
Abstract
Purpose
Axenfeld-Rieger syndrome (ARS) is an autosomal dominant disorder characterized by ocular anterior segment abnormalities. In the current study, we describe clinical and genetic findings in a Chinese ARS pedigree.
Methods
An ARS pedigree was recruited and patients were given comprehensive ophthalmic examinations and general physical examinations. DNA from the proband II:2 was used for exome sequencing. Sanger sequencing was utilized to identify and validate PITX2 variations. qPCR and western blotting were performed to detect PITX2 expression in immortalized peripheral blood lymphocytes.
Results
All affected family members showed typical ocular abnormalities, including iris atrophy, corectopia, shallow anterior chamber, complete or partial angle closure, and advanced glaucoma. They also exhibited systemic anomalies, such as microdontia, hypodontia, and redundant periumbilical skin. A heterozygous splice-site variation c.390 + 1G > A in PITX2, which might lead to a truncated PITX2 protein (p.Val131IlefsX127), was found in the proband. Sanger sequencing validated that the variation completely co-segregated with the ARS phenotype within this family and was absent in 100 unrelated controls. Western blotting revealed that the nuclear PITX2 protein was significantly decreased in patients compared with controls. Nonetheless, there was no significant difference in the total PITX2 protein level, consistent with qPCR results showing no alteration in PITX2 mRNA levels in the patient group.
Conclusions
PITX2 c.390 + 1G > A (p.Val131IlefsX127) was a novel genetic etiology of the ARS pedigree. The mutation leads to decreased nuclear PITX2, indicating lower transcriptional activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Axenfeld-Rieger syndrome (ARS) is an autosomal dominant hereditary disease with highly genetic heterogeneity [1]. The major candidate genes are paired-like homeodomain 2 (PITX2), located at 4q25, which plays a fundamental role in genetic control of ocular anterior segment development [2], and forkhead box C1 (FOXC1), located at 6p25, which acts as an important regulator of cell migration and differentiation during embryogenesis [3]. Mutations in these two genes are found in 40%–70% of ARS patients [4, 5]. PITX2 mutations are commonly associated with ocular, dental, and umbilical anomalies, whereas FOXC1 mutations are mostly associated with isolated ocular or ocular, heart, and/or hearing defects [6]. Other genetic causes, such as a mutation at locus 13q14 [7] might also be associated with ARS.
Clinical phenotypes of the eye in ARS patients are heterogeneous, which can present as posterior embryotoxon, iridogoniodysgenesis, polycoria, corectopia, or iris stromal hypoplasia. Glaucoma, secondary to maldevelopment of aqueous humor drainage structures, is the most serious consequence of ARS; approximately 50% of ARS patients progress to glaucoma [8]. Onset of secondary glaucoma typically occurs before the teenage years [9].
The current study describes the clinical phenotypes and genetic characterization of a Chinese family with ARS who harbor a PITX2 c.390 + 1G > A (p.Val131IlefsX127) mutation. Western blotting and quantitative real-time PCR (qPCR) indicated decreased levels of nuclear PITX2 protein in peripheral blood lymphocytes (PBL), but total PITX2 levels were unchanged.
Material and methods
Subjects
A Chinese Han pedigree with four affected individuals was recruited and peripheral blood samples were collected. The study adhered to the principles of the declaration of Helsinki; clinical protocols were approved by the Second Xiangya Hospital Institutional Review Board. Written informed consent was obtained from all included subjects.
Patients underwent basic ophthalmic examinations that comprised evaluation of best-corrected visual acuity (BCVA), Goldman applanation tonometry, slit-lamp biomicroscopic examination, and fundus examination. Other necessary examinations were also performed, including fundoscopy (non-mydriatic retinal camera, TRC-NW300; Topcan, Itabashi-ku, Tokyo, Japan), anterior segment photography (SL-D4; Topcan, Itabashi-ku), ultrasound biomicroscopy (UBM), and assessment of visual field defects (Humphrey Visual Field Analyzer; Carl Zeiss Humphrey Systems, Dublin, CA, USA).
Targeted exome sequencing
Genomic DNA was extracted according to the standard phenol–chloroform method. DNA of the proband II-2 was used for 90-cycles of paired-end exome sequencing performed on Illumina HiSeq2500 Analyzers (Illumina, San Diego, California, USA). Base-calling and calculation of quality values for every base were performed by pipeline software version 1.3.4 (Illumina). Reads were aligned to human reference genome GRCh37 using the Burrows-Wheeler Aligner (BWA; http://bio-bwa.sourceforge.net/) [10]. Single nucleotide variants (SNVs) and insertions and deletions (indels) were identified with SOAPsnp (http://soap.genomics.org.cn/soapsnp.html) and samtools (http://samtools.sourceforge.net/). SNVs and indels with read depth ≥ 8 × and quality ≥ 30 were reserved for subsequent analyses. Polymorphic SNVs were excluded based on the dbSNP database and the 1000 genomes annotation. SNVs and indels affecting coding sequences were annotated with Annotate Variation (ANNOVAR, http://annovar.openbioinformatics.org).
Sanger sequencing of implicated gene and splice site prediction
Sanger sequencing of the PCR amplicons was used to validate the pathogenic PITX2 (NM_001204397) mutation identified via exome sequencing. Sequences of the primers used for PCR were: forward primer (F), 5′-GACGGGAAAGTGTGTGTGTTT-3′, and reverse primer (R), 5′-GAGGGAACTGTAATCTCGCAAC-3′. PCR was performed in a 10 µl reaction mixture using 2 × Takara Taq™ HS Perfect Mix (Takara Biotechnology, Dalian, China). Amplification conditions consisted of an initial denaturation at 94 °C for 30 s, followed by 33 cycles of denaturation at 94 °C for 5 s, annealing at 60 °C for 20 s, and extension at 72 °C for 20 s. Final extension was performed at 72 °C for 7 min. NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/) was used to predict the donor and acceptor splice sites of the mutant PITX2 sequence.
Construction of immortal peripheral blood lymphocytes (PBL)
The mixture, which contained 2 mL heparin sodium treated blood, 2 mL RPMI-1640 basic medium and Ficoll-Paque PLUS (GE Healthcare, Chicago, IL, USA), were centrifuged at 2500 rpm for 10 min to isolate lymphocytes from human peripheral blood. The lymphocytes layer was carefully extracted with a Pasteur pipet and rinsed with RPMI-1640 basic medium three times. Following 1000 rpm centrifugation for 5 min, the pellet containing lymphocytes was resuspended in 1.5 mL lymphocyte culture medium containing 75% RPMI-1640 and 25% FBS (Gibco, NY, USA) and cultured in a T25 flask at 37 °C in a 5% CO2 atmosphere. B95-8 cells were cultured in a T25 flask for 5–7 days without changing the medium. Cells and medium were collected in a 15 ml conical tube and cryopreserved in −80 °C. When using EB virus, the cellular mixtures were frozen and thawed repeatedly three times, followed by centrifugation at 3000 rpm for 3 min. The supernatant was then collected for PBL infection by adding 200 µg/ml CyA (0.1 ml; Sigma-Aldrich, St. Louis, MO, USA) and 1.5 ml B95-8 cell derived EB virus into the primary PBL. Lymphocyte culture medium was added to cells depending on their growth until the medium volume reached 20 ml, after which half of the medium was replaced every 2–4 days. After culturing for 3–4 weeks, an immortal PBL line was obtained.
RNA extraction and qPCR measurement
Total RNA was extracted from immortal lymphocytes derived from two patients and three controls using Trizol reagent (Life Technologies, NY, USA). cDNA was synthesized from 1 μg total RNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, MA, USA). qPCR was performed on an Applied Biosystems® StepOne™ Plus Real-Time PCR System (Thermo Scientific, Waltham, MA, USA) with Maxima SYBR Green qPCR Master Mixes (Thermo Scientific). Relative expression of PITX2 was normalized to β-actin and analyzed by the comparative Ct method. Primers used for qPCR were: PITX2 (after the mutant site): forward primer, 5′-TACCTGTCCCTGTCACTCTTGA-3′ and reverse primer, 5′-AAGAACCCCTCCAATAAGGAAA-3′, β-actin, forward primer, 5′-CACGATGGAGGGGCCGGACTCATC-3′ and reverse primer, 5′-TAAAGACCTCTATGCCAACACAGT-3′. A two-tailed Student’s t test was used to determine the significance of differences between the two groups. Data is presented as mean ± SD.
Western Blotting and nuclear extraction
Nuclear extraction from PBL was performed using the NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific). Cell lysates and PBL nuclei were prepared with SDS sample buffer (63 mM Tris–HCl, 10% Glycerol, 2% SDS, 0.0025% Bromophenol Blue, pH 6.8) supplemented with 1% protease inhibitor cocktails (Sigma-Aldrich, St. Louis, MO, USA). After ultra-sonication, the lysate was centrifuged at 13,000 rpm for 10 min at 4 °C to collect the supernatant. The protein concentration was measured using the Pierce BCA™ protein assay kit (Thermo Scientific). Total cellular (20 ug) and nuclear (40 ug) protein were loaded onto SDS-PAGE gels. Proteins were transferred to a PVDF membrane, blocked with 5% non-fat milk in 0.1% PBST (0.1% Triton-X 100 in 1 × PBS), and blotted with primary antibodies (anti-PITX2 antibody, Abcam, Cambridge, UK; anti-PCNA antibody, Boster Biotechnology, China; anti-GAPDH antibody, Sigma-Aldrich) overnight at 4 °C. After washing three times in 0.1% PBST for 30 min, membranes were incubated with secondary HRP-conjugated antibodies (Kangwei biotech, China) for 1 h at room temperature. After washing with 0.1% PBST three times for 10 min each, membranes were treated with HRP substrates and visualized on a FluorChem FC3 system (ProteinSimple, California, USA).
Results
Clinical findings
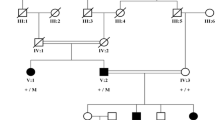
We described a three-generation pedigree (Fig. 1a) in which four of seven individuals were diagnosed with Axenfeld-Rieger syndrome (ARS). The proband II:2 in our study is a 26-year-old female who presented with gradual dimness of vision over five years. Her best-corrected visual acuity was 0.2 in the right eye and 0.1 in the left eye. Her intraocular pressure (IOP) was 29 mmHg eye and 25 mmHg in the right and left eye, respectively. She presented with small cornea, iris atrophy, corectopia, polycoria, and synechia in both eyes (Fig. 1b, Fig. 2). Moreover, she had typical craniofacial features of ARS, such as prominent forehead, telecanthus, and a flattened mid-face with a broad, flat nasal bridge (Fig. 1b), thin upper lip, hypodontia and microdontia (Fig. 1c). Her elder son (III:1) who is five years old was diagnosed with secondary glaucoma at age three. His intraocular pressure was 13 mmHg and 12 mmHg in the right and left eye, respectively, and he was being treated with 0.5% Timolol twice a day. He also had clinical features typical of ophthalmic and craniofacial disorders (Fig. 1d, e) as well as redundant peri-umbilical skin in his abdomen (Fig. 1f). The proband’s father (I:1), who is now 52 years old, had had no light perception for 10 years and was diagnosed with secondary glaucoma at age 36. His IOP was 31 mmHg and 32 mmHg in the right and left eye, respectively, and he was being treated with three IOP-lowering medicines. He also had clinical features that could be diagnosed as ARS. There were no clinical features in other individuals in the pedigree.

General phenotypes of the proband. a The AR pedigree. Roman numerals refer to generations and individuals within a generation were numbered from left to right. Proband is noted with an arrow. Filled symbols refer to ARS patients, open symbols refer to unaffected individuals. b The ocular features of the proband. c Intraoral view of the proband. d The ocular features of the patient III:1. e Intraoral view of patient III:1. f Protuberant umbilicus in patient III:1

Clinical ocular symptoms of the proband. The bilateral eyes have small corneas, irises atrophy, corectopia, polycoria, and synechia. OD, right eye; OS, left eye
Exome sequencing identified PITX2 c.390 + 1G > A (p.Val131IlefsX127) as the genetic basis of ARS
To identify the possible genetic mutation within the pedigree, we chose DNA from the proband II:2 for exome sequencing. A heterozygous c.390 + 1G > A substitution in the splice donor site of PITX2 intron 6 was detected. All family members were then screened for a mutation at this site via Sanger sequencing. The same was detected in I:1, III:1 and III:2 (Fig. 3a). All but III:2, who showed a normal phenotype, were diagnosed with ARS. Since III:2 is only 3 years old and the disease usually occurs after the teenage years, it is possible that he could exhibit an ARS phenotype as he ages. The c.390 + 1G > A mutation was not detected in any of 100 control individuals from outside the pedigree.

DNA sequence analysis of the PITX2 c.390 + 1G > A (p.Val131IlefsX127) mutation. a Sanger sequencing results of the pedigree. The arrow refers to the mutant base. b The splice site change caused by DNA mutation. c.390 + 1G > A ruins the former splice donor site in c.390 and introduces a new splice donor site in c.390 at c.390 + 1165, which brings part of intron 6 into the mutant transcript (Fig 3b). c The splice site changes give rise to a shift in the reading frame and introduce a stop codon at position 258
NetGene2 was used to predict splice site changes resulting from the mutation detected in the pedigree. As a result of the mutation, the guanine at c.390 + 1 of PITX2 is replaced by an adenine, causing the insertion of partial intron 6 into the mutant transcript (Fig. 3b), which is predicted to create a shift in the reading frame and introduce a stop codon at position 258 (Fig. 3c). This genetic result suggests that PITX2 c.390 + 1G > A may be the etiology of the ARS in this family.
Decreased nuclear PITX2 expression level
To further explain the effect of the mutant PITX2 protein in the ARS phenotype, we constructed immortal PBL lines from all seven family members (four patients and three normal members) as well as from one unrelated normal control, to assay the expression level and localization of PITX2. As is shown in Fig. 4c, qPCR of PBL mRNA showed no significant difference in PITX2 transcription between patients and controls (1.075 ± 0.022 and 1.032 ± 0.035, respectively; p = 0.2652). Moreover, immunoblotting (Fig. 4a and b) suggested no significant differences among patients and controls in the total cellular PITX2 protein level (1.094 ± 0.187 and 1 ± 0.129, respectively; p = 0.4004), which is accordance with our qPCR results. Considering that PITX2 is a crucial transcriptional factor in ocular development and needs to translocate to the cellular nucleus for normal function, we detected the PITX2 protein level in the nucleus. The PITX2 mutation led to decreased nuclear PITX2 protein levels compared to controls (0.0552 ± 0.00808 and 1.000 ± 0.143, respectively; p = 0.0008) (Fig. 4a, 4b). Taken together, the dislocation of PITX2 protein might cause insufficient functional PITX2 protein in nucleus, which in turn caused ocular anterior segment developmental abnormalities.

Decreased nuclear PITX2 expression in patients. a Immunoblotting shows that total PITX2 levels in patients and controls are not significantly different. b The quantitative results of immunoblotting. c qPCR result shows that there is no difference between patients and controls in PITX2 transcription in immortalized peripheral blood lymphocytes. ***, p < 0.001; ns, no significant difference
Discussion
ARS, which is caused by mutations of either PITX2 or FOXC1, exhibits considerable phenotypic heterogeneity. We now report an ARS pedigree with cosegregating mutations in PITX2 that was associated with typical anterior segment phenotypes, glaucoma, and craniofacial features. The age of optical symptoms presentation was younger in the second generation (II:2 and III-1) than the first (I:2). The IOP was easily controlled in all patients, indicating that the phenotype resulting from this mutation is relatively mild.
PITX2 is a homeodomain transcription factor that regulates the development of the ocular anterior segment [11], branchial arches [12], heart [13], and pituitary [14]. PITX2 mutations have been associated with a series of anterior segment malformations including ARS, Peter’s-like anomaly, iris hypoplasia/iridogoniodysgenesis syndrome, and ring dermoid of the cornea [5]. It has been reported to be a key effector of anterior segment patterning through retinoic acid and Wnt signaling [15]. In developing murine eyes, the expression pattern of PITX2 is initiated in the periocular mesenchyme, retained in mesenchyme derived corneal endothelium and TM, and finally restricted to the angle [16, 17]. PITX2 and FOXC1 might form a complex within the nucleus on the chromatin [18]. The PITX2 c.390 + 1G > A mutation we detected in this study is predicted to produce a truncated protein, which leads to the ARS pathology. However, in the current study, we observed both normal levels both mRNA and protein expression from PITX2 in patient-derived PBLs. It is probable that the normal copy of the PITX2 gene can compensate for gene expression. Moreover, the mutant PITX2 present in the pedigree exerted a dominant negative effect over the wild-type allele. While there was no difference in the level of cellular PITX2 in affected and unaffected pedigree members, those with the c.390 + 1G > A mutation had lower levels nuclear PITX2. As the subcellular localization of PITX2 is required for ocular development, the lower levels functional protein in the nucleus may be the cause of ARS phenotype in our pedigree.
In summary, we report a novel heterozygous splice site mutation in PITX2 in a family with variable ocular phenotypes. A correlation between the PITX2 mutation and phenotypic severity was observed, and a disturbed PITX2 nuclear localization was identified in affected individuals. Further study is warranted to determine the cause of the impaired PITX2 protein nuclear localization in this patient cohort.
References
Hjalt TA, Semina EV (2005) Current molecular understanding of Axenfeld-Rieger syndrome. Expert Rev Mol Med 7:1–17
Kumar J, Moses K (1997) Transcription factors in eye development: a gorgeous mosaic? Genes Dev 11:2023–2028
Kaur K, Ragge NK, Ragoussis J (2009) Molecular analysis of FOXC1 in subjects presenting with severe developmental eye anomalies. Mol Vis 15:1366–1373
Weisschuh N, De Baere E, Wissinger B (2011) Tumer Z. Axenfeld-Rieger syndrome. Eur J Hum Genet, Clinical utility gene card for, p 19
Reis LM, Tyler RC, Volkmann Kloss BA, Schilter KF, Levin AV, Lowry RB, Zwijnenburg PJ, Stroh E, Broeckel U, Murray JC, Semina EV (2012) PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur J Hum Genet 20:1224–1233
Yang Y, Wang X, Zhao Y, Qin M (2018) A novel 4q25 microdeletion encompassing PITX2 associated with Rieger syndrome. Oral Dis 24:1247–1254
Phillips JC, del Bono EA, Haines JL, Pralea AM, Cohen JS, Greff LJ, Wiggs JL (1996) A second locus for Rieger syndrome maps to chromosome 13q14. Am J Hum Genet 59:613–619
Seifi M, Walter MA (2018) Axenfeld-Rieger syndrome. Clin Genet 93:1123–1130
Souzeau E, Siggs OM, Zhou T, Galanopoulos A, Hodson T, Taranath D, Mills RA, Landers J, Pater J, Smith JE, Elder JE, Rait JL, Giles P, Phakey V, Staffieri SE, Kearns LS, Dubowsky A, Mackey DA, Hewitt AW, Ruddle JB, Burdon KP, Craig JE (2017) Glaucoma spectrum and age-related prevalence of individuals with FOXC1 and PITX2 variants. Eur J Hum Genet 25:1290
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Evans AL, Gage PJ (2005) Expression of the homeobox gene Pitx2 in neural crest is required for optic stalk and ocular anterior segment development. Hum Mol Genet 14:3347–3359
Liu W, Selever J, Lu MF, Martin JF (2003) Genetic dissection of Pitx2 in craniofacial development uncovers new functions in branchial arch morphogenesis, late aspects of tooth morphogenesis and cell migration. Development 130:6375–6385
Tessari A, Pietrobon M, Notte A, Cifelli G, Gage PJ, Schneider MD, Lembo G, Campione M (2008) Myocardial Pitx2 differentially regulates the left atrial identity and ventricular asymmetric remodeling programs. Circ Res 102:813–822
Ai D, Wang J, Amen M, Lu MF, Amendt BA, Martin JF (2007) Nuclear factor 1 and T-cell factor/LEF recognition elements regulate Pitx2 transcription in pituitary development. Mol Cell Biol 27:5765–5775
Gage PJ, Qian M, Wu D, Rosenberg KI (2008) The canonical Wnt signaling antagonist DKK2 is an essential effector of PITX2 function during normal eye development. Dev Biol 317:310–324
Gage PJ, Camper SA (1997) Pituitary homeobox 2, a novel member of the bicoid-related family of homeobox genes, is a potential regulator of anterior structure formation. Hum Mol Genet 6:457–464
Hjalt TA, Semina EV, Amendt BA, Murray JC (2000) The Pitx2 protein in mouse development. Dev Dyn 218:195–200
Berry FB, Lines MA, Oas JM, Footz T, Underhill DA, Gage PJ, Walter MA (2006) Functional interactions between FOXC1 and PITX2 underlie the sensitivity to FOXC1 gene dose in Axenfeld-Rieger syndrome and anterior segment dysgenesis. Hum Mol Genet 15:905–919
Acknowledgements
We would like to thank the family and normal individuals for participating in this study. We would also like to thank the members of the Department of Ophthalmology, the Second Xiangya Hospital, China for helpful discussions.
Funding
The study was supported by the National Natural Science Foundation of China (No.81700837, No.81402247), Funding of Department of Science and Technology, Hunan (No. 2015TP2007), 2019 China Scholarship Council (No.201906370215) and Postgraduate Independent Exploration and Innovation Project of Central South University (No. 2018zzts258). The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
FZ, LSZ and KL conceived and wrote the paper. FZ and MDC collected patient’s information. YTY and XCD taken the picture of patients and made figures. LH and JMS revised the manuscript. FZ and KL reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Second Xiangya Hospital research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, F., Zhang, L., He, L. et al. A PITX2 splice-site mutation in a family with Axenfeld-Rieger syndrome leads to decreased expression of nuclear PITX2 protein. Int Ophthalmol 41, 1503–1511 (2021). https://doi.org/10.1007/s10792-021-01704-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10792-021-01704-5