
Abstract
Electrochemical properties of magnetite nanoparticles supported on carbon paste electrodes were investigated by means of cyclic voltammetry. The measurements were performed for bare and coated with citrate magnetite nanoparticles in aqueous solutions of various electrolytes: NaCl, NaClO4, and Na2HPO4. Cyclic voltammetry curves obtained on bare and citrate functionalized Fe3O4 nanoparticles are rather similar indicating that the electrochemical properties of the magnetite nanoparticles are not significantly affected by the citrate coating. Cyclic voltammetry scans reveal the formation of metallic iron below −1.2 V. The defective structure of the oxide phases formed by oxidation of metallic iron seems to play an important role in the chemisorption of chloride ions and their subsequent oxidation.
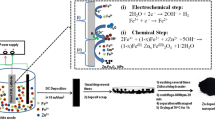
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Magnetite (Fe3O4) is interesting from the electrochemical point of view since it contains iron ions in + 2 and + 3 formal oxidation states. Fe3O4 can be oxidized to Fe2O3 (hematite, α-Fe2O3, and maghemite, γ-Fe2O3) and to various iron oxyhydroxides (goethite, α-FeOOH, akaganeite, β-FeOOH, and lepidocrocite γ-FeOOH) or reduced to Fe(OH)2 and even to metallic iron depending on the applied potential, pH and type of supporting electrolyte [1]. Fe3O4 is suitable to be investigated by electrochemical techniques since it is a good conductor. Most of the studies on magnetite films were performed aimed at understanding the corrosion mechanisms of iron and iron alloys in various environments [2,3,4]. The investigation of the redox behavior of magnetite and other iron oxide nanoparticles (NPs) is also prompted by the prospect to prepare these nanostructured materials by exploiting the oxidation of iron electrodes [5]. Electrodes based on Fe3O4 NPs have potential applications in various fields such as electricity storage [6], electrocatalysis [7, 8], and chemical sensors [9]. Magnetite NPs can be functionalized with a variety of molecules to prepare stable dispersions in aqueous or non-aqueous environments. If the coating is able to modify the electrochemical properties of Fe3O4 NPs, this possibility could be of particular interest for the application as chemical sensors. The adsorption of citrate anions on the surface of magnetite NPs allows one to obtain aqueous dispersions stabilized by the electrostatic repulsions between negatively charged particles [10,11,12]. Since the pioneering works of Allen et al. [13, 14] electrochemical studies of magnetite NPs have been carried out on electrodes consisting of Fe3O4 particles mixed with carbon paste (CP) [15]. In a limited number of studies cyclic voltammetry (CV) measurements were performed on dispersions of magnetite NPs or on NPs directly supported on other types of electrodes [16,17,18].
In the present work, we investigated by means of CV the redox properties of Fe3O4 NPs supported on carbon paste electrodes (CPEs). The measurements were performed on bare and coated with citrate magnetite NPs at neutral (or slightly basic) pH in solutions of NaCl, NaClO4, and Na2HPO4. Most of the results reported in the literature on the electrochemical properties of magnetite were obtained in solutions at extreme acid or basic pH values [19, 20], whereas the measurements shown in this work were performed at neutral pH at which the dissolution of Fe3O4 should not occur. This is one of the novelties of the present work. The aim of this study was twofold. One objective was to observe the influence of the functionalization with citrate ions on the electrochemical properties of Fe3O4 NPs and the other one was to highlight influence of the supporting electrolytes on the electrochemical processes occurring at the solid-solution interface. These aspects are relevant in view of the various potential electrochemical applications of magnetite NPs.
2 Experimental
Analytical research grade (Sigma Aldrich) products were used in the experiments.
The preparation of Fe3O4 NPs, the functionalization with citrate as well as their characterization are described in detail in ref. [21]. Shortly, magnetite NPs were synthesized by the co-precipitation method from solutions of FeCl3⋅6H2O and Fe (NH4)2(SO4)2⋅6H2O (1:2 Fe(II)/Fe(III) molar ratio) in double distilled water (DDW) by adding a solution of NaOH. The reaction solution was deaerated by N2 bubbling and the reaction was carried out at 60 °C. The black precipitate of magnetite NPs was separated from the liquid phase by using a magnet, washed several times with DDW and dried in a flux of N2. The functionalization of Fe3O4 NPs with citrate was carried out following two different methods. In the “ex-situ” procedure citrate ions were adsorbed on magnetite NPs by immersing the NPs in a sodium citrate solution. In the “in-situ” procedure, the functionalization of magnetite NPs was obtained by co-precipitation in the presence of citrate. In our previous works bare and citrate coated magnetite NPs were characterized by Fourier Transform Infrared (FTIR) spectroscopy, Dynamic Light Scattering (DLS), X-ray Diffraction (XRD), and X-ray Photoelectron Spectroscopy (XPS) [21, 22].These studies showed that the “in-situ” procedure leads to citrate coated magnetite NPs which have a significantly smaller size (4 nm) than those prepared following the “ex-situ” method (10 nm), almost the same size of bare magnetite NPs [21]. DLS measurements indicated that magnetite NPs form aggregates with a size around 100–200 nm.
Materials and apparatus for electrochemical measurements have been described elsewhere [23]. A Ag|AgCl|KCl (3 M) electrode and a platinum wire were used as the reference and as the auxiliary electrodes, respectively. The potential values reported in text and figures are relative to that of the Ag|AgCl|KCl 3 M electrode (0.21 V with respect to the normal hydrogen electrode, NHE, potential). All experiments were carried out at room temperature. The scan rate of the potential was 50 mVs−1, unless otherwise specified. Measurements were performed in DDW solutions of NaCl (0.5 M), NaClO4 (0.5 M), and Na2HPO4 (0.10 M). We did not use buffered solutions to avoid possible effects on the electrochemical properties due to the adsorption of components of the buffer on the NPs. The solutions were deaerated by bubbling N2 into the cell for at least 15 min. The working electrode was prepared by mixing carbon paste (BASi®) and Fe3O4 NPs in a mortar until a homogenous mixture was obtained. The composition of the mixture used in the experiments was around 20 weight % of magnetite. A portion of the prepared mixture was packed into an electrode for carbon paste and the surface of the electrode was thoroughly rinsed with DDW. An electrode prepared in the same way but without adding Fe3O4 NPs served as a blank. Before each experiment, the electrode surface was renewed by a gentle polishing.
3 Results and discussion
3.1 Cyclic voltammetry of bare magnetite NPs
The first seven CV curves of a CPE modified with Fe3O4 NPs, immersed in a 0.5 M NaCl solution are shown in Fig. 1, top panel. Since the range of potential is rather wide, CV curves were measured for a CPE without NPs to check for possible contributions of side processes to the current. The comparison of the CV curves of the Fe3O4/CPE with those of the CPE electrode shows that the contribution to the measured current due to reduction of H+ or to oxidation of oxygen of water is negligible with respect to the processes involving Fe3O4 NPs (Fig. 1S, supplementary info). The CV curves of the magnetite modified CPE are a characterized by broad peaks resulting from the contributions of several processes to the observed features. Indeed, according to the iron Pourbaix diagram under these conditions several electron transfer processes probably overlap [24]. A tentative assignment of the observed peaks to specific reactions is based on the redox potentials reported in the literature [20, 25]. However, since the measurements were performed at different pH values, on various types of magnetite samples (films, particles etc.) the assignment of the peaks in the voltammograms is not unambiguous [20]. Actually, the reduction of Fe3O4 to Fe(OH)2 and ultimately to metallic iron is most likely occurring at potentials too close to be discriminated. In the scan #1 the potential is varied from 0 to + 1.2 V and no peak is observed. When the potential is reversed from + 1.2 to −1.4 V, a broad cathodic peak is detectable at ca. −0.5 V which could be due to the reduction of Fe(III) to Fe(II) (scan #2). A more intense cathodic peak is observed at ca. −1.0 V. Features in this region of potentials were reported in previous studies by CV of magnetite NPs [16], magnetite electrodes [20] and epitaxial magnetite films [26]. This peak was ascribed to the reduction of iron ions in magnetite to metallic iron according to the reaction:

Top panel: CV curves of a CPE with bare magnetite NPs. Middle panel: CV curves of a CPE with citrate functionalized magnetite NPs with the “ex-situ” procedure. CV curves of a CPE in a sodium citrate solution (1 × 10–2 M) are also shown (black thin lines). Bottom panel: CV of a CPE electrode with citrate functionalized magnetite NPs prepared with the “in-situ” procedure. Scans #1, #3, #5, and #7 are in the positive potential direction (anodic scans). Scans #2, #4, and 6 are in the negative direction (cathodic scans). The supporting electrolyte was NaCl 0.5 M. The potentials are relative to that of the Ag|AgCl|KCl 3 M electrode. In all the measurements the potential scan rate was 50 mVs−1
Since the measurements reported in the literature were performed at basic pH the peak attributed to the formation of metallic iron can appear to a different potential in the present work in which the CV curves were acquired at neutral or close to neutral pH.
When the potential is reversed from -1.4 to + 1.2 V oxidation peaks are observed in subsequent cycles (scans #3, #5, and #7). The broad feature after the onset potential E = −1.2 V can be attributed to the oxidation of metallic iron to Fe(II). It is likely that Fe(II) is present as Fe(OH)2 at the surface of the NPs [20]. The other two peaks can be ascribed to the oxidation of Fe(OH)2 to Fe(III) compounds such as Fe(OH)3 and Fe2O3. The origin of the peak at ca. + 0.9 V will be discussed later. When the potential is scanned from + 1.2 to −1.4 V, the cathodic peak moves from -1.0 V in scan #2 to less negative values in scans #4 and #6. At the same time, the current of this peak decreases. A decrease of the current magnitude at −1.4 V (i.e., at the inversion potential) upon increasing the number of scans, is also observed. The observed variation may be attributed either to a change of conductivity of the NPs after reduction of the oxides or to a slowdown of the oxidation during the first scans. The decrease of the current at the inversion potential parallels the decrease of the cathodic peak intensity at ca. −1.0 V. The evolution of the CV curves over the last 7 scans is shown in Fig. 2, top panel. The peak positions and intensities do not vary significantly as the number of scans is increased and a steady-state condition is reached (Figs. 2 and 3, top panel).

Top panel: CV curves of a CPE electrode with bare magnetite NPs. Middle panel: CV curves of a CPE electrode with citrate functionalized magnetite NPs prepared with the “ex-situ” procedure. Bottom panel: CV of a CPE electrode with citrate functionalized magnetite NPs prepared with the “ex-situ” procedure. Scans #15, #17, #19 are anodic scans. Scans #14, #16, #18, and #20 are cathodic scans. The supporting electrolyte was NaCl 0.5 M

Complete set of CV scans of a CPE electrode with bare magnetite NPs (top panel), a CPE electrode with citrate functionalized magnetite NPs prepared with the “ex-situ” procedure (middle panel); a CPE electrode with citrate functionalized magnetite NPs prepared with the “in-situ” procedure (bottom panel). The supporting electrolyte was NaCl 0.5 M
3.2 CV of magnetite citrate coated NPs
The first seven CV curves of the CPE modified with citrate coated magnetite NPs immersed in a 0.5 M NaCl solution are shown in Fig. 1, middle and bottom panels. The CV curves of the citrate coated magnetite NPs prepared following the two procedures described above exhibit only minor differences. The main difference is the current which is larger for the “in-situ” NPs than for the “ex-situ.” These curves are also similar to those measured for bare magnetite NPs. CV curves acquired under the same experimental conditions for a solution of sodium citrate do not exhibit any redox peak (Fig. 1, middle panel, thin black line). Indeed studies of citrate adsorption on Pt electrodes did not show any peaks attributable to redox reactions of citrate ions [27, 28]. Thus, we can conclude that there is no significant change of the electrochemical properties either of the magnetite NPs or of the citrate ions after adsorption. The observed minor differences between the CV curves of the citrate functionalized magnetite NPs and those of the bare NPs could be due to the different size of the magnetite NPs. As shown in a previous study, citrate coated Fe3O4 prepared following the “in-situ” procedure have a size smaller than that of both bare NPs and of those functionalized with citrate according to the “ex-situ” procedure [21].
3.3 Influence on the CV curves of chloride ions in solution
In all the measurements there is a peak around + 0.9 V in the anodic curves which cannot be explained taking into account the redox reactions of iron [25]. The oxidation of Fe(III) to ferrate (FeO42−) occurs at higher potentials and ferrates are stable only at high pH [29]. On the other hand, we found that the area under this peak depends on the fraction of magnetite NP in the CPE as shown by the linear variation of the area under the peak at + 0.9 V with the weight fraction of magnetite NPs (Fig. 4). Incidentally, this behavior indicates that magnetite NPs are homogenously dispersed in the mixture with CP. CV curves were acquired with various scan rates of the potential. The peak position shifts monotonically from + 0.85 to + 1.03 V when the potential scan rate is increased from 10 to 200 mV s−1 while the peak current shows a linear dependence on the potential scan rate (Fig. 5). The linear dependence of the peak current as function of the potential scan rate indicates that the oxidized species are adsorbed at the surface of the electrode [30, 31].

Area (mV A) of the peak at 0.9 V as a function of the weight fraction of magnetite NPs mixed with CP. The linear fitting of the data (R = 0.97) is shown

Current (mA) of the peak at ca. 0.9 V as a function of the scanning rate (mVs−1). The linear fitting of the data (R = 0.99) is shown
In literature, an oxidation peak at ca. + 0.9 V was observed in the CV curves measured for fine-grained blast furnace sludges in a CPE with 1 M HCl as electrolyte [32]. Although the origin of this feature was not discussed in that paper, it could be ascribed to the oxidation of chloride ions adsorbed on the iron oxides in the sludge since it was observed when the CV measurements were performed in HCl solutions. Similarly, in the present case, the oxidation of adsorbed Cl− ions could be responsible for the peak at around + 0.9 V in the CV curves. In order to verify this hypothesis, CV experiments were repeated with other supporting electrolytes, not containing chloride ions. Actually the CV curves obtained with the Fe3O4/CPE in a solution of NaClO4 do not show the oxidation peak at + 0.9 V (Fig. 6). On the other hand, this peak appears when chloride ions are present and the potential has reached values at which the formation of metallic iron occurs (Fig. 6). A further evidence for the assignment of the anodic peak around + 0.9 V to the oxidation of chloride ions is provided by the CV curves in solutions of Na2HPO4 as supporting electrolyte in which this peak is not observed (Fig. 7, top panel). Neither this peak nor that due to the formation of metallic iron are detectable after NaCl was added to the phosphate solution (Fig. 7, bottom panel). This observation suggests that phosphate ions have an effect on the formation of metallic iron which, in turn, plays an important role in chloride ion oxidation. The CV curves measured in solutions of Na2HPO4 are similar to those acquired in a buffer solution of Na2HPO4 and KH2PO4 (Fig. 2S, supplementary info). Thus, it seems that in the present case the use of buffered solutions does not change significantly the measured CV curves. It is worth noting that the peak ascribed to the oxidation of chloride ions appears only when in the previous cathodic scans the potential has reached values low enough to form metallic iron. This threshold potential is around −1.2 V for solutions containing NaClO4 as supporting electrolyte and NaCl (Fig. 9). These results suggest that chloride ions are not chemisorbed at the surface of the pristine magnetite NPs since no oxidation peak is observed in the first anodic scan. On the contrary they are chemisorbed after reduction of the iron oxides to Fe(0) and its subsequent re-oxidation to Fe(II) and Fe(III). The defective structure of the iron oxides produced by the electrochemical oxidation of metallic iron could explain why chloride ions are chemisorbed only after a reduction scan at potentials negative enough to form metallic iron [33]. In order to verify this hypothesis an investigation “in-situ” by spectroscopic and/or microscopy techniques would be needed. XRD measurements that we performed on the electrode material after the CV measurements did not reveal any change in the crystal structure of the magnetite NPs. We observed that in the absence of chloride ions in solution with NaClO4 as supporting electrolyte the peak ascribed to the formation of metallic Fe is not detectable. The formation iron oxychlorides (for instance FeOCl) or iron chloride complexes requiring less negative reduction potentials than iron oxides and hydroxides could explain this result. A faster kinetics of the iron chloride complexes in solution might also explain why the cathodic peak is observed in the presence of chloride. Finally, the observation of the chloride oxidation peak in the CV curves of citrate coated magnetite NPs is consistent with the partial coverage of the surface of the NPs with citrate ions found by XPS [22]. Different shifts of this peak and variations of the peak current as a function of the number of scans are observed by comparing the CV curves of the three samples. In the case of bare Fe3O4 NPs the peak position does not change upon increasing the number of scans, whereas the peak intensity increases (Figs. 1 and 3, top panel). The increase of the current with the number of scans can be explained considering the increase of the area occupied by the iron oxides (and/or hydroxides) formed after oxidation of metallic iron, a surface on which the adsorption of chloride ions occurs.

CV curves of a CPE electrode with bare Fe3O4 NPs in a solution of NaClO4 as supporting electrolyte (top panel) and in a solution of NaClO4 with NaCl (bottom panel). Scans #1, #3, and 5 are in the positive direction. Scans #2, #4, and 6 are in the negative direction

CV curves of a CPE electrode with bare Fe3O4 NPs in a Na2HPO4 solution. Top panel: without Cl−. Bottom panel: with NaCl. Scans #1 and #3 are in the positive direction. Scan #2 is in the negative direction

CV curves of a CPE electrode with bare Fe3O4 NPs in a NaClO4 and NaCl solution. The curves were measured at various minimum values of the cathodic scan. Scan2 #1 (black curve) and #3 (blue curve) are in the positive direction. Scan #2 (red curve) is in the negative direction
Variations of the position of chloride oxidation peak are observed also in the case of the citrate coated NPs upon increasing the number of scans. For the “ex-situ” sample this peak shifts from + 0.89 V in the first scans to + 0.97 V in the last scans (Figs. 2 and 3, middle panel). In the CV curves of the “in-situ” sample the peak exhibits an opposite trend, shifting from + 0.91 V to + 0.86 V upon increasing the number of scans (Figs. 2 and 3, bottom panel). As in the case of bare magnetite NPs, the CV curves tend to a steady-state condition with increasing the number of scans (Figs. 2 and 3). These variations are more difficult to interpret than in the case of bare magnetite NPs (Figs. 1–3). It is likely that the adsorption of citrate ions occurs on different crystallographic planes of the NPs depending on the preparation procedure. Hence, upon increasing the number of CV scans, zones with different structural and compositional defects on which chloride ions adsorb can develop.
The oxidation of adsorbed chloride ions can lead to the formation of Cl2 [34]:
However the formation of other oxidation products such as Cl2O or ClO− is also possible:
4 Conclusion
In this work CV measurements were performed on CPE modified with magnetite NPs in solutions of various supporting electrolytes at neutral (or slightly basic) pH. Under this condition it was possible to prevent (or at least to reduce) the dissolution of Fe3O4 which occurs in particular at low pH. The CV curves measured for bare and citrate functionalized Fe3O4 NPs are similar indicating that the electrochemical properties of the magnetite NPs are not significantly affected by the adsorption of citrate ions. CV scans suggest the formation of metallic iron for applied potentials below −1.2 V in NaCl solutions. The adsorption of chloride ions on the electrode surface appears to be significantly influenced by the redox cycle of magnetite NPs. The defective structure of the oxide phases formed by the oxidation of metallic iron is probably responsible for the chemisorption of chloride ions. In future studies electrochemical impedance spectroscopy will be used to elucidate the different electron transfer processes.
References
Rochelle RM, Scherwertmann U (2006) The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses. Weinheim, Wiley-VCH.
Song I, Gervasio D, Payer JH (1996) Electrochemical behaviour of iron and iron oxide thin films in alkaline (1 M KOH) aqueous solution: a voltammetry study for cathodic instability of coating/metal interface. J Appl Electrochem 26:1045–1052. https://doi.org/10.1007/BF00242199
Eliyan FF, Kish JR, Alfantazi A (2015) Voltammetric analysis on the formation of Fe(OH)2 and FeCO3, and on the reactivity of passivation of steel in carbonate solutions. J Mater Eng Perform 24:2473–2480. https://doi.org/10.1007/s11665-015-1525-y
White AF, Peterson ML, Hochella MF (1994) Electrochemistry and dissolution kinetics of magnetite and ilmenite. Geochim Cosmochimic Acta 58:1859–1875. https://doi.org/10.1016/0016-7037(94)90420-0
Ramimoghadam D, Bagheri S, Hamid SBA (2014) Progress in electrochemical synthesis of magnetic iron oxide nanoparticles. J Magn Magn Mater 368:207–229. https://doi.org/10.1016/j.jmmm.2014.05.015
Lu L, Peng M, Wu L, Dong Y, You G, Duan Y, Yang W, He L, Liu X (2021) Progress in iron oxides based nanostructures for applications in energy storage. Nanoscale Res Lett 16:138. https://doi.org/10.1186/s11671-021-03594-z
Dong CD, Chen CW, Hung CM (2016) Preparation, physicochemical, and electrochemical properties of magnetite electrodes for methanol electrocatalytic oxidation in an alkaline medium. Desalination Water Treat 57(29404):29410. https://doi.org/10.1080/19443994.2016.1204561
Ola O, Ullah H, Chen Y, Thummavichai K, Wang N, Zhu Y (2021) DFT and experimental studies of iron oxide-based nanocomposites for efficient electrocatalysis. J Mater Chem C 9:6409–6417. https://doi.org/10.1039/d1tc01022k
Kulpa-Koterwa A, Ossowski T, Niedziałkowski P (2021) Functionalized Fe3O4 nanoparticles as glassy carbon electrode modifiers for heavy metal ions detection—a mini review. Materials 14:772. https://doi.org/10.3390/ma14247725
Liu J, Sun Z, Deng Y, Zou Y, Li C, Guo X, Xiong L, Gao Y, Li F, Zhao D (2009) Highly water-dispersible biocompatible magnetite particles with low cytotoxicity stabilized by citrate groups. Angew Chem Int Ed 48:5875–5879. https://doi.org/10.1002/anie.200901566
Szekeres M, Tóth IY, Illés E, Hajdú A, Zupkó I, Farkas K, Oszlánczi G, Tiszlavicz L, Tombácz E (2013) Chemical and colloidal stability of carboxylated core-shell magnetite nanoparticles designed for biomedical applications. Int J Mol Sci 14:14550–14574. https://doi.org/10.3390/ijms140714550
Granath T, Mandel K, Löbmann P (2021) Overcoming the inhibition effects of citrate: precipitation of ferromagnetic nanoparticles with tunable morphology, magnetic properties, and surface charge via ferrous citrate oxidation. Part Part Syst Charact 38:2100098. https://doi.org/10.1002/ppsc.2021100098
Allen PD, Hampson NA, Bignold GJ (1979) The electrodissolution of magnetite: Part I. The electrochemistry of Fe3O4/C discs-potentiodynamic experiments. J Electroanal Chem 99:299. https://doi.org/10.1016/S0022-0728(79)80094-7
Allen PD, Hampson NA, Bignold GJ (1980) The electrodissolution of magnetite: Part II. The oxidation of bulk magnetite. J Electroanal Chem 111:223. https://doi.org/10.1016/S0022-0728(80)80042-8
Rodríguez-López A, Torres-Torres D, Mojica-Gomez J, Estrada-Arteaga C, Antaño-López R (2011) Characterization by electrochemical impedance spectroscopy of magnetite nanoparticles supported on carbon paste electrode. Electrochim Acta 56:8078–8084. https://doi.org/10.1016/j.electacta.2010.11.039
Accogli A, Bertoli L, Panzeri E, Gibertini E, Pesce R, Bussetti G, Magagnin L (2021) Electrochemical characterization of magnetite (Fe3O4) nanoaggregates in acidic and alkaline solutions. ACS Omega 6:26880–26887. https://doi.org/10.1021/acsomega.1c03142
Bradley MJ, Tratnyek PG (2019) Electrochemical characterization of magnetite with agarose-stabilized powder disk electrodes and potentiometric methods. ACS Earth Space Chem 3:688–699. https://doi.org/10.1021/acsearthspacechem.8b00200
Roberts JJP, Westgard JA, Cooper LM, Murray RW (2014) Solution voltammetry of 4 nm magnetite iron oxide nanoparticles. J Am Chem Soc 136:10783–10789. https://doi.org/10.1021/ja505562p
Fetisov VB, Ermakov AN, Belysheva GM, Fetisov AV, Kamyshov VM, Brainina KZ (2004) Electrochemical dissolution of magnetite in acid solutions. J Solid State Electrochem 8:565–571. https://doi.org/10.1007/s10008-004-0499-8
Monteiro JF, Ivanova YA, Kowalevsky AV, Ivanou DK, Frade JR (2016) Reduction of magnetite to metallic iron in strong alkaline medium. Electrochim Acta 193:284–292. https://doi.org/10.1016/electacta.2016.02.058
Atrei A, Mahdizadeh FF, Baratto MC, Scala A (2021) Effect of citrate on the size and the magnetic properties of primary Fe3O4 nanoparticles and their aggregates. Appl Sci 11:6974. https://doi.org/10.3390/app11156974
Atrei A, Lesiak B, Toth J (2022) Magnetite nanoparticles functionalized with citrate: a surface science study by XPS and ToF-SIMS. Appl Surf Sci 602:154366. https://doi.org/10.1016/j.apsusc.2022.154366
Vinogradov MM, Nelyubina YV, Corsini M, Fabrizi de Biani F, Kudinov AR, Loginov DA (2017) Thioether iron complexes [(X-SMe-7,8–C2B9H10)Fe(C6H6)] (X = 9 or 10) as synthons of neutral ferracarborane fragments. Eur J Inorg Chem 38:4627–4634. https://doi.org/10.1002/ejic.201700333
Huang LF, Rondinelli JM (2019) Reliable electrochemical phase diagram of magnetic transition metals and related compounds from high-throughput ab initio calculations. npj Mate Degrad 3:26. https://doi.org/10.1038/s41529-019-0088-z
Pang SC, Chin SF, Anderson MA (2007) Redox equilibria of iron oxides in aqueous-based magnetite dispersions: effect of pH and redox potential. J Colloid Interface Sci 311:94–101. https://doi.org/10.1016/j.jcis.2007.02.058
He Z, Gudavarthy RV, Koza JA, Switzer JA (2011) Room temperature electrochemical reduction of epitaxial magnetite films to epitaxial iron films. J Am Chem Soc 133:12358–12361. https://doi.org/10.1021/ja203975z
Berkh O, Burstein L, Shacham-Diamand Y, Gileadi E (2011) The chemical and electrochemical activity of citrate on Pt electrodes. J Electrochem Soc 158:F85–F91. https://doi.org/10.1149/1.3560572
Gonzalez-Pena OI, Chapman TW, Vong YM, Antano-Lopez R (2008) Study of adsorption of citrate on Pt by CV and EQCM. Electrochim Acta 53:5549–5554. https://doi.org/10.1016/j.electacta.2008.02.104
Macova Z, Bouzek K, Hives J, Sharma VK, Terryn RJ, Baum JC (2009) Research progress in the electrochemical synthesis of ferrate(VI). Electrochem Acta 54:2673–2683. https://doi.org/10.1016/j.electacta.2008.11.034
Elgrishi N, Rountree KJ, McCarthy BD, Rountree ES, Eisenhart TT, Dempsey HL (2018) A practical beginner’s guide to cyclic voltammetry. J Chem Educ 95:197–206. https://doi.org/10.1021/acs,jchemed.7b00361
Zanello P, Fabrizi de Biani F, Nervi C (2012) Inorganic electrochemistry: theory, practice and application, 2nd edn. RSC Publishing
Novak V, Raska P, Matysek D, Kostura B (2018) Electrochemical characterization of fine-grained blast furnace sludge after acid leaching using carbon paste electrodes. J Solid State Electrochem 22:3457–3466. https://doi.org/10.1007/s10008-018-4056-2
Schmuki P, Büchler M, Virtanen S, Isaaxs HS, Ryan MP, Böhni H (1999) Passivity of iron in alkaline solutions studied by in situ XANES and a laser reflection technique. J Electrochem Soc 146:2097–2102. https://doi.org/10.1149/1.1391897
Wang Y, Liu Y, Wiley D, Zhao S, Tang Z (2021) Recent advances in electrocatalytic chloride oxidation for chlorine gas production. J Mater Chem A 9:18974–18993. https://doi.org/10.1039/d1ta02745j
Acknowledgements
This publication was produced with the co-funding European Union—Next Generation EU, in the context of The National Recovery and Resilience Plan, Investment 1.5 Ecosystems of Innovation, Project Tuscany Health Ecosystem (THE), CUP: B83C22003920001. The authors thank Simone Veglianti for the help with the measurements.
Funding
Open access funding provided by Università degli Studi di Siena within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
M.C., A.A. performed the measurements M.C., F.F.D., A.A. wrote and reviewed the main manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Corsini, M., Fabrizi de Biani, F. & Atrei, A. Electrochemical properties of magnetite nanoparticles supported on carbon paste electrode in various electrolyte solutions: A study by cyclic voltammetry. J Appl Electrochem 54, 1557–1565 (2024). https://doi.org/10.1007/s10800-023-02056-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10800-023-02056-x