
Abstract
In the present study, five different DNA extraction procedures were examined to determine their effectiveness for extracting DNA suitable for NGS applications. This included two silica-membrane spin column kits, phenol:chloroform, and two CTAB-based methods. Spectrophotometric and fluorimetric measurements as well as standard gel electrophoresis were used as criteria for evaluating the quantity and quality of the isolated DNA prior to the sequencing. Herein, the method of establishing and maintaining axenic Euglena cultures is also presented. The modified CTAB-based method proved to be highly efficient. In terms of DNA quantity and purity (according to the absorbance ratios), the chosen method resulted in DNA of high molecular weight and quality, which fulfills the library construction requirements. Genomic DNA of Euglena hiemalis (CCAP 1224/35) and E. longa (CCAP 1204-17a) isolated using the suggested protocol had been successfully sequenced on the Illumina HiSeq platform. A modified, rapid CTAB-based method of total DNA isolation from Euglena has been described. In terms of the DNA quantity and quality, the protocol devised involving the washing step with DMSO:acetonitrile proved superior to the commonly used, commercially manufactured kits and isolation with phenol:chloroform. The method is also less labor-intensive and time-consuming than the traditional CTAB-based protocol.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Euglenids (Euglenida) are unicellular, free-living phytoflagellates, widespread in various aquatic environments (Zakryś et al. 2017). Their plastids, enclosed by three membranes, are a secondary acquisition from the Pyramimonas-related green alga. Thus, the euglenids are an interesting case for studying the evolution of organelles (Turmel et al. 2009; Hrdá et al. 2012; Zakryś et al. 2017). Despite these organisms (especially Euglena gracilis) being utilized as an object of intensive laboratory studies on photosynthetic capacity, phototaxis, and metabolic and gene expression pathways and having even been proposed as an attractive feedstock for biodiesel and biomass production, knowledge about the organization of their genetic material remains very limited (Milanowski et al. 2014; Yoshida et al. 2016; Ebenezer et al. 2017; Li et al. 2017; Tomiyama et al. 2017). With the advent of the next-generation sequencing (NGS) platforms, investigation of the genomes of many algae species has become more affordable than ever before (Saint-Marcoux et al. 2015; Tan et al. 2015; Gawryluk et al. 2016; Yurchenko et al. 2016). However, the efforts aimed at sequencing Euglena nuclear genomes have remained challenging and, to date, an incomplete task (Milanowski et al. 2016; Ebenezer et al. 2017). It is known that the nuclear genomes of euglenids are large, highly repetitive and complex, therefore difficult to study. Only the draft genome assembly of E. gracilis has been published so far (Ebenezer et al. 2017). The preliminary data indicate that the genome of this organism ranges around from 1.4 up to 2 Gbp (giga base pairs) (Ebenezer et al. 2017). Therefore, obtaining sufficient data to provide adequate coverage enabling genome assembly requires extraction of particularly high-quality DNA sample. The DNA material suitable for NGS analysis should be characterized by high molecular weight with an A260/280 ratio between 1.8 and 2.0 (Healey et al. 2014; Lucena-Aguilar et al. 2016). The sample should be free from co-precipitating contaminant substances, such as proteins, polysaccharides, waxes, and photosynthetic pigments, which tether or obstruct NGS DNA library preparation (Healey et al. 2014).
The difficulty of DNA isolation from eukaryotic microalgae is a well-known and often reported issue (Eland et al. 2012; Tear et al. 2013; Maneeruttanarungroj and Incharoensakdi 2016). Many lineages have developed, apart from the cell wall, unique surface structures, such as frustules, loricae, or mucilage sheaths, frequently enriched by some highly resistant compounds (Barsanti et al. 2001; Popper et al. 2014). The cells of euglenids are covered with a complex structure called the pellicle. It is organized in a series of overlapping, proteinaceous strips enveloped by the plasma membrane and supported on the microtubule corset (Leander et al. 2007; Zakryś et al. 2017). The pellicle strips run along the entire length of the cell in an imbricated manner, which allows them to slide against each other. This arrangement enables dynamic changes in cell shape, called metaboly or euglenoid movement (Zakryś et al. 2017). Furthermore, euglenids may also secrete protective, mucilaginous material. It accumulates under culture conditions as clumps of mucus composed of mostly glycosylated polypeptides and insoluble gelatinous polysaccharides (Cogburn and Schiff 1984). The abovementioned features render euglenids recalcitrant to cell disruption.
Most of the DNA extraction methods currently applied for DNA isolation from euglenids were developed and optimized for different organisms—i.e. plants, yeast, and mammals. These protocols tend to obtain relatively small amounts of DNA, which is often highly diluted, contaminated, and prone to tearing. The use of physical cell disruption methods, including liquid homogenization, sonication, or grinding in liquid nitrogen, may result in obtaining highly fragmented DNA. Although such DNA samples may still be used as a template for PCR, followed by standard sequencing, the method is inapplicable for whole genome study purposes. Furthermore, cell strains acquired from the collections of cultures or isolated from environmental samples are usually contaminated with bacteria and fungi. Mechanical separation of the cells, such as micromanipulation and/or equilibrium centrifugation, followed by subsequent passages in liquid media is often not enough to establish axenic Euglena cultures (Gilbert 1970; Jones et al. 1973). Moreover, supplementing the liquid growth media with antibiotics (especially those newly developed) is usually harmful for euglenids. These agents have a stronger effect on Euglena cells at lower concentrations than on bacteria or fungi. Many antibiotics, routinely used in purification of algal and protozoan cultures, such as streptomycin, kanamycin, and neomycin, permanently bleach photosynthetic euglenids (Droop 1967; Jones et al. 1973; Tucci et al. 2010). Their chloroplasts become aberrant and are gradually diluted out of dividing cells, which eventually leads to a hereditary loss of plastids. Such changed cells have a limited life span and exhibit other atypical traits, i.e., improper divisions (Ebringer 1964). Thus, they cannot be regarded as a reflection of the natural state and used for de novo next-generation sequencing. It is particularly important to strive for elimination of contaminants prior to the stage of sample preparation. Foreign DNA admixtures can lead to wrong or confusing results in the assembly of the desired genome, particularly when reference data is absent (Langdon 2014; Merchant et al. 2014; Strong et al. 2014; Gruber 2015).
Herein, we present the comparison of five commonly used DNA isolation protocols and culture purification method developed while working on the de novo genome sequencing of the two Euglena species: photoautotrophic Euglena hiemalis (CCAP 1224/35) and secondarily heterotrophic E. longa (CCAP 1204-17a). The lack of a rapid and efficient method for pure, high-quality genomic DNA extraction from Euglena species has led us towards attempting to optimize a method for the isolation of highly concentrated DNA, suitable for next-generation sequencing purposes.
Methods
Cell strains and growth conditions
Cultures of Euglena gracilis Z (SAG 1224-5/25), Euglena hiemalis (CCAP 1224/35), and Euglena longa (CCAP 1204-17a) were cultivated statically in the Cramer-Myers medium (Cramer and Myers 1952), supplemented with ethanol (0.8% v/v) and aqueous soil extract (1% v/v). Cells were grown at 18 °C under white light exposure (16:8-h light/dark cycle, ca. 27 μmol photons m−2 s−1).
Disc diffusion antibiotic sensitivity testing
In order to determine the antibiotic susceptibility of contaminant organisms, the agar diffusion test was performed (Bauer et al. 1966). The initial, non-axenic cultures of euglenids were plated on Tryptone Soya Yeast Extract Agar (TSYEA; BTL) supplemented with amphotericin B (1% v/w; Sigma). Then, the antibiotic-impregnated paper discs (Oxoid) were placed on the plates and left to incubate. Our previous experience has shown that antibiotics affecting DNA or protein synthesis, particularly those recently developed, are more lethal for euglenids at lower concentrations than for their bacterial and/or fungal contaminants. Therefore, they were not taken into account in this study. Various agents inhibiting bacterial cell wall synthesis, such as ampicillin (25 μg), cefotaxime (30 μg), fosfomycin (50 μg), gentamicin (30 μg), penicillin (25 μg), rifampicin (30 μg), trimethoprim (2.5 μg), and vancomycin (30 μg), were used in the antibiotic screening, as the least harmful for the cells of euglenids. Cefotaxime and vancomycin—the compounds generating the largest zones of inhibition—were chosen for the final purification procedure.
Culture purification
The initial cultures of euglenids were mechanically pre-purified by centrifugation (2500×g, 30 s, RT) and washing with distilled water (each time the supernatant was discarded). The procedure was repeated as long as the amount of bacteria observed under the microscope was visibly decreasing. Such prepared cultures were diluted and streaked on solid Cramer-Myers medium supplemented with mineral medium (5% v/v) (Starr 1964), aqueous soil extract (5% v/v), and amphotericin B (1% v/w; Sigma), and agarised with TSYEA (1% v/w; BTL). In order to obtain zones with decreasing concentrations of antibiotics, only two discs, one for each of the selected compounds (cefotaxime and vancomycin, respectively), were placed on the opposite sides of the plate (supplementary Figure S1, supplementary material online). Grown Euglena colonies (visible under the microscope as bacteria-free and alive) were subsequently restreaked on the same medium for further purification. To increase survivability of the Euglena cells, the antibiotic discs were placed on the plates every second passage. The procedure was repeated until the axenic algal cultures were obtained. Afterwards, they were transferred to the liquid medium and constantly monitored for bacterial and fungal presence.
Genomic DNA extraction protocols
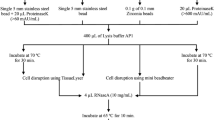
Five DNA extraction methods were evaluated in this study. The DNA was isolated from all three species (E. gracilis, E. longa, E. hiemalis) in pentaplicates with each extraction protocol. The initial experimental steps remained the same in all cases. A total volume of 10 mL of liquid cultures in the logarithmic growth phase was centrifuged (5000×g, 5 min, RT) and rinsed with nuclease-free water three times to completely remove the residues of the growth medium. Washed Euglena cells were then resuspended, aliquoted (1 mL), and centrifuged. Then, each of the cell pellets (± 50 mg) was processed in accordance with the chosen method’s requirements. Finally, the DNA was eluted or resuspended in 100 μL of nuclease-free water (GE Healthcare).
Extraction with commercial silica-membrane column kits
Two commercially manufactured kits, designed for quick purification of genomic DNA—DNeasy Blood & Tissue (Qiagen) and DNeasy Plant (Qiagen)—were tested. In the case of the DNeasy Blood & Tissue kit, the spin column protocol designed for purification of total DNA from animal blood/cultured cells was applied, whereas in the case of DNeasy Plant kit, the TissueRuptor protocol with liquid nitrogen was used. All steps were performed strictly as described in the instructions provided by the manufacturer. In both cases, on-column RNAse A (100 mg mL−1, 4 μL, 2 min, RT; Qiagen) digestion was carried out.
Extraction with phenol:chloroform
Genomic DNA was isolated with standard ethanol precipitation following phenol:chloroform:isoamyl alcohol (24:25:1 v/v; AppliChem) treatment, according to the previously described, albeit slightly modified protocol (Psifidi et al. 2010; Green and Sambrook 2017). Specifically, 1 mL of lysis buffer containing 10 mM Tris HCl (pH = 7.5), 1 mM EDTA, 50 mM NaCl, 0.2% SDS (v/w), and 1 mg of proteinase K (Qiagen) was used to digest Euglena cells for 1.5 h at 56 °C. The lysate extraction was performed twice with 1 mL of phenol:chloroform:isoamyl alcohol (24:25:1 v/v; AppliChem). Following the first extraction step, the aqueous phase was treated with RNAse A (100 mg mL−1, 4 μL; Qiagen) and incubated at 37 °C for 30 min with periodic, gentle mixing. Afterwards, the extraction was repeated. Then, 2.5 volume of absolute ethanol (AppliChem) and 0.1 volume of 3 M sodium acetate (pH = 5.2) were added and the DNA was precipitated at − 20 °C for 1.5 h. The sample was spun at 12,000×g, 10 min, RT, and the DNA pellet was washed twice with 70% ethanol. Finally, the pellet was air-dried and resuspended in nuclease-free water.
Extraction with traditional CTAB method
The cetyltrimethylammonium bromide (CTAB; AppliChem) DNA isolation was performed strictly as described elsewhere (Allen et al. 2006). The volumes of utilized reagents were downscaled according to the amount of the initial Euglena biomass.
Extraction with modified CTAB method
The CTAB-based, rapid DNA extraction protocol (Healey et al. 2014) was slightly modified. Prior to the proper isolation, the cell pellets were washed with ice-cold DMSO:acetonitrile (1:1 v/v; AppliChem). This step was introduced to perforate the pellicle (and to facilitate the penetration of the lysis buffer into the cells), as well as to remove the photosynthetic pigments, polysaccharides, and wax esters which hinder nucleic acids isolation and purification (Rosenberg 1967; Mederic et al. 1987; Barsanti et al. 2001; del Campo et al. 2010; Healey et al. 2014). Also, the RNAse A (100 mg mL−1, 4 μL; Qiagen) treatment was extended to 30 min in comparison to the guidelines provided by Healey et al. (2014).
DNA integrity assessment
The integrity of the DNA samples obtained using each tested extraction method was examined through standard gel electrophoresis (Psifidi et al. 2015). In detail, 5 μL of each DNA extract was analyzed in a 1.5% agarose gel stained with 0.5% Midori Green (Nippon), run in 1× TAE buffer. DNA bands were visualized using the ChemiDoc UV transilluminator (Bio-Rad).
DNA purity and yield
For each of the applied extraction procedures, the concentration and purity of the recovered DNA were assessed spectrophotometrically with NanoPhotometer NP80 (Implen). An Abs260/280 ratio was used to evaluate protein contamination while an Abs260/230 ratio was used to determine organic solvents contamination. Afterwards, for each species/isolation method, one sample (with the best parameters) was selected based on the absorbance values and subjected to fluorimetric measurements. Concentration of the DNA in those samples was further examined using the High Sensitivity DNA Assay implemented by Qubit 3.0 fluorometer (Thermo Scientific). Each time, 1.5 μL (absorbance) or 1 μL (fluorescence) of DNA sample (or nuclease-free water as a blank solution) was used during the sample assessment. Each measurement was performed twice, and obtained values were averaged. Total DNA yield was calculated based on DNA concentration derived from the NanoPhotometer and Qubit results calculated together with the total volume of the DNA extract.
Application of the isolated DNA in high throughput sequencing
Based on the average values of the above parameters, out of five extraction methods tested, the most effective and robust one was selected. In order to evaluate its application in NGS library construction and sequencing, single isolates of E. hiemalis and E. longa, which exhibited optimal parameters of concentration and quality, were chosen for further manipulations. The DNA prepared for sequencing was stored in − 20 °C no longer than a few days, avoiding its exposure to temperature amplitudes.
Preparation of a pair-end reads (PE150) library was carried out externally using NEBNext DNA Library Prep Master Mix Set for Illumina (NEB) and sequenced commercially on an HiSeq4000 instrument (Genomed, Warsaw, Poland). The quality of the DNA library was assessed using a 2100 Bioanalyzer (Agilent). Additionally, the quality of raw sequencing reads after trimming (removal of library adaptors) was analyzed using the FastQC software (Andrews 2010).
Results and discussion
DNA concentration and quality
The mean values of the absorbance ratios for each of the extraction methods tested and statistical comparisons between them are shown in (Fig.1a; supplementary Table S2, supplementary graph S3, supplementary material online). The silica membrane-based DNeasy Blood & Tissue kit enabled the extraction of the DNA with the overall (across all species and samples) mean A260/280 ratio of 2.07 ± 0.06 and the overall mean A260/230 ratio of 2.07 ± 0.13 across the Euglena species. Isolation with the other commercial kit—DNeasy Plant—produced samples with the overall mean A260/280 ratio of 1.86 ± 0.091 and the overall mean A260/280 ratio of 1.36 ± 0.47, i.e., highly contaminated with organic compounds, most probably chaotropic salts. Such a significant amount of admixtures may hinder downstream applications. Hence, the DNA samples extracted with this kit may be considered unsuitable for NGS analysis. Both CTAB-based isolation protocols resulted in samples with the mean absorbance ratios slightly higher than with the DNeasy Blood & Tissue kit, yet still satisfactory: the traditional method with the overall mean A260/280 ratio of 2.04 ± 0.12 and the overall mean A260/280 ratio of 2.02 ± 0.22, while the rapid method—with the overall mean A260/280 ratio of 2.03 ± 0.05 and overall mean A260/280 ratio of 1.97 ± 0.09. The phenol:chloroform method yielded samples with the following mean absorbance ratios across the species: A260/280 of 2.03 ± 0.12 and A260/280 of 2.20 ± 0.64, respectively.

Summary statistics of the a A260/280 (dark gray) and A260/230 (light gray) ratios measured with the spectrophotometer (NP80, Implen) and b mean values of the DNA concentrations across all species together for five methods tested. Bars represent mean values of illustrated parameters and whiskers represent standard deviation (SD). DNA isolation methods were tested in pentaplicates (five repetitions for each method and species). Measurements were taken twice for each sample (n = 15)
In terms of the DNA concentration, as evidenced by spectrophotometry (Fig. 1b; supplementary Table S2, supplementary material online), the isolation with DNeasy Plant kit turned out to be the least efficient (with the mean DNA concentration of 15.78 ± 0.89 ng μL−1 in all samples), while isolation with CTAB-based methods proved to be the most efficient. Traditional protocol yielded the mean DNA concentration of 301.56 ± 156.27 ng μL−1 in all samples and the modified rapid protocol resulted in a mean DNA concentration of 498.61 ± 184.63 ng μL−1 in all samples, respectively.
In most cases, the fluorimetric assessment of the samples (Fig. 2; supplementary Table S4, supplementary material online) with the best absorbance ratios indicated a slightly higher mean DNA concentration (CQ—supplementary Table S4, supplementary material online) than spectrophotometry (C—supplementary Table S2, supplementary material online). However, it has likewise confirmed that the DNeasy Plant kit was the least efficient. It resulted in mean DNA concentration (CQ) of 19.22 ± 8.89 ng μL−1 across the species, compared to the overall mean spectrophotometric result (C, 18.17 ± 3.66 ng μL−1). The phenol:chloroform method, slightly more efficient than the previous one, allowed samples to be obtained with a mean DNA concentration (CQ) of 88 ± 51.45 ng μL−1 across the species, compared to the overall mean spectrophotometric result (C, 68.88 ± 36.19 ng μL−1). The second spin column kit, DNeasy Blood & Tissue, yielded samples with a mean DNA concentration (CQ) of 327.67 ± 3.79 ng μL−1 across the species, compared to the overall mean spectrophotometric result (C, 218.22 ± 57.35 ng μL−1). While traditional CTAB-based protocol resulted in samples with a mean DNA concentration (CQ) of 330 ± 179.92 ng μL−1 across the species, compared to the overall mean spectrophotometric result (C, 364.67 ± 184.17 ng μL−1), the modified CTAB-based method, the most efficient according to fluorimetric results, yielded samples with a mean DNA concentration of 368.67 ± 42.00 ng μL−1 across the species, compared to the overall mean spectrophotometric result (C, 329.37 ± 81.52 ng μL−1).

Mean values of the DNA concentration measured spectrophotometrically (NP80, Implen; dark gray) and fluorimetrically (Qubit 3.0, Thermo Scientific; light gray) across samples with the best A260/280 and A260/230 ratios from all species together for five methods tested. Bars represent mean values of illustrated parameters and whiskers represent standard deviation (SD). Measurements were taken twice for each sample (n = 3)
The spectrophotometric method does not allow to determine whether the material is degraded or not and whether the sample is contaminated (Psifidi et al. 2015). Meanwhile, the fluorimeter measures the concentration of an intact (undenatured and not fragmented), double-stranded DNA. Discordance between these two methods (Fig. 2) was to be expected, since similar findings have been reported (O’Neill et al. 2011; Nakayama et al. 2016; Hussing et al. 2018). Results obtained herein further support discrepancy between spectrophotometry and fluorimetry. However, contradictory evidence has been provided as well (Haque et al. 2003; Foley et al. 2011). Notwithstanding this, it is assumed that the optimal workflow for quality assessment is to control the presence of potential contaminants with a spectrophotometer (NanoPhotometer) and subsequently to quantify double-stranded DNA with a fluorometer (Qubit) (Simbolo et al. 2013). Applying both simultaneously certainly provides more accurate information about the examined sample than any method separately. Therefore, we recommend this approach, especially in the case of de novo genome sequencing.
The CTAB methods (both traditional and simplified) gave the best results in terms of DNA integrity and concentration. This has been proven not only through the assessment of the spectrophotometry and fluorimetry results (Figs. 1 and 2; supplementary graph S3, supplementary material online) but also by standard electrophoresis of the samples (Fig. 3). The rapid CTAB-based method presented provides a considerable improvement over the previously described protocols of DNA extraction from Euglena species. Whereas methods using commercial spin column kits allowed much smaller DNA amounts to be obtained, they remained clean and rather intact. A common problem encountered during the isolation with spin column kits is the insufficient quantity of DNA in the final sample. Therefore, the whole extraction should be performed in several repetitions, and/or the material ought to be pooled together (further concentrated). A major drawback to this procedure is the possible deterioration of the sample quality due to further precipitation and resuspension steps. According to the results obtained in this study, none of the evaluated extraction kits allowed the amount of DNA needed for NGS applications to be isolated. However, it is worth mentioning that the DNeasy Blood & Tissue kit proved to be significantly more efficient than the DNeasy Plant kit (Figs. 1 and 3; supplementary graph S3, supplementary material online). This one turned out to be the worst performer across the methods evaluated in this study. On the other hand, phenol:chloroform isolation produced the most sheared and poor-quality DNA, which is definitely not suitable for NGS applications (Fig. 3).

Evaluation of DNA integrity. Agarose gel (1.5% v/w) electrophoresis of DNA of euglenids extracted by five methods. EGZ—E. gracilis; HIE—E. hiemalis, LNG—E. longa, M—molecular weight marker GeneRuler 1kb Plus (Thermo Scientific). In order to compare the performance of different DNA extraction methods, the same volume of samples with different concentrations was applied (5 μL per lane). The spectrophotometrically/fluorimetrically assessed concentrations of the samples are listed in supplementary tables S2 (bold) and S4
According to the above results, two samples obtained with the modified CTAB-based protocol, one of E. hiemalis and one of E. longa, respectively, were chosen and further subjected for library preparation followed by high-throughput sequencing. The reason behind the decision to exclusively use the DNA isolated with the modified CTAB-based method for high-throughput sequencing, without including isolates deriving from other methods, was mainly based on the satisfactory parameters of the DNA integrity and purity, but also the substantially higher yield, compared to, e.g., DNeasy Blood & Tissue method. As mentioned previously, the nuclear genomes of euglenids are unexpectedly large and complex, for single-celled organisms. Therefore, it was necessary to obtain a highly concentrated DNA template to ensure sufficient coverage of reads for assembled genome sequences.
High-throughput sequencing quality
The genomic DNA samples were transferred to the external company (Genomed, Warsaw, Poland) for library preparation and sequencing using the Illumina HiSeq 4000 platform. Samples successfully passed standard quality control measures, which require high molecular weight of genomic DNA, purity of polysaccharide as well as RNA and protein contamination, and an A260/280 ratio ranging between 1.8 and 2.2.
The E. hiemalis sequencing library produced over 240,000,000 reads, whereas E. longa—more than 300,000,000 reads of a very good quality, both confirmed with the FastQC analysis (Fig. 4). In either case, the average value of PHRED quality scores (for Illumina 1.9 encoding) across bases in the reads ranged between 32 and 40, with the majority of the nucleotides expressing the highest score (Fig. 4, blue line indicates average score values).

Overview of the range of PHRED quality score values (Illumina 1.9 encoding) across all bases for 150PE HiSeq Illumina reads at each position in the E. hiemalis (H) and E. longa (L) fastq file (1 and 2 for each species graphs). For each position a box-whisker plot is drawn. Central red line—median value, yellow box—inter-quartile range (25–75%), upper and lower whiskers—10 and 90% points, blue line—mean quality
For enzymatic reactions, such as PCR, the demands regarding DNA material condition are less strict those for NGS standard libraries preparations. High-throughput sequencing requires DNA not only in large amounts but also in very good quality, meaning lack of organic/inorganic contamination. Column methods can be ineffective in such applications, since usually each step involves washing or filtering. This supplementary caution is certainly welcome; however, it may decrease the final DNA extraction yield (Healey et al. 2014). In the case of E. hiemalis and E. longa, it proves to be a serious problem, and without a more effective protocol of DNA isolation, it would eventually be impossible to obtain a fine sequencing library.
It has been previously discussed elsewhere that the DNA of both high concentration and quality is more stable and degrades more slowly, even when stored for a longer period of time (Psifidi et al. 2015). Eventually, this supports extra future NGS runs (when more data is needed), since it is better to carry out sequencing using the isolates from the same extraction course to eliminate potential bias and save time for additional laborious laboratory exercise.
The method of isolation is crucial for particular experimental design and the modified CTAB-based protocol presented proved to be efficient for whole genome NGS library construction and then sequencing. Isolation has been effective in both phototrophic and non-phototrophic species; therefore, we believe it can be universally applied. This is particularly important due to certain differences in the cell wall composition across the euglenids—especially the spatial structure of the pellicle and the amount or distribution of mucus, as well as the presence or absence of photosynthetic dyes and secondary metabolites.
Conclusions
In this study, a comparison between five various procedures of DNA extraction for whole genome sequencing of euglenids was conducted. So far, no such analyses for Euglena species have been carried out. In the era of growing interest in the genomics of algae, the results presented herein are of high practical significance. An efficient, fast, and reliable procedure grounded on a modified CTAB-based isolation protocol was proposed as the most suitable method of extracting euglenids NGS-suitable genomic DNA.
Furthermore, an efficient Euglena culture purification method was proposed herein. The use of discs impregnated with cefotaxime and vancomycin, respectively, together with amphotericin B, allowed the axenic strains to be established. The described experimental approach was aimed at maintaining the euglenids cells in the best possible condition with simultaneous elimination of bacterial and fungal contaminants. The culture purification step performed prior to the sequencing was crucial for the de novo genome sequencing and assembly. Subsequent passages did not show the presence of any other organisms in the cultures purified with the developed method. Therefore, the methodology described is effective and can be successfully applied for other euglenids.
References
Allen GC, Flores-Vergara MA, Krasynanski S, Kumar S, Thompson WF (2006) A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat Protoc 1:2320–2325
Andrews S (2010) FastQC A Quality Control tool for High Throughput Sequence Data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 26 January 2018
Barsanti L, Vismara R, Passarelli V, Gualtieri P (2001) Paramylon (β-1,3-glucan) content in wild type and WZSL mutant of Euglena gracilis. Effects of growth conditions. J Appl Phycol 13:59–65
Bauer AW, Kirby WM, Sherris JC, Turck M (1966) Antibiotic susceptibility testing by a standardized single disk method. Am J Clin Pathol 45:493–496
Cogburn JN, Schiff JA (1984) Purification and properties of the mucus of Euglena gracilis (Euglenophyceae). J Phycol 20:533–544
Cramer M, Myers J (1952) Growth and photosynthetic characteristics of Euglena gracilis. Arch Microbiol 17:384–402
Droop MR (1967) A procedure for routine purification of algal cultures with antibiotics. Br Phycol Bull 3:295–297
Ebenezer TE, Carrington M, Lebert M, Kelly S, Field MC (2017) Euglena gracilis genome and transcriptome: organelles, nuclear genome assembly strategies and initial features. In: Schwartzbach S, Shigeoka S (eds) Euglena: biochemistry, cell and molecular biology. Springer, Cham, pp 125–140
Ebringer L (1964) Bleaching of Euglenas by antibiotics— a specific form of antagonism in Actinomycetes. Folia Microbiol (Praha) 35:249–255
Eland LE, Davenport R, Mota CR (2012) Evaluation of DNA extraction methods for freshwater eukaryotic microalgae. Water Res 46:5355–5364
Foley C, O’Farrelly C, Meade KG (2011) Technical note: Comparative analyses of the quality and yield of genomic DNA from invasive and noninvasive, automated and manual extraction methods. J Dairy Sci 94:3159–3165
Gawryluk RMR, del Campo J, Okamoto N, Strassert JFH, Lukeš J, Richards TA, Worden AZ, Santoro AE, Keeling PJ (2016) Morphological identification and single-cell genomics of marine diplonemids. Curr Biol 26:3053–3059
Gilbert JJ (1970) Monoxenic cultivation of the rotifer Brachionus calyciflorus in a defined medium. Oecologia 4:89–101
Green MR, Sambrook J (2017) Isolation of high-molecular-weight DNA using organic solvents. Cold Spring Harb Protoc 2017:pdb.prot093450
Gruber K (2015) Here, there, and everywhere: from PCRs to next-generation sequencing technologies and sequence databases, DNA contaminants creep in from the most unlikely places. EMBO Rep 16:898–901
Haque KA, Pfeiffer RM, Beerman MB, Struewing JP, Chanock SJ, Bergen AW (2003) Performance of high-throughput DNA quantification methods. BMC Biotechnol 3:20
Healey A, Furtado A, Cooper T, Henry RJ (2014) Protocol: a simple method for extracting next-generation sequencing quality genomic DNA from recalcitrant plant species. Plant Methods 27:21
Hrdá Š, Fousek J, Szabová J, Hampl V, Vlček Č (2012) The plastid genome of Eutreptiella provides a window into the process of secondary endosymbiosis of plastid in euglenids. PLoS One 7:e33746
Hussing C, Kampmann ML, Mogensen HS, Børsting C, Morling N (2018) Quantification of massively parallel sequencing libraries—a comparative study of eight methods. Sci Rep 8:1110
Jones K, Rhodes ME, Evans SC (1973) The use of antibiotics to obtain axenic cultures of algae. Br Phycol J 8:185–196
del Campo EM, del Hoyo A, Casano LM, Martínez-Alberola F, Barreno E (2010) A rapid and cost-efficient DMSO-based method for isolating DNA from cultured lichen photobionts. Taxon 59:588–591
Langdon WB (2014) Mycoplasma contamination in the 1000 Genomes Project. BioData Min 7:3
Leander BS, Esson HJ, Breglia SA (2007) Macroevolution of complex cytoskeletal systems in euglenids. BioEssays 29:987–1000
Li M, Muñoz HE, Goda K, Di Carlo D (2017) Shape-based separation of microalga Euglena gracilis using inertial microfluidics. Sci Rep 7:10802
Lucena-Aguilar G, Sánchez-López AM, Barberán-Aceituno C, Carrillo-Ávila JA, López-Guerrero JA, Aguilar-Quesada R (2016) DNA source selection for downstream applications based on DNA quality indicators analysis. Biopreserv Biobank 14:264–270
Maneeruttanarungroj C, Incharoensakdi A (2016) Rapid method for DNA isolation from a tough cell wall green alga Tetraspora sp. CU2551. World J Microbiol Biotechnol 32:99
Mederic C, Bertaux O, Rouzeau JD, Valencia R (1987) Isolation of high molecular weight DNA from whole Euglena cells. Protoplasma 141:139–148
Merchant S, Wood DE, Salzberg SL (2014) Unexpected cross-species contamination in genome sequencing projects. PeerJ 2:e675
Milanowski R, Karnkowska A, Ishikawa T, Zakryś B (2014) Distribution of conventional and nonconventional introns in tubulin (α- and β-) genes of euglenids. Mol Biol Evol 31:584–593
Milanowski R, Gumińska N, Karnkowska A, Ishikawa T, Zakryś B (2016) Intermediate introns in nuclear genes of euglenids—are they a distinct type? BMC Evol Biol 16:49
Nakayama Y, Yamaguchi H, Einaga N, Esumi M (2016) Pitfalls of DNA quantification using DNA-binding fluorescent dyes and suggested solutions. PLoS One 11:e0150528
O’Neill M, McPartlin J, Arthure K, Riedel S, McMillan ND (2011) Comparison of the TLDA with the Nanodrop and the reference Qubit System. J Phys 307:1–6
Popper ZA, Ralet M-C, Domozych DS (2014) Plant and algal cell walls: diversity and functionality. Ann Bot 114:1043–1048
Psifidi A, Dovas CI, Banos G (2010) A comparison of six methods for genomic DNA extraction suitable for PCR-based genotyping applications using ovine milk samples. Mol Cell Probes 24:93–98
Psifidi A, Dovas CI, Bramis G, Lazou T, Russel CL, Arsenos G, Banos G (2015) Comparison of eleven methods for genomic DNA extraction suitable for large-scale whole-genome genotyping and long-term DNA banking using blood samples. PLoS One 10:e0115960
Rosenberg A (1967) Galactosyl diglycerides: their possible function in Euglena chloroplasts. Science 157:1191–1196
Saint-Marcoux D, Proust H, Dolan L, Langdale JA (2015) Identification of reference genes for real-time quantitative PCR experiments in the liverwort Marchantia polymorpha. PLoS One 10:e0118678
Simbolo M, Gottardi M, Corbo V, Fassan M, Mafficini A, Malpeli G, Lawlor RT, Scarpa A (2013) DNA qualification workflow for next generation sequencing of histopathological samples. PLoS One 8:e62692
Starr RC (1964) The culture collection of algae at Indiana University. Am J Bot 51:1013–1044
Strong MJ, Xu G, Morici L, Splinter Bon-Durant S, Baddoo M, Lin Z, Fewell C, Taylor CM, Flemington EK (2014) Microbial contamination in next generation sequencing: implications for sequence-based analysis of clinical samples. PLoS Pathog 10:e1004437
Tan B, Ng C, Nshimyimana JP, Loh LL, Gin KY-H, Thompson JR (2015) Next-generation sequencing (NGS) for assessment of microbial water quality: current progress, challenges, and future opportunities. Front Microbiol 6:1027
Tear CJ, Lim C, Wu J, Zhao H (2013) Accumulated lipids rather than the rigid cell walls impede the extraction of genetic materials for effective colony PCRs in Chlorella vulgaris. Microb Cell Factories 12:106
Tomiyama T, Kurihara K, Ogawa T, Maruta T, Ogawa T, Ohta D, Sawa Y, Ishikawa T (2017) Wax ester synthase/diacylglycerol acyltransferase isoenzymes play a pivotal role in wax ester biosynthesis in Euglena gracilis. Sci Rep 7:13504
Tucci S, Vacula R, Krajcovic J, Proksch P, Martin W (2010) Variability of wax ester fermentation in natural and bleached Euglena gracilis strains in response to oxygen and the elongase inhibitor flufenacet. J Eukaryot Microbiol 57:63–69
Turmel M, Gagnon MC, O’Kelly CJ, Otis C, Lemieux C (2009) The chloroplast genomes of the green algae Pyramimonas, Monomastix, and Pycnococcus shed new light on the evolutionary history of prasinophytes and the origin of the secondary chloroplasts of euglenids. Mol Biol Evol 26:631–648
Yoshida Y, Tomiyama T, Maruta T, Tomita M, Ishikawa T, Arakawa K (2016) De novo assembly and comparative transcriptome analysis of Euglena gracilis in response to anaerobic conditions. BMC Genomics 17:182
Yurchenko T, Ševčíková T, Strnad H, Butenko A, Eliáš M (2016) The plastid genome of some eustigmatophyte algae harbours a bacteria-derived six-gene cluster for biosynthesis of a novel secondary metabolite. Open Biol 6:160249
Zakryś B, Milanowski R, Karnkowska A (2017) Evolutionary origin of Euglena. In: Schwartzbach S, Shigeoka S (eds) Euglena: biochemistry, cell and molecular biology. Springer, Cham, pp 3–14
Acknowledgements
We would like to thank Dr. Anna Karnkowska for supporting NGS data analysis. The study was carried out at the Biological and Chemical Research Centre, University of Warsaw, established within the project co-financed by European Union from the European Regional Development Fund under the Operational Programme Innovative Economy, 2007–2013.
Funding
This work was funded by grant 2015/19/B/NZ8/00166 from the National Science Centre, Poland.
Author information
Authors and Affiliations
Contributions
NG was responsible for the concept and design of experiments. The experiments were performed by NG, HW, PH (DNA isolation), and BZ (establishing of axenic cultures). NG and MP analyzed the data and wrote the paper. RM supervised all experiments, analyses, and writing of the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gumińska, N., Płecha, M., Walkiewicz, H. et al. Culture purification and DNA extraction procedures suitable for next-generation sequencing of euglenids. J Appl Phycol 30, 3541–3549 (2018). https://doi.org/10.1007/s10811-018-1496-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10811-018-1496-0