
Abstract
Botryococcus braunii CCALA778 is a green microalga that can produce large amounts of extracellular carbohydrates and therefore is a potential host for industrial applications such as materials, food and pharmaceutical products. The downside of B. braunii is its slow growth and therefore, improvements on the biomass productivity or carbohydrate production will make this microalga more attractive for industrial exploitation. Microalgae grow naturally in the presence of bacteria and these can be beneficial or antagonistic. In outdoor cultivation systems, contamination by bacteria is common. The role or effects of bacteria present in B. braunii CCALA778 are not yet fully elucidated. We used UV-C treatment to reduce bacterial abundance in B. braunii CCALA778 cultures and 16S rRNA gene amplicon MiSeq sequencing for bacterial community analysis. The effect of the reduced amount of bacteria on biomass growth and production of extracellular carbohydrates was analysed. It is shown that UV-C treatment can reduce the bacterial population substantially without harming B. braunii. Bacteria removed by UV-C were antagonistic to B. braunii CCALA778 as in their absence production of biomass and extracellular carbohydrates was enhanced significantly. CCALA778 treated with UV-C accumulated 826 ± 61 mg L−1 of extracellular carbohydrates by day 15 compared with 422 ± 135 mg L−1 accumulated extracellular carbohydrates in the untreated culture.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Botryococcus braunii is a green microalga that can produce long-chain hydrocarbons and high amounts of extracellular carbohydrates (termed here exopolysaccharides) depending on the strain used (Allard and Casadevall 1990; Fernandes et al. 1989; Banerjee et al. 2002; Metzger and Largeau 2005; Gouveia et al. 2017). Hydrocarbons derived from B. braunii can be hydrocracked and transformed into aviation turbine fuel (Hillen et al. 1982). Carbohydrates have a range of industrial uses, including thickeners, stabilizers and gelling agents in food products, as well as in the pharmaceutical and cosmeceutical applications (Buono et al. 2012; Donot et al. 2012; Borowitzka 2013).
Botryococcus braunii is a colony-forming microalga found in fresh and brackish water across the world. Depending on what type of hydrocarbons are produced, B. braunii is subclassified into four chemical races, designated A, B, L and S (Kawachi et al. 2012). Some strains of B. braunii can produce up to 86% hydrocarbons on biomass dry weight basis (Brown et al. 1969), whereas the carbohydrate producing strains can produce up to 4.5 g L−1 of exopolysaccharides (EPS) into the medium. The EPS from B. braunii is mainly composed of galactose and minor quantities of other sugars as fucose, rhamnose, glucose and uronic acid residues (Fernandes et al. 1989; Lupi et al. 1994; Díaz Bayona and Garcés 2014). The drawback of B. braunii is that it is a slow-growing microorganism and therefore it is not currently attractive for industrial applications.
In natural aquatic environments, microalgae coexist with bacteria due to the presence of exopolysaccharide substances surrounding the microalgae (Bell and Mitchell 1972; Ramanan et al. 2015). The interaction between microalgae and bacteria can be mutually beneficial as well as antagonistic in nature (Cole 1982; Cooper and Smith 2015; Segev et al. 2016). The early work from Chirac and colleagues (Chirac et al. 1982), in which they describe the presence of Pseudomonas sp. and Flavobacterium sp. in two strains of B. braunii, concluded that bacteria were not essential for the production of hydrocarbons. However, it was shown that numerous bacteria could exert a beneficial or an antagonistic effect on the growth yields and hydrocarbon content (Chirac et al. 1982). One study using B. braunii Ba10 strain revealed the presence of rod-shaped bacteria in the rim of the colony aggregations of the B. braunii Ba10 strain. The bacteria were proposed to be growth promoting and closely related to Hyphomonadaceae (Tanabe et al. 2015). Few studies have looked at the role of bacteria on the EPS-producing strains of B. braunii and although bacteria were observed in association with the growth of B. braunii, no conclusions were drawn (Fernandes et al. 1989; Allard and Casadevall 1990; Lupi et al. 1991). Questions such as are the bacteria important for the growth of B. braunii or do the bacteria exert any effects on the EPS produced by B. braunii still need to be elucidated. The objective of this study is to understand the potential role of bacteria on the growth and on the production of EPS by B. braunii CCALA778.
To study the role or effects of bacteria present in microalgae cultures, it is necessary to remove them. Several approaches are commonly used such as centrifugation, washing and sieving methods, and the use of antibiotics (Bruckner and Kroth 2009; Lee et al. 2015; Han et al. 2016). Another method to remove bacteria commonly used in environmental technology, medical practices and the pharmaceutical industry is the use of UV (Schmidt and Kauling 2007; Carney and Lane 2014; Yen et al. 2014). Ultraviolet light C (UV-C) is in the range of 200–280 nm. Especially the UV range between 250 and 270 nm is strongly absorbed by nucleic acids (Dai et al. 2012). Studies which are in progress to test the treatment of infectious diseases with UV-C show promising results in killing prokaryotic microorganisms while keeping eukaryotic mammalian cells viable. It was found that the UV-C doses necessary to damage mammalian cells were ten times higher than the doses necessary to inactivate Escherichia coli (which is considered to be one of the most resistant bacterial species to UV). UV exposure has also been successfully used to control bacterial abundance with microalgae such as Tetraselmis sp. (Sharma et al. 2014).
In this study we used batch cultures of B. braunii CCALA778 obtained from a culture collection. We investigated the bacterial community present and its effects on biomass productivity as well as on the production of total carbohydrates and the EPS fraction of CCALA778 under laboratory conditions. UV-C treatment was used to reduce the bacterial abundance of CCALA778 and comparisons were made with the untreated CCALA778. 16S rRNA gene analysis was used to determine the bacterial abundance, as well as to assess the efficiency of the UV-C method in reducing the bacteria present with CCALA778. Biomass dry weight, EPS, colony size, total carbohydrates and carbohydrate composition were compared between UV and non-UV treated cultures.
Materials and Methods
Strain and media preparation
Botryococcus braunii CCALA778 strain (race A) was obtained from the Culture Collection of Autotrophic Organisms (Trebon, Czech Republic) and transferred to Erlenmeyer flasks with modified CHU 13 medium with the following composition: 400 mg L−1 KNO3, 200 mg L−1 MgSO4.2H2O, 108 mg L−1 CaCl2.2H2O, 104.8 mg L−1 K2HPO4, 20 mg L−1 Fe-Na2EDTA, 9.4 μg L−1 Na2O4Se, 2.86 mg L−1 H3BO3, 1.8 mg L−1 MnSO4.4H2O, 220 μg L−1 ZnSO4.7H2O, 90 μg L−1 CoSO4.7H2O, 80 μg L−1 CuSO4.5H2O, 60 μg L−1 Na2MoO4.2H2O, 10 μL L−1 H2SO4. Final pH was adjusted to pH 7.2 with NaOH. NaHCO3 was added to a final concentration of 5 mM. Botryococcus braunii CCALA778 was kept at a light intensity of 100 μmol photons m−2 s−1, light:dark photoperiod 18:6 h, ambient air concentration of CO2, temperature of 23 °C and mechanical shaking at 90 rpm.
UV-C treatment
UV-C treated (UV)
Botryococcus braunii CCALA778 cells were plated on square modified CHU 13 agar plates after dilution to 4.4×103–4.4×105 cells per plate. In the first instance, UV treatment was performed to create viable mutants of B. braunii that could be grown from single cells. This approach appeared unsuccessful but upon UV treatment, the bacterial load was remarkably reduced which led to further UV experiments. To increase the chance of removing all bacteria, B. braunii cells were exposed to a UV-C dose of 82,500 μJ cm−2 (15–20% survival) using the CL1000 Ultraviolet Crosslinker. After 24 h incubation in the dark, the plates were transferred to a 25 °C incubator with 50 μmol photons m−2 s−1 light and a light:dark photoperiod of 18:6 h. After 3–4 weeks, colonies without a visible bacterial halo were suspended in 5 μL modified CHU 13 medium and spotted on modified CHU 13 agar plates. After 3 weeks, colonies without visible bacterial contamination were transferred to a new modified CHU 13 agar plate. The last step was repeated one more time before selected colonies were suspended in 200 μL liquid modified CHU 13 medium and used as an inoculum for growth. Hereafter, the UV treated CCALA778 was kept for 6 months in similar environmental conditions as the non-UV treated parental strain.
UV treated with added bacteria (UV+Bac)
Preparation of the UV+Bac treated CCALA778 was done in the following way: Firstly, cultivable bacteria were isolated from the untreated CCALA778 by streaking 1 mL of the culture broth onto lysogeny broth (LB) agar. Next, the plates were incubated at room temperature for 3 days before storage at 4 °C. At the start of the experiment (day 0), 3 mL of modified CHU 13 medium was pipetted onto the Petri dish containing the cultivable bacteria and mixed thoroughly with a sterile rod creating a bacteria mix with a cloudy colour. Next, 2 mL of the bacteria mix was pipetted and added to an Erlenmeyer flask containing approximately 120 mL of UV treated CCALA778.
Experimental design
UV treated, UV+Bac and untreated CCALA778 culture flasks were prepared with actively growing cells such that the initial dry weight was 0.5 g L−1. The cultures were grown in Infors HT Multriton incubators in 250-mL conical flasks and a volume of 120 mL modified CHU 13 was used as medium (as described above). The temperature was set at 23 °C, with 2.5% CO2 enriched air and shaking at 90 rpm. Illumination was provided by Phillips lamps FL-Tube L 36 W/77, with 150 μmol photons m−2 s−1, and a light:dark photoperiod of 18:6 h. The Erlenmeyer flasks were capped with aeraseal sterile film (Alphalabs). The experiment was conducted in triplicate and samples were taken at days 0, 3, 6, 9, 12 and 15.
Biomass dry weight
Five-millilitre aliquots of culture broth were filtered with a pre-weighed GF/D glass-fibre membrane (Whatman). The GF/D filters with biomass were dried at 100 °C for 24 h and weighed after cooling to room temperature in a desiccator. The biomass amount was determined by subtraction. This weight minus the pre-weighed membrane corresponds to the total biomass in the 5 mL aliquot.
Exopolysaccharide content
Culture broth (1000 μL) was pipetted into a 1.5-mL Eppendorf tube and stored at − 20 °C until further analysis. To separate the EPS from the cellular biomass, the stored 1000 μL was centrifuged for 3 min at 21,000×g to concentrate the cells, and 500 μL of the supernatant was carefully transferred to a separate Eppendorf tube. The supernatant was lyophilized using a Sublimator 2x3x3 from ZIRBUS technology for 28 h and stored at − 20 °C until further analysis. The EPS was determined using the method first reported by Dubois et al. (1956). In brief, the lyophilized samples were hydrolysed by adding 500 μL of 2.5 M HCl and incubated for 3 h while mixing every hour, and neutralized thereafter by adding 500 μL of 2.5 M NaOH solution. A centrifugation step of 1 min at 21,000×g was used to pellet potential bacterial cell debris and the hydrolysed sample was stored at − 20 °C. For analysing the EPS content, 50 μL of the stored hydrolysed sample was added to 450 μL of demineralized water. Gently, 500 μL of 5% (w/v) phenol in water solution was pipetted into the tube followed by 2.5 mL of 96% (v/v) concentrated sulphuric acid added directly onto the liquid surface, and the mixture was incubated at room temperature for 10 min. Hereafter, the tube was placed in a 35 °C water bath for 30 min, while mixing every 5 min. Subsequently, the sample was left at room temperature for 5 min prior to absorbance measurements. A dilution series of a standard stock solution, which contained a total of 1 g L−1 of monosaccharides, with a ratio of 8/15 D-galactose, 4/15 D-fucose and 3/15 D-glucose, was prepared. These monosaccharide ratios are the main constituents of B. braunii CCALA778 carbohydrates (Gouveia et al. 2017). The absorbance of the standards and samples was measured at 486 nm and the EPS content was determined.
Total carbohydrate content and composition
Hydrolysis of algae samples was performed as described previously (Gouveia et al. 2017). The carbohydrate composition was determined by high-performance anion exchange chromatography (HPAEC) using an ICS-3000 Ion Chromatography HPLC system equipped with a CarboPac PA-1 column (250 × 2 mm) in combination with a CarboPac PA guard column (25 × 2 mm) and a pulsed electrochemical detector in pulsed amperometric detection mode (Dionex, USA) at 20 °C as reported by Gilbert-López et al. (2017). Elution was performed at 28 °C for the separation of glucose and glucosamine.
Colony size measurements
Three millilitres of culture broth, for all time points, was harvested for colony size measurements using a Mastersizer 2000 (Malvern Instruments) with the following settings: particle refractive index of 1.5, dispersant refractive index of 1.33 and obscuration levels set for 1 to 2%. The volume of culture broth used was dependent on the biomass concentration at the different time points. The size of the colonies is expressed in Volume Weighted Mean D[4,3] = \( \frac{\sum {n_{\mathrm{i}}{d}_{\mathrm{i}}}^4}{\sum {n}_{\mathrm{i}}{d_{\mathrm{i}}}^3} \), where ni is the number of particles with diameter di.
Confocal imaging
Cultures of algae growing in log phase were stained for DNA presence and analysed with confocal microscopy to visualize algal nuclei, but also checked for bacterial presence. A comparison was made for 9 days grown UV treated, UV+Bac and untreated CCALA778 cultures. DNA staining was performed by incubating cells in growth medium for > 5 min in a mixture of DAPI fixative with a final concentration of 2 μg mL−1. Staining was achieved in the presence of 2% (v/v) para-formaldehyde and 0.025% (v/v) glutaraldehyde as fixatives combined with 0.01% (w/v) Triton X-100 to allow dye penetration through the EPS and into the cells. Thin coverslip-slide mountings were made of the cells. Confocal images were obtained with a Zeiss Confocal Laser Scanning Microscope 510 with an Axiovert 200M equipped with a 63 × 1.4 NA Plan-Apo objective. DAPI was excited with an Ar diode laser of 405 nm (30 mW) at 10% and the emission was collected at BP 420–490 nm. Corresponding bright field images were omitted. The representative selected images were increased in image resolution and FL-brightness (+ 10 to 15%), and aligned in Photoshop 2015CC.
DNA extraction and 16S rRNA gene amplification
On day 0 and day 15, 5 mL of liquid culture broth was sampled from all Erlenmeyer flasks and stored at − 80 °C. The sample was spun down at 4816×g for 5 min and the supernatant was discarded. Next, 5 mL of sterile dH2O was added and the centrifugation step was repeated, followed by the removal of the supernatant. In the previous step, the dH2O was added to assist with the removal of EPS that could obstruct DNA extraction. The pellet was suspended in 1 mL of S.T.A.R buffer (Roche, USA) and transferred to a 2-mL sterilized tube with 0.1mm zirconia/silica beads (Biospecs) and 5 glass beads of 3 mm. Cells were subjected to three rounds of homogenization of 60 s at a speed of 5500 rpm with a Precellys Beadbeater (Bertin Technologies). Hereafter, the samples were heated for 5 min at 95 °C and 300 rpm to improve the breakdown of any polymeric substances around the colony matrix of CCALA778. After the heating step, the samples were centrifuged for 5 min at 21,000×g at + 4 °C. Then, DNA was extracted using the Maxwell 16 Tissue LEV Total RNA purification kit (Promega, USA) with aid of a Maxwell 16 instrument (Promega, USA). The purity and quantity of DNA was examined by electrophoresis on a 1% (w/v) agarose gel and measured with a Nanodrop (ND1000, Thermo Fisher Scientific). The extracted DNA was stored at − 20 °C until further use.
Amplicons from the V1-V2 region of 16S rRNA genes were generated by a two-step PCR strategy. For the first step, the forward primer 27F-DegS (5′GTTYGATYMTGGCTCAG 3′ where M = A or C; R = A or G; W = A or T; Y = C or T) and an equimolar mixture of reverse primers 338R I (5′GCWGCCTCCCGTAGGAGT 3′) and 338R II (5′ GCWGCCACCCGTAGGTGT 3′ where M = A or C; R = A or G; W = A or T; Y = C or T) were used. Eighteen base pair Universal Tags 1 and 2 (Unitag1 = GAGCCGTAGCCAGTCTGC; Unitag2 = GCCGTGACCGTGACATCG) were appended at the 5′ end of the forward and reverse primers respectively (Daims et al. 1999; van den Bogert et al. 2011; Tian et al. 2016). The first PCR mix (50 μL) contained 10 μL of 5× HF buffer (Thermo Scientific, the Netherlands), 1 μL of dNTP Mix (10 mM; Promega, the Netherlands), 1 U of Phusion Hot Start II High-Fidelity DNA polymerase (Thermo Scientific), 1 μM of Unitag1-27F-DegS forward primer, 1 μM of Unitag2-338R I and II reverse primers, 10 μL of template DNA and 27.5 μL of nuclease-free water. Amplification included an initial denaturation at 98 °C for 30 s, 25 cycles of denaturation at 98 °C for 10 s, annealing at 56 °C for 20 s and elongation at 72 °C for 20 s, and a final extension at 72 °C for 10 min. The PCR product size was examined by 1% gel electrophoresis. The second PCR mix (100 μL) contained 62 μL of nuclease-free water, 5 μL of PCR1 product, 20 μL of 5× HF buffer, 2 μL of dNTP Mix, 2 U of Phusion Hot Start II High-Fidelity DNA polymerase, 500 nM of a forward primer (Unitag1) and reverse primer (Unitag2) appended with an 8 nt sample-specific barcode for each sample. Amplification included an initial denaturation at 98 °C for 30 s, 5 cycles of denaturation at 98 °C for 10 s, annealing at 52 °C for 20 s and elongation at 72 °C for 20 s; and a final extension at 72 °C for 10 min. The concentration of PCR products was quantified with a Qubit Fluorometer (Life Technologies, Germany) in combination with the dsDNA BR Assay kit (Invitrogen, USA). Purified products were then pooled in equimolar amounts of 134 ng μL−1 and sequenced on a MiSeq platform (GATC-Biotech, Germany).
Processing of MiSeq data
Illumina sequencing data was analysed using the NG-Tax pipeline (Ramiro-Garcia et al. 2016). Briefly, in the first step, paired-end libraries were combined and only read pairs with perfectly matching primers and barcodes were retained. The second step was operational taxonomic unit (OTU) picking, and for each sample, an OTU table was created with the most abundant sequences, using a minimum user-defined relative abundance threshold of 0.1%. Thirdly, OTUs were subjected to non-reference–based chimera checking. Lastly, remaining sequences were clustered into OTUs at the 98.5% identity level. Taxonomic assignment of the OTUs was done using the UCLUST algorithm (Edgar 2010) and the Silva_126_SSU Ref database, containing 1,675,819 unique full-length 16S rRNA gene sequences. The 16S rRNA gene dataset obtained in this study is deposited in the Sequence Read Archive (SRA), NCBI with accession number SRP107368.
Microbial community analyses
For the interpretation of the microbial community data on phylum level, the OTU abundance table was converted to relative abundance and visualized as a bar chart using Microsoft Office Excel 2010. OTUs assigned to chloroplasts are grouped together under chloroplasts from B. braunii.
Statistical analysis
A univariate analysis of variance (one-way ANOVA) was carried out to compare the biomass dry weight and average colony size of the B. braunii untreated, UV treated and UV+Bac. For biomass dry weight, the final days of the experiment, at day 12 and day 15, were selected because for industrial cultivation, the total productivity is the most interesting parameter. For the average colony size, all time points were selected for the different treatments. ANOVA was carried out using R open source software (version 3.1.1) with the aov function and the post hoc Tukey’s test with the TukeyHSD function. All statistical analyses were done at a significance level of 0.05.
Results
The effects of the UV-C treatment on the abundance of bacteria in B. braunii CCALA778 are shown in Fig. 1. The UV treated CCALA778 showed almost 100% of the 16S rRNA genes assigned to chloroplast both at days 0 and 15. The relative abundance of bacteria in the UV treated CCALA778 was less than 1% in most samples with the highest relative abundance being 3% for one replicate at day 15. In contrast, the untreated CCALA778 showed 70% of the 16S rRNA genes assigned to bacterial origin for both days 0 and 15. The UV+Bac treated CCALA778 showed higher relative abundance of bacteria compared with the UV treated CCALA778 and less compared with the untreated CCALA778. In both untreated and UV+Bac treated CCALA778 at days 0 and 15, the dominant phylum was Proteobacteria followed by Bacteroidetes, with the exception of one replicate of UV+Bac treated CCALA778 at day 0 in which the abundance of Bacteroidetes was higher than Proteobacteria. The phyla Planctomycetes, Firmicutes and Actinobacteria were detected in lower relative abundance for both UV+Bac and untreated cultures. Zooming further on the bacterial composition, 25 bacteria species were detected with relative abundance equal to or above 0.1% in the untreated CCALA778 at day 0 (Supplement Fig 1). Two bacteria with high relative abundance (16–30%) were identified at genus level: Sphingopyxis sp. and Devosia sp. In the UV+Bac treated CCALA778, only 8 bacteria species were detected at day 0 from the initial 25 present in the untreated CCALA778. The 8 bacteria species detected do not include the two most abundant ones, Sphingopyxis sp. and Devosia sp., found in the untreated CCALA778. In the UV treated CALA778 at day 0, three bacterial species were detected: Phyllobacteriaceae, Dyadobacter sp. and Chitinophagaceae were also detected in the untreated and UV+Bac CCALA778 at day 0. At day 15, the untreated CCALA778 microbial composition remained fairly similar to the composition at day 0. In the UV+Bac CCALA778 at day 15, the addition of Devosia sp. and Shinella sp. was detected. These two bacteria were found among the top 10 most abundance bacteria in the untreated CCALA778. For the UV treated CCALA778, we observed the disappearance of 3 bacteria species detected at day 0 and the appearance of 3 new ones in lower relative abundance compared with the 3 species found at day 0. These were Phenylobacterium sp., Staphylococcus sp. and an unclassified bacterium.

Relative abundance of bacteria compared with Botryococcus braunii strain CCALA778. Green colour represents CCALA778 quantified by the chloroplast 16S rRNA gene. Remaining colours represent the different bacterial phyla present in each sample. Each bar represents an individual sample with n = 3 per untreated, UV and UV+bacteria (Bac) added treatment
Confocal microscopy confirmed the reduction of bacterial abundance after the UV treatment (Fig. 2). Clumps of bacteria were observed in the untreated and UV+Bac CCALA778 (Fig. 2a, c), while the UV treated CCALA778 showed no bacterial DNA staining (Fig. 2b).

DAPI staining of Botryococcus braunii CCALA778 and bacterial DNA. DAPI accumulates in the DNA of the nuclei of algae (indicated with an arrow), and of the bacteria (indicated with an arrowhead) for a untreated CCALA778, b UV treated CCALA778 and c UV+bacteria (Bac) CCALA778. Scale bars are 10 μm
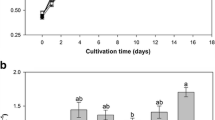
Figure 3 a shows the growth curve of B. braunii for all treatments of the CCALA778 cultures. In the first 3 days, a decrease was observed in the biomass dry weight of UV treated and UV+Bac treated CCALA778, followed by a fast growth between days 3 and 6. From day 9 onwards, the UV treated CCALA778 showed higher biomass compared with UV+Bac treated and untreated CCALA778. For both the untreated and UV+Bac treated CCALA778 biomass dry weight was similar between days 9 and 12, with UV+Bac treated CCALA778 showing a slight increase by day 15 over the untreated CCALA778. ANOVA analysis at day 12 and day 15 revealed that UV treated CCALA778 biomass accumulation was significantly higher than UV+Bac treated and untreated CCALA778 (Table 1).

Botryococcus braunii strain CCALA778 growth and cell morphology after UV-C treatment compared with untreated and UV+bacteria (Bac) treated. a Growth as biomass dry weight over time. b Colony size, measured as volume weighted mean. Some treatment points are overlapping. Error bars represent standard deviation (n = 3)
A larger average colony size was observed for the UV treated CCALA778 (Fig. 3b) compared with the untreated and UV+Bac treated CCALA778. At day 0, the average colony size for the UV treated CCALA778 was 75 μm with a standard deviation of ± 3.5 μm compared with 57 ± 7.8 μm for the UV+Bac treated CCALA778 and 50 ± 3.4 μm for the untreated CCALA778. From day 0 to day 9, all treatments showed a decrease in average colony size. From days 9 to 15, all culture made a switch from decreasing to increasing average colony size. The UV treated CCALA778 especially showed a large increase of average colony size when compared with the UV+Bac and untreated CCALA778. ANOVA analysis revealed that UV treated CCALA778 colony size was significantly larger than untreated CCALA778 across all time points except for day 6 and significantly larger than UV+Bac treated CCALA778 across all time points except day 3 and day 6 (Table 2).
Total carbohydrate content was determined for all treatments at day 9, day 12 and day 15 (Fig. 4). All conditions showed an increase of total carbohydrate content per dry weight over time. Untreated CCALA778 had the lowest amounts of total carbohydrates per dry weight biomass at day 9, day 12 and day 15. On the other hand, UV treated CCALA778 showed higher total carbohydrate per dry weight biomass over the UV+Bac treated and untreated CCALA778, except at day 9, in which UV+Bac treated CCALA778 had the highest ratio.

Total carbohydrate content of Botryococcus braunii strain CCALA778 per dry biomass at days 9, 12 and 15. Error bars represent standard deviation (n = 3)
The amount of EPS measured for all culture conditions over time is shown in Fig. 5. All conditions showed an increase of EPS over time with UV treated CCALA778 having by far the highest amount. From day 0 until day 15, the accumulation of EPS was higher in UV treated CCALA778 when compared with the UV+Bac and untreated CCALA778, reaching 826 ± 61 mg L−1 at day 15. The highest EPS production by UV+Bac treated and untreated CCALA778 was 481 ± 31 and 422 ± 135 mg L−1, respectively.

Accumulation of EPS of Botryococcus braunii strain CCALA778 over time. Error bars represent standard deviation (n = 3)
The carbohydrate composition of the biomass and EPS was similar for all different conditions at days 9, 12 and 15 (Fig. 6). All treatments showed similar carbohydrate composition with D-galactose (Gal) and D-glucose (Glu) as the most abundant carbohydrate fractions, followed by D-mannose (Man) and D-fucose (Fuc). In lower quantities, D-arabinose (Ara), D-rhamnose (Rha), D-glucosamine (GlcN), D-xylose (XvL), D-galacturonic acid (GlaA) and D-glucuronic acid (GlcA) were detected. In all treatments, both the relative amount of Gal and Man monomers decreased over time while Glu increased.

Total carbohydrate composition of Botryococcus braunii strain CCALA778. a Untreated culture. b UV treated culture. c UV+bacteria added (Bac) culture. Error bars represent standard deviation (n = 3)
Discussion
UV-C treatment was successful in reducing the abundance of bacteria present in CCALA778 and thus it is a promising method for outdoor cultivation systems as those systems are susceptible to bacterial contamination (Lee 2001; Wang et al. 2013). In recent years, this method has also been shown to control and reduce bacteria from other microalgae such as Tetraselmis sp. (Sharma et al. 2014). Although it was not possible to create an UV treated CCALA778 100% free of bacteria, it is likely that further UV-C treatment upon UV treated CCALA778 could yield an axenic culture. Two bacterial species which resisted UV-C treatment were identified as Dyadobacter sp. and Phenylobacterium sp. Dyadobacter sp. has been isolated from glacial ice and glacial ice is known to be an environment with high UV exposure, which could explain why the Dyadobacter sp. in our culture resisted UV-C treatment (Liu et al. 2014). Alternative treatments to remove Dyadobacter sp. from the culture can be applied. For example, in previous work [unpublished data], it was shown that an antibiotic mix of penicillin and rifampicin was successful in removing these bacteria from B. braunii CCALA778 culture.
The results suggest that bacteria could interfere with the growth of CCALA778 as shown in Fig. 3 a, for instance, depicting the highly reduced bacteria abundance seen of UV treated CCALA778 cultures. Here, significantly higher biomass accumulation was found from day 9 onwards compared with the untreated CCALA778. UV+Bac biomass accumulation was reduced to similar densities as the untreated CCALA778. An explanation of why the presence of bacteria reduced the growth of CCALA778 could be nutrient competition. One study investigating the nutrient-mediated interactions between algae and bacteria showed that phosphorus utilization by bacteria can occur when organic carbon is present, proliferating bacteria growth (Liu et al. 2012).
Reduction of bacterial abundance also created significant changes in the average colony size of CCALA778. UV treated CCALA778 showed a significant larger average colony size when compared with the untreated CCALA778 and the UV+Bac treated CCALA778. CCALA778 also displayed a rhythmic cycle with decreasing and increasing colony size over time. From our results, the mechanisms behind this cycle are not clear but we hypothesize that high EPS content, as present in the UV treated CCALA778, leads to a larger colony size. In the study by Zhang and Kojima (1998), they observed a positive correlation between colony size and light availability in which colony size decreased with decreasing light availability. They also observed a decrease in EPS content over time which could also contribute to a decrease in colony size. In another recent study, it was shown that an iron(III)-rich medium positively influenced the size and shape of colonies with B. braunii Bot-22 strain and that addition of glucose enhanced the effect of iron(III) (Tanoi et al. 2014).
UV treated CCALA778 showed higher total carbohydrate per dry biomass by day 15 while the untreated CCALA778 showed the lowest (Fig. 4). It is likely that the presence of bacteria was responsible for the lower amounts of total carbohydrates per dry biomass found in the untreated CCALA778. This is probably best supported with the results seen in the accumulation of EPS over time in Fig. 5. The untreated CCALA778, containing a higher abundance of bacteria compared with the UV treated CCALA778 (Fig. 1), showed lower amounts of EPS. Also when bacteria were added to the UV treated culture, UV+Bac treated CCALA778 showed a reduction of EPS, suggesting that the bacteria were responsible for the decrease of EPS. In a previous study using the B. braunii strain ACOI58, the role of bacteria in the production of EPS accumulation could not be determined, although similar attempts were also tried by introducing bacteria present in the ACOI58 strain to the B. braunii UTEX 572 strain (Fernandes et al. 1989). The composition of the carbohydrates produced by CCALA778 remained unchanged between treatments, indicating that the presence or absence of bacteria did not affect the composition of carbohydrates. Similar compositions of carbohydrates present with B. braunii are reported in five other strains (Allard and Casadevall 1990). This suggests that UV treatment can be used for the reduction of the bacterial load when cultivating CCALA778 as it does not change the EPS composition.
The role of the remaining 1–3% bacteria that survived the UV treatment could not be answered. This is because there is a lack of a complete axenic B. braunii CCALA778 that can act as a control. Questions such as are the remaining bacteria antagonistic or beneficial to the growth and EPS production of B. braunii CCALA778? Is the measured increase in biomass and EPS production, a consequence of removal of most antagonistic bacteria, or from the presence of the beneficial bacteria that remained in the culture after treatment and which are no longer masked by the presence of the detrimental bacteria? Although we cannot answer these questions completely, we took a probabilistic approach for the interpretation of the results. Firstly, we assume randomness of the bacterial removal by UV meaning that the UV treatment reaches all bacteria species equally and therefore the removal treatment of bacteria is totally random. Secondly, when we interpret the microbial community (Sup Fig. 1), we do not see similarities in the bacterial species present between days 0 and 15. If the bacteria were responsible for any beneficial effects on the growth or EPS production of B. braunii CCALA778, we would expect to see a steady relative abundance and ratio of the respective bacteria (Eigemann et al. 2013; Ramanan et al. 2015).
A second question that remains is the side effects of UV treatment. Can we exclude possible mutation effects to both B. braunii and the bacteria? This unfortunately is difficult to assess. Genome sequencing and comparison would have to be carried out which is particularly difficult to do with B. braunii because of the high bacterial load present with most B. braunii strains. One point we take is the fact that the UV treated CCALA778 was 6 months old prior to the experiment leading us to assume that the culture was stable. Another point is that B. braunii is an extremely robust green microalgae able to survive desiccation for long periods of time and extreme low temperatures (Demura et al. 2014). For these reasons, we assume that no mutation occurred to B. braunii.
We infer that B. braunii CCALA778 does not need bacteria to grow or to produce EPS in the quantities that it is known for. Rather, their presence is undesirable as they reduce EPS accumulation. Botryococcus braunii remains a slow-growing microalga and attempts to improve its growth are needed for industrial applications.
Conclusions
From this study, we conclude that most of the bacteria present with the microalga B. braunii CCALA778 used in this study are antagonistic. We showed that by reducing the bacterial abundance in B. braunii CCALA778 using UV, the biomass growth and EPS accumulation increased. The carbohydrate composition of B. braunii CCALA778 was not affected by the presence or absence of bacteria. Thus, UV-C treatment is an effective method to reduce the bacterial load in the B. braunii CCALA778 strain.
References
Allard B, Casadevall E (1990) Carbohydrate composition and characterization of sugars from the green microalga Botryococcus braunii. Phytochemistry 29:1875–1878
Banerjee A, Sharma R, Chisti Y, Banerjee UC (2002) Botryococcus braunii: a renewable source of hydrocarbons and other chemicals. Crit Rev Biotechnol 22:245–279
Bell W, Mitchell R (1972) Chemotactic and growth responses of marine bacteria to algal extracellular products. Biol Bull 143:265–277
Borowitzka MA (2013) High-value products from microalgae—their development and commercialisation. J Appl Phycol 25:743–756
Brown AC, Knights BA, Conway E (1969) Hydrocarbon content and its relationship to physiological state in the green alga Botryococcus braunii. Phytochemistry 8:543–547
Bruckner CG, Kroth PG (2009) Protocols for the removal of bacteria from freshwater benthic diatom cultures. J Phycol 45:981–986
Buono S, Langellotti AL, Martello A, Bimonte M, Tito A, Carola A, Apone F, Colucci G, Fogliano V (2012) Biological activities of dermatological interest by the water extract of the microalga Botryococcus braunii. Arch Dermatol Res 304:755–764
Carney LT, Lane TW (2014) Parasites in algae mass culture. Front Microbiol 5:278
Chirac C, Casadevall E, Largeau C, Metzger P (1982) Effect of algal strain and of associated bacteria on hydrocarbon productivity from Botryococcus braunii. Comptes Rendus L’Academie Sci Ser III 295:671–674
Cole JJ (1982) Interactions between bacteria and algae in aquatic ecosystems. Annu Rev Ecol Syst 13:291–314
Cooper MB, Smith AG (2015) Exploring mutualistic interactions between microalgae and bacteria in the omics age. Curr Opin Plant Biol 26:147–153
Dai T, Vrahas MS, Murray CK, Hamblin MR (2012) Ultraviolet C irradiation: an alternative antimicrobial approach to localized infections. Expert Rev Anti-Infect Ther 10:185–195
Daims H, Brühl A, Amann R, Schleifer KH, Wagner M (1999) The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: development and evaluation of a more comprehensive probe set. Syst Appl Microbiol 22:434–444
Demura M, Ioki M, Kawachi M, Nakajima N, Watanabe MM (2014) Desiccation tolerance of Botryococcus braunii (Trebouxiophyceae, Chlorophyta) and extreme temperature tolerance of dehydrated cells. J Appl Phycol 26:49–53
Díaz Bayona KC, Garcés LA (2014) Effect of different media on exopolysaccharide and biomass production by the green microalga Botryococcus braunii. J Appl Phycol 26:2087–2095
Donot F, Fontana A, Baccou JC, Schorr-Galindo S (2012) Microbial exopolysaccharides: main examples of synthesis, excretion, genetics and extraction. Carbohydr Polym 87:951–962
Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F (1956) Colorimetric method for determination of sugars and related substances. Anal Chem 28:350–356
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461
Eigemann F, Hilt S, Salka I, Grossart HP (2013) Bacterial community composition associated with freshwater algae: species specificity vs dependency on environmental conditions and source community. FEMS Microbiol Ecol 83:650–663
Fernandes HL, Tomé MM, Lupi FM, Fialho AM, Sá-Correia I, Novais JM (1989) Biosynthesis of high concentrations of an exopolysaccharide during the cultivation of the microalga Botryococcus braunii. Biotechnol Lett 11:433–436
Gilbert-López B, Mendiola JA, van den Broek LAM, Houweling-Tan B, Sijtsma L, Cifuentes A, Herrero M, Ibáñez E (2017) Green compressed fluid technologies for downstream processing of Scenedesmus obliquus in a biorefinery approach. Algal Res 24:111–121
Gouveia JD, Ruiz J, van den Broek LAM, Hesselink T, Peters S, Kleinegris DMM, Smith AG, van der Veen D, Barbosa MJ, Wijffels RH (2017) Botryococcus braunii strains compared for biomass productivity, hydrocarbon and carbohydrate content. J Biotechnol 248:77–86
Han J, Wang S, Zhang L, Yang G, Zhao L, Pan K (2016) A method of batch-purifying microalgae with multiple antibiotics at extremely high concentrations. Chin J Oceanol Limnol 34:79–85
Hillen LW, Pollard G, Wake LV, White N (1982) Hydrocracking of the oils of Botryococcus braunii to transport fuels. Biotechnol Bioeng 24:193–205
Kawachi M, Tanoi T, Demura M, Kaya K, Watanabe MM (2012) Relationship between hydrocarbons and molecular phylogeny of Botryococcus braunii. Algal Res 1:114–119
Lee H-G, Shin S-Y, Jin L, Yoo C, Srivastava A, La H-J, Ahn C-Y, Kim H-S, Oh H-M (2015) Establishment and maintenance of an axenic culture of Ettlia sp. using a species-specific approach. Biotechnol Bioprocess Eng 20:1056–1063
Lee Y-K (2001) Microalgal mass culture systems and methods: their limitation and potential. J Appl Phycol 13:307–315
Liu H, Zhou Y, Xiao W, Ji L, Cao X, Song C (2012) Shifting nutrient-mediated interactions between algae and bacteria in a microcosm: evidence from alkaline phosphatase assay. Microbiol Res 167:292–298
Liu Y, Hu A, Shen L, Yao T, Jiao N, Wang N, Xu B (2014) Draft genome sequence of Dyadobacter tibetensis type strain (Y620-1) isolated from glacial ice. Stand Genomic Sci 9:883–892
Lupi FM, Fernandes HML, Sá-Correia I, Novais JM (1991) Temperature profiles of cellular growth and exopolysaccharide synthesis by Botryococcus braunii Kütz. UC 58. J Appl Phycol 3:35–42
Lupi FM, Fernandes HML, Tomé MM, Sá-Correia I, Novais JM (1994) Influence of nitrogen source and photoperiod on exopolysaccharide synthesis by the microalga Botryococcus braunii UC 58. Enzym Microb Technol 16:546–550
Metzger P, Largeau C (2005) Botryococcus braunii: a rich source for hydrocarbons and related ether lipids. Appl Microbiol Biotechnol 66:486–496
Ramanan R, Kang Z, Kim B-H, Cho D-H, Jin L, Oh H-M, Kim H-S (2015) Phycosphere bacterial diversity in green algae reveals an apparent similarity across habitats. Algal Res 8:140–144
Ramiro-Garcia J, Hermes GDA, Giatsis C, Sipkema D, Zoetendal EG, Schaap PJ, Smidt H (2016) NG-Tax, a highly accurate and validated pipeline for analysis of 16S rRNA amplicons from complex biomes. F1000Research 5:1791
Schmidt S, Kauling J (2007) Process and laboratory scale UV inactivation of viruses and bacteria using an innovative coiled tube reactor. Chem Eng Technol 30:945–950
Segev E, Wyche TP, Kim KH, Petersen J, Ellebrandt C, Vlamakis H, Barteneva N, Paulson JN, Chai L, Clardy J, Kolter R (2016) Dynamic metabolic exchange governs a marine algal-bacterial interaction. eLife 5:e17473
Sharma K, Li Y, Schenk PM (2014) UV-C-mediated lipid induction and settling, a step change towards economical microalgal biodiesel production. Green Chem 16:3539–3548
Tanabe Y, Okazaki Y, Yoshida M, Matsuura H, Kai A, Shiratori T, Ishida K, Nakano S, Watanabe MM (2015) A novel alphaproteobacterial ectosymbiont promotes the growth of the hydrocarbon-rich green alga Botryococcus braunii. Sci Rep 5:10467
Tanoi T, Kawachi M, Watanabe MM (2014) Iron and glucose effects on the morphology of Botryococcus braunii with assumption on the colony formation variability. J Appl Phycol 26:1–8
Tian L, Scholte J, Borewicz K, van den Bogert B, Smidt H, Scheurink AJW, Gruppen H, Schols HA (2016) Effects of pectin supplementation on the fermentation patterns of different structural carbohydrates in rats. Mol Nutr Food Res 60:2256–2266
van den Bogert B, de Vos WM, Zoetendal EG, Kleerebezem M (2011) Microarray analysis and barcoded pyrosequencing provide consistent microbial profiles depending on the source of human intestinal samples. Appl Environ Microbiol 77:2071–2080
Wang H, Zhang W, Chen L, Wang J, Liu T (2013) The contamination and control of biological pollutants in mass cultivation of microalgae. Bioresour Technol 128:745–750
Yen S, Sokolenko S, Manocha B, Blondeel EJM, Aucoin MG, Patras A, Daynouri-Pancino F, Sasges M (2014) Treating cell culture media with UV irradiation against adventitious agents: minimal impact on CHO performance. Biotechnol Prog 30:1190–1195
Zhang K, Kojima E (1998) Effect of light intensity on colony size of microalga Botryococcus braunii in bubble column photobioreactors. J Ferment Bioeng 86:573–576
Acknowledgements
We thank Dr. Norbert C.A. de Ruijter Manager Wageningen Light Microscopy Centre (WLMC) for assisting with the end-to-end process of making the confocal microscope images used in this study.
Funding
This project is carried out with financial support from the European Community under the seventh framework programme (Project SPLASH, contract nr. 311956).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PNG 299 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

{kind=link}
Cite this article
Gouveia, J.D., Moers, A., Griekspoor, Y. et al. Effect of removal of bacteria on the biomass and extracellular carbohydrate productivity of Botryococcus braunii. J Appl Phycol 31, 3453–3463 (2019). https://doi.org/10.1007/s10811-019-01847-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-019-01847-0