed since growth of small follicles to secondary/multi-laminar follicles is already observed after this first step while the remaining steps of the procedure are technically challenging and lead to morphologically abnormal cumulus oocyte complexes [54].
Expression of Aurora B was not detected in ovarian cortex tissue, either by Western blot or immunohistochemistry. Aurora C was absent from small follicles and only detectable in oocytes at more advanced stages of follicular development. Inhibition of Aurora B/C is therefore unlikely to influence the capacity of ovarian cortex tissue to restore fertility, as this is thought to depend mainly on the pool of small follicles [22, 41]. Restoration of ovarian activity and subsequent recruitment of these follicles after autotransplantation takes between 3.5 and 6.5 months [21]. An effect on these newly recruited follicles by any remaining GSK1070916 in the graft is not to be expected in view of its low molecular weight (0.5 kDa), promoting rapid removal during the extensive washing steps following the purging procedure and massive dilution of the inhibitor directly after transplantation in the surrounding patients’ fluids and tissues.
In mice, Aurora C is essential for proper meiotic cell division during spermatogenesis. Female Aurkb and Aurkc single or double knockout female mice, however, are still fertile [60]. In humans, homozygous AURKC mutation is associated with male infertility, but women carrying this mutation were reported to be fertile [11]. This indicates that, in contrast to spermatogenesis, oogenesis might not require AURKC. Taken together this indicates our ex vivo purging protocol is unlikely to impair the function of the ovarian cortex tissue after autotransplantation.
The results in this paper indicate that purging by targeting the mitotic catastrophe signalling pathway in CML and AML metastases-contaminated ovarian cortex tissue intended for fertility preservation purposes is possible by a short-term incubation with GSK1070916, a specific AURKB/C inhibitor. In addition to the techniques already available to prevent reseeding of CML/AML through transplantation of contaminated ovarian tissue, purging with GSK1070916 may further contribute to the safety of fertility restoration in leukaemia patients currently considered to be at high risk for ovarian involvement.
Data availability
Not applicable.
Code availability
Not applicable.
References
Abir R, Aviram A, Feinmesser M, Stein J, Yaniv I, Parnes D, et al. Ovarian minimal residual disease in chronic myeloid leukaemia. Reprod BioMed Online. 2014;28:255–60.
Amorim CA, Shikanov A. The artificial ovary: current status and future perspectives. Future Oncol. 2016;12:2323–32.
Bastings L, Beerendonk CC, Westphal JR, Massuger LF, Kaal SE, van Leeuwen FE, et al. Autotransplantation of cryopreserved ovarian tissue in cancer survivors and the risk of reintroducing malignancy: a systematic review. Hum Reprod Update. 2013;19:483–506.
Bastings L, Westphal JR, Beerendonk CC, Bekkers RL, Zusterzeel PL, Hendriks JC, et al. Clinically applied procedures for human ovarian tissue cryopreservation result in different levels of efficacy and efficiency. J Assist Reprod Genet. 2016;33:1605–14.
Bockstaele L, Tsepelidis S, Dechene J, Englert Y, Demeestere I. Safety of ovarian tissue autotransplantation for cancer patients. Obstet Gynecol Int. 2012;2012:495142.
Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol. 2003;4:842–54.
Cheung CH, Sarvagalla S, Lee JY, Huang YC, Coumar MS. Aurora kinase inhibitor patents and agents in clinical testing: an update (2011 - 2013). Expert Opin Ther Pat. 2014;24:1021–38.
Cronin KA, Lake AJ, Scott S, Sherman RL, Noone AM, Howlader N, et al. Annual report to the nation on the status of cancer, part I: national cancer statistics. Cancer. 2018;124:2785–800.
Diaz-Garcia C, Herraiz S, Such E, Andres MDM, Villamon E, Mayordomo-Aranda E, et al. Dexamethasone does not prevent malignant cell reintroduction in leukemia patients undergoing ovarian transplant: risk assessment of leukemic cell transmission by a xenograft model. Hum Reprod. 2019;34:1485–93.
Dieterich K, Soto Rifo R, Faure AK, Hennebicq S, Ben Amar B, Zahi M, et al. Homozygous mutation of AURKC yields large-headed polyploid spermatozoa and causes male infertility. Nat Genet. 2007;39:661–5.
Dieterich K, Zouari R, Harbuz R, Vialard F, Martinez D, Bellayou H, et al. The Aurora Kinase C c.144delC mutation causes meiosis I arrest in men and is frequent in the North African population. Hum Mol Genet. 2009;18:1301–9.
Dittrich R, Lotz L, Fehm T, Krussel J, von Wolff M, Toth B, et al. Xenotransplantation of cryopreserved human ovarian tissue--a systematic review of MII oocyte maturation and discussion of it as a realistic option for restoring fertility after cancer treatment. Fertil Steril. 2015;103:1557–65.
Dolmans MM. Recent advances in fertility preservation and counseling for female cancer patients. Expert Rev Anticancer Ther. 2018;18:115–20.
Dolmans MM, Donnez J. Indications for fertility preservation in women from malignant diseases to benign conditions to age-related fertility decline. Minerva Ginecol. 2018;70:402–7.
Dolmans MM, Iwahara Y, Donnez J, Soares M, Vaerman JL, Amorim CA, et al. Evaluation of minimal disseminated disease in cryopreserved ovarian tissue from bone and soft tissue sarcoma patients. Hum Reprod. 2016;31:2292–302.
Dolmans MM, Luyckx V, Donnez J, Andersen CY, Greve T. Risk of transferring malignant cells with transplanted frozen-thawed ovarian tissue. Fertil Steril. 2013;99:1514–22.
Dolmans MM, Marinescu C, Saussoy P, Van Langendonckt A, Amorim C, Donnez J. Reimplantation of cryopreserved ovarian tissue from patients with acute lymphoblastic leukemia is potentially unsafe. Blood. 2010;116:2908–14.
Donnez J, Dolmans MM. Ovarian cortex transplantation: 60 reported live births brings the success and worldwide expansion of the technique towards routine clinical practice. J Assist Reprod Genet. 2015;32:1167–70.
Donnez J, Dolmans MM. Fertility preservation in women. N Engl J Med. 2017;377:1657–65.
Donnez J, Dolmans MM, Diaz C, Pellicer A. Ovarian cortex transplantation: time to move on from experimental studies to open clinical application. Fertil Steril. 2015;104:1097–8.
Donnez J, Dolmans MM, Pellicer A, Diaz-Garcia C, Sanchez Serrano M, Schmidt KT, et al. Restoration of ovarian activity and pregnancy after transplantation of cryopreserved ovarian tissue: a review of 60 cases of reimplantation. Fertil Steril. 2013;99:1503–13.
Donnez J, Jadoul P, Squifflet J, Van Langendonckt A, Donnez O, Van Eyck AS, et al. Ovarian tissue cryopreservation and transplantation in cancer patients. Best Pract Res Clin Obstet Gynaecol. 2010;24:87–100.
Drexler HG, MacLeod RA. Leukemia-lymphoma cell lines as model systems for hematopoietic research. Ann Med. 2003;35:404–12.
Dutta-Simmons J, Zhang Y, Gorgun G, Gatt M, Mani M, Hideshima T, et al. Aurora kinase A is a target of Wnt/beta-catenin involved in multiple myeloma disease progression. Blood. 2009;114:2699–708.
Estrugo D, Fischer A, Hess F, Scherthan H, Belka C, Cordes N. Ligand bound beta1 integrins inhibit procaspase-8 for mediating cell adhesion-mediated drug and radiation resistance in human leukemia cells. PLoS One. 2007;2:e269.
Fellmeth JE, Gordon D, Robins CE, Scott RT Jr, Treff NR, Schindler K. Expression and characterization of three Aurora kinase C splice variants found in human oocytes. Mol Hum Reprod. 2015;21:633–44.
Fu J, Bian M, Jiang Q, Zhang C. Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res. 2007;5:1–10.
Gavish Z, Peer G, Roness H, Cohen Y, Meirow D. Follicle activation and 'burn-out' contribute to post-transplantation follicle loss in ovarian tissue grafts: the effect of graft thickness. Hum Reprod. 2014;29:989–96.
Gellert SE, Pors SE, Kristensen SG, Bay-Bjorn AM, Ernst E, Yding Andersen C. Transplantation of frozen-thawed ovarian tissue: an update on worldwide activity published in peer-reviewed papers and on the Danish cohort. J Assist Reprod Genet. 2018;35:561–70.
Gerritse R, Beerendonk CC, Westphal JR, Bastings L, Braat DD, Peek R. Glucose/lactate metabolism of cryopreserved intact bovine ovaries as a novel quantitative marker to assess tissue cryodamage. Reprod BioMed Online. 2011;23:755–64.
Gerstl B, Sullivan E, Chong S, Chia D, Wand H, Anazodo A. Reproductive outcomes after a childhood and adolescent young adult cancer diagnosis in female cancer survivors: a systematic review and meta-analysis. J Adolesc Young Adult Oncol. 2018;7:627–42.
Goldenson B, Crispino JD. The aurora kinases in cell cycle and leukemia. Oncogene. 2015;34:537–45.
Gougeon A, Testart J. Germinal vesicle breakdown in oocytes of human atretic follicles during the menstrual cycle. J Reprod Fertil. 1986;78:389–401.
Grondahl ML, Borup R, Vikesa J, Ernst E, Andersen CY, Lykke-Hartmann K. The dormant and the fully competent oocyte: comparing the transcriptome of human oocytes from primordial follicles and in metaphase II. Mol Hum Reprod. 2013;19:600–17.
Hardwicke MA, Oleykowski CA, Plant R, Wang J, Liao Q, Moss K, et al. GSK1070916, a potent Aurora B/C kinase inhibitor with broad antitumor activity in tissue culture cells and human tumor xenograft models. Mol Cancer Ther. 2009;8:1808–17.
Kandoussi I, Lakhlili W, Taoufik J, Ibrahimi A. Docking analysis of verteporfin with YAP WW domain. Bioinformation. 2017;13:237–40.
Kawamura K, Cheng Y, Suzuki N, Deguchi M, Sato Y, Takae S, et al. Hippo signaling disruption and Akt stimulation of ovarian follicles for infertility treatment. Proc Natl Acad Sci U S A. 2013;110:17474–9.
Kawamura K, Kawamura N, Hsueh AJ. Activation of dormant follicles: a new treatment for premature ovarian failure? Curr Opin Obstet Gynecol. 2016;28:217–22.
Kawasaki A, Matsumura I, Miyagawa J, Ezoe S, Tanaka H, Terada Y, et al. Downregulation of an AIM-1 kinase couples with megakaryocytic polyploidization of human hematopoietic cells. J Cell Biol. 2001;152:275–87.
Keros V, Xella S, Hultenby K, Pettersson K, Sheikhi M, Volpe A, et al. Vitrification versus controlled-rate freezing in cryopreservation of human ovarian tissue. Hum Reprod. 2009;24:1670–83.
Kim S, Lee Y, Lee S, Kim T. Ovarian tissue cryopreservation and transplantation in patients with cancer. Obstet Gynecol Sci. 2018;61:431–42.
Kimmins S, Crosio C, Kotaja N, Hirayama J, Monaco L, Hoog C, et al. Differential functions of the Aurora-B and Aurora-C kinases in mammalian spermatogenesis. Mol Endocrinol. 2007;21:726–39.
Koeffler HP, Golde DW. Human myeloid leukemia cell lines: a review. Blood. 1980;56:344–50.
Kollareddy M, Zheleva D, Dzubak P, Brahmkshatriya PS, Lepsik M, Hajduch M. Aurora kinase inhibitors: progress towards the clinic. Investig New Drugs. 2012;30:2411–32.
Komlodi-Pasztor E, Sackett DL, Fojo AT. Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res. 2012;18:51–63.
Kristensen SG, Liu Q, Mamsen LS, Greve T, Pors SE, Bjorn AB, et al. A simple method to quantify follicle survival in cryopreserved human ovarian tissue. Hum Reprod. 2018;33:2276–84.
Langbeen A, Jorssen EP, Granata N, Fransen E, Leroy JL, Bols PE. Effects of neutral red assisted viability assessment on the cryotolerance of isolated bovine preantral follicles. J Assist Reprod Genet. 2014;31:1727–36.
Laronda MM, Rutz AL, Xiao S, Whelan KA, Duncan FE, Roth EW, et al. A bioprosthetic ovary created using 3D printed microporous scaffolds restores ovarian function in sterilized mice. Nat Commun. 2017;8:15261.
Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–5.
Lovitt CJ, Shelper TB, Avery VM. Advanced cell culture techniques for cancer drug discovery. Biology (Basel). 2014;3:345–67.
Masters JR. Human cancer cell lines: fact and fantasy. Nat Rev Mol Cell Biol. 2000;1:233–6.
Mc Gee MM. Targeting the mitotic catastrophe signaling pathway in cancer. Mediat Inflamm. 2015;2015:146282.
McGrath JP, Williamson KE, Balasubramanian S, Odate S, Arora S, Hatton C, et al. Pharmacological inhibition of the histone lysine demethylase KDM1A suppresses the growth of multiple acute myeloid leukemia subtypes. Cancer Res. 2016;76:1975–88.
McLaughlin M, Albertini DF, Wallace WHB, Anderson RA, Telfer EE. Metaphase II oocytes from human unilaminar follicles grown in a multi-step culture system. Mol Hum Reprod. 2018;24:135–42.
McLaughlin M, Kinnell HL, Anderson RA, Telfer EE. Inhibition of phosphatase and tensin homologue (PTEN) in human ovary in vitro results in increased activation of primordial follicles but compromises development of growing follicles. Mol Hum Reprod. 2014;20:736–44.
McNeish I, Anthoney A, Loadman P, Berney D, Joel S, Halford SER, et al. A phase I pharmacokinetic (PK) and pharmacodynamic (PD) study of the selective aurora kinase inhibitor GSK1070916A. J Clin Oncol. 2013;31:2525.
Meirow D, Hardan I, Dor J, Fridman E, Elizur S, Ra'anani H, et al. Searching for evidence of disease and malignant cell contamination in ovarian tissue stored from hematologic cancer patients. Hum Reprod. 2008;23:1007–13.
Moy C, Oleykowski CA, Plant R, Greshock J, Jing J, Bachman K, et al. High chromosome number in hematological cancer cell lines is a negative predictor of response to the inhibition of Aurora B and C by GSK1070916. J Transl Med. 2011;9:110.
Mulder CL, Eijkenboom LL, Beerendonk CCM, Braat DDM, Peek R. Enhancing the safety of ovarian cortex autotransplantation: cancer cells are purged completely from human ovarian tissue fragments by pharmacological inhibition of YAP/TAZ oncoproteins. Hum Reprod. 2019;34:506–18.
Nguyen AL, Drutovic D, Vazquez BN, El Yakoubi W, Gentilello AS, Malumbres M, et al. Genetic interactions between the aurora kinases reveal new requirements for AURKB and AURKC during oocyte meiosis. Curr Biol. 2018;28:3458–3468.e3455.
Peek R, Bastings L, Westphal JR, Massuger LF, Braat DD, Beerendonk CC. A preliminary study on a new model system to evaluate tumour-detection and tumour-purging protocols in ovarian cortex tissue intended for fertility preservation. Hum Reprod. 2015;30:870–6.
Peters IT, Stegehuis PL, Peek R, Boer FL, van Zwet EW, Eggermont J, et al. Noninvasive detection of metastases and follicle density in ovarian tissue using full-field optical coherence tomography. Clin Cancer Res. 2016;22:5506–13.
Pors SE, Ramlose M, Nikiforov D, Lundsgaard K, Cheng J, Andersen CY, et al. Initial steps in reconstruction of the human ovary: survival of pre-antral stage follicles in a decellularized human ovarian scaffold. Hum Reprod. 2019;34:1523–35.
Rodriguez-Iglesias B, Novella-Maestre E, Herraiz S, Diaz-Garcia C, Pellicer N, Pellicer A. New methods to improve the safety assessment of cryopreserved ovarian tissue for fertility preservation in breast cancer patients. Fertil Steril. 2015;104:1493–1502.e1491–1492.
Rosendahl M, Andersen MT, Ralfkiaer E, Kjeldsen L, Andersen MK, Andersen CY. Evidence of residual disease in cryopreserved ovarian cortex from female patients with leukemia. Fertil Steril. 2010;94:2186–90.
Rosendahl M, Greve T, Andersen CY. The safety of transplanting cryopreserved ovarian tissue in cancer patients: a review of the literature. J Assist Reprod Genet. 2013;30:11–24.
Salama M, Anazodo A, Woodruff TK. Preserving fertility in female patients with hematological malignancies: a multidisciplinary oncofertility approach. Ann Oncol. 2019;30:1760–75.
Schleedoorn MJ, Peppelman M, van Erp PEJ, Beerendonk CCM, Nelen W, Braat DDM, et al. Assessment of reflectance confocal microscopy for non-invasive selection of optimal ovarian cortex fragments for autotransplantation. Reprod BioMed Online. 2019;38:999–1009.
Shapira M, Meirow D, Raanani H, Roness H. Impact of first-line cancer treatment on follicle quality in cryopreserved ovarian samples. Hum Reprod. 2020;35:1248–9.
Siegel R, Miller K, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68.
Silber SJ, DeRosa M, Goldsmith S, Fan Y, Castleman L, Melnick J. Cryopreservation and transplantation of ovarian tissue: results from one center in the USA. J Assist Reprod Genet. 2018;35:2205–13.
Soares M, Saussoy P, Maskens M, Reul H, Amorim CA, Donnez J, et al. Eliminating malignant cells from cryopreserved ovarian tissue is possible in leukaemia patients. Br J Haematol. 2017;178:231–9.
Spears N, Lopes F, Stefansdottir A, Rossi V, De Felici M, Anderson RA, et al. Ovarian damage from chemotherapy and current approaches to its protection. Hum Reprod Update. 2019.
Teh A, Izzati UZ, Mori K, Fuke N, Hirai T, Kitahara G, et al. Histological and immunohistochemical evaluation of granulosa cells during different stages of folliculogenesis in bovine ovaries. Reprod Domest Anim. 2018;53:569–81.
Telfer EE, McLaughlin M, Ding C, Thong KJ. A two-step serum-free culture system supports development of human oocytes from primordial follicles in the presence of activin. Hum Reprod. 2008;23:1151–8.
Vitrat N, Cohen-Solal K, Pique C, Le Couedic JP, Norol F, Larsen AK, et al. Endomitosis of human megakaryocytes are due to abortive mitosis. Blood. 1998;91:3711–23.
Wang C, Zhu X, Feng W, Yu Y, Jeong K, Guo W, et al. Verteporfin inhibits YAP function through up-regulating 14-3-3sigma sequestering YAP in the cytoplasm. Am J Cancer Res. 2016;6:27–37.
Xiao S, Zhang J, Romero MM, Smith KN, Shea LD, Woodruff TK. In vitro follicle growth supports human oocyte meiotic maturation. Sci Rep. 2015;5:17323.
Yasen M, Mizushima H, Mogushi K, Obulhasim G, Miyaguchi K, Inoue K, et al. Expression of Aurora B and alternative variant forms in hepatocellular carcinoma and adjacent tissue. Cancer Sci. 2009;100:472–80.
Zekri A, Ghaffari SH, Yaghmaie M, Estiar MA, Alimoghaddam K, Modarressi MH, et al. Inhibitor of Aurora kinase B induces differentially cell death and polyploidy via DNA damage response pathways in neurological malignancy: shedding new light on the challenge of resistance to AZD1152-HQPA. Mol Neurobiol. 2016;53:1808–23.
Zhang Y, Nagata Y, Yu G, Nguyen HG, Jones MR, Toselli P, et al. Aberrant quantity and localization of Aurora-B/AIM-1 and survivin during megakaryocyte polyploidization and the consequences of Aurora-B/AIM-1-deregulated expression. Blood. 2004;103:3717–26.
Zovko A, Yektaei-Karin E, Salamon D, Nilsson A, Wallvik J, Stenke L. Montelukast, a cysteinyl leukotriene receptor antagonist, inhibits the growth of chronic myeloid leukemia cells through apoptosis. Oncol Rep. 2018;40:902–8.
Zver T, Alvergnas-Vieille M, Garnache-Ottou F, Roux C, Amiot C. A new method for evaluating the risk of transferring leukemic cells with transplanted cryopreserved ovarian tissue. J Assist Reprod Genet. 2015;32:1263–6.
Acknowledgements
We would like to thank Melissa Hillebrandt-Roeffen for expert technical assistance and Joanna in ‘t Hout for the help in conducting the statistical analysis.
Funding
Unconditional funding was received from Merck B.V. (The Netherlands), an affiliate of Merck KGaA, Darmstadt, Germany (Number A19-1294).
Author information
Authors and Affiliations
Contributions
The project and experiments were designed by R.P, L.L.E., D.D.M.B. and C.C.M.B. Experiments were performed by L.L.E., R.P., J.L. and T.K.. L.L.E and R.P. drafted the original manuscript. All authors (L.L.E., C.L.M., B.A.R., N.M.M., J.L., T.K, D.D.M.B., C.C.M.B. and R.P.) critically reviewed and revised the manuscript and approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethics committee of the Radboud university medical centre, Nijmegen, The Netherlands. Written informed consent was obtained from all patients.
Consent for publication
Not applicable.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(PNG 4032 kb).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eijkenboom, L., Mulder, C., van der Reijden, B. et al. Purging human ovarian cortex of contaminating leukaemic cells by targeting the mitotic catastrophe signalling pathway. J Assist Reprod Genet 38, 1571–1588 (2021). https://doi.org/10.1007/s10815-021-02081-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10815-021-02081-9