
Abstract
Purpose
To describe the ocular manifestations in a cohort of patients with systemic sarcoidosis (SS). Recent advances in the pathophysiology, diagnosis, and therapy of SS are also discussed.
Methods
Data from 115 Italian patients diagnosed between 2005 and 2016 were retrospectively reviewed. All but the first 17 patients underwent a comprehensive ophthalmologic examination. The diagnosis was based on clinical features, the demonstration of non-caseating granulomas in biopsies from involved organs, and multiple imaging techniques. Data on broncho-alveolar lavage fluid analysis, calcemia, calciuria, serum angiotensin-converting enzyme levels and soluble interleukin-2 receptor levels were retrieved when available.
Results
Ocular involvement, detected in 33 patients (28.7%), was bilateral in 29 (87.9%) and the presenting feature in 13 (39.4%). Anterior uveitis was diagnosed in 12 patients (36.4%), Löfgren syndrome and uveoparotid fever in one patient each (3%), intermediate uveitis in 3 patients (9.1%), posterior uveitis in 7 (21.2%), and panuveitis in 9 (27.3%). First-line therapy consisted of corticosteroids, administered as eyedrops (10 patients), sub-Tenon’s injections (1 patient), intravitreal implants (9 patients), or systemically (23 patients). Second-line therapy consisted of steroid-sparing immunosuppressants, including methotrexate (10 patients) and azathioprine (10 patients). Based on pathogenetic indications that tumor necrosis factor (TNF)-α is a central mediator of granuloma formation, adalimumab, targeting TNF-α, was employed in 6 patients as a third-line agent for severe/refractory chronic sarcoidosis.
Conclusion
Uveitis of protean type, onset, duration, and course remains the most frequent ocular manifestation of SS. Diagnostic and therapeutic advancements have remarkably improved the overall visual prognosis. An ophthalmologist should be a constant component in the multidisciplinary approach to the treatment of this often challenging but intriguing disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sarcoidosis is a chronic, multisystem, non-caseating granulomatous disease of unknown etiology. Its clinical course, natural history, and outcome are remarkably variable, as in approximately one-third of patients the disease has a tendency to wax and wane. Although sarcoidosis differs in patients of different ethnicities in terms of its clinical variability and visual outcome [1, 2], among those in whom clinical expression is characterized by acute onset with erythema nodosum and/or asymptomatic bilateral hilar lymphadenopathy the course is usually self-limiting and spontaneous resolution, mostly within the first 2–3 years after the diagnosis, occurs in one-third to half of the patients [3, 4]. However, those with an insidious onset and slow progression will eventually develop a multi-systemic disorder that includes lung involvement and multiple extra-pulmonary lesions [5].
Sarcoidosis is a major cause of ocular inflammation. The prevalence of ocular manifestations in patients with systemic sarcoidosis (SS) has been reported as 12.9% [6], 23% [7], 26% [8], and up to 79.2% [9]. However, given the strikingly protean spectrum of clinical manifestations, the lower rates may reflect, in addition to true geographic and ethnic variations, a possibly missed diagnosis in patients with mild ocular signs and symptoms. Ocular manifestations may be the presenting sign of sarcoidosis in 11–30% of patients [10, 11] who will later develop extra-ocular disease, but ocular findings may be recognized at any time during the course of SS [12, 13].
Although all segments of the eye and its adnexa can be involved, the most common sarcoidosis-related ocular disease is uveitis that may result in permanent visual impairment if not timely diagnosed and properly treated [14,15,16]. However, across the entire spectrum of uveitides (caused by several systemic diseases), the proportion of patients with sarcoid uveitis is variably reported as 2.4% [17], 3% [18], 6.4% [19], and up to 15% [20], again reflecting ethnicity, geography, and diagnostic challenges.
The purpose of this study was to describe the ocular manifestations in a cohort of patients with chronic SS who were diagnosed, treated and followed-up in a cooperative interdisciplinary approach.
Materials and methods
This is a retrospective, cross-sectional, observational analysis of the medical records of patients who sometime between 2005 and 2016 were examined at the university hospital of the University of Bari, Italy. The population of this cohort study consisted of 115 Caucasian patients from Apulia and other regions of southern Italy. The study was the result of a collaboration between the hospital’s Ophthalmology and Internal Medicine departments. Prior to the study period, 17 patients had been diagnosed with SS in the Internal Medicine department but had not reported visual disturbances, such that an ophthalmological examination was not requested by the examining physician. Later, two patients in whom ocular manifestations were the presenting feature of sarcoidosis underwent a thorough (ocular and extra-ocular) investigation, which led to our decision that all subsequent SS patients should undergo both an ophthalmological and a general internal medicine examination, whether or not they complained of ocular symptoms. Our study population of 115 patients thus consisted of the17 initially examined patients, the two patients examined thereafter and 96 subsequent patients, all of whom received a thorough examination to achieve a definite diagnosis of SS and establish the extent of organ involvement.
In addition, to provide a comprehensive description of ocular sarcoidosis, collaborations were conducted with the Department of Uveitis and Ocular Pathology, Sankara Nethralaya, Chennai, India, and the Department of Ophthalmology, Helsinki University Hospital, Finland. Both institutions contributed to the design and interpretation of the study and provided representative images of sarcoid uveitis.
The University’s Institutional Review Board approved the study and, given the retrospective nature of the study, which was based on a case records review, waived the need for patients’ written informed consent to study enrollment.
Since no widely accepted gold standard test is available for the diagnosis of sarcoidosis, in our cohort of patients SS was diagnosed on the basis of compatible clinical features, the demonstration of non-caseating granulomas in biopsies of involved organs, and typical radiological findings. All patients received a complete physical examination and work-up in order to assess the baseline extent and severity of the disease as well as the clinical phenotype.
Pulmonary sarcoidosis was confirmed by mediastinoscopy in 61 patients. Endobronchial ultrasound-guided transbronchial fine-needle aspiration of lymph nodes in the hilar, mediastinal or both locations was performed in 47 patients with radiological evidence of pulmonary involvement (41%). Rapid on-site cytological assessment led to the diagnosis in most cases, with additional lymph nodes and/or transbronchial lung biopsies performed when the diagnosis was still uncertain. Broncho-alveolar lavage (BAL) fluid was examined in 55 patients. Elevated cellularity (> 15% lymphocytes) and a CD4/CD8 ratio > 3.5 have a high specificity (93–96%) but a low sensitivity (53–59%) for sarcoidosis [21].
Eighty-four biopsies were carried out in 78 of the 115 patients (68%). The biopsy sites were lymph nodes (n = 34 patients), liver (n = 19), lymph nodes plus liver (n = 2), bronchial mucosa (n = 13), skin (n = 9), lacrimal gland (n = 3), skin plus lacrimal gland (n = 1), conjunctiva (n = 2), and lacrimal gland plus conjunctiva (n = 1). Second biopsies were performed when the results of the first were doubtful or inconclusive. 18F-fluorodeoxyglucose positron emission tomography in combination with a computed tomography scan (18F-FDG PET/CT) was used to detect occult sites of disease and to pinpoint the organs more suitable for diagnostic biopsy [22, 23].
Serum levels of angiotensin-converting enzyme (ACE), measured by a spectrophotometric kinetic assay, were available for 89 patients, none of whom was taking ACE inhibitors at the time of examination. In a 2016 report, the measurement of soluble interleukin-2 receptor (sIL-2R) was recommended as a useful screening marker for sarcoidosis in patients with uveitis of any type [24]. Since the patients in our study were enrolled between 2005 and 2016, only in the 11 patients with a more recent diagnosis were sIL-2R levels measured by an enzyme-linked immunosorbent assay (ELISA). Three of those patients had ocular involvement.
Serum and urine calcium concentrations, measured using a colorimetric method, were retrieved in 95 patients (including all 33 with ocular sarcoidosis) and in 78 patients (including 27 with ocular sarcoidosis), respectively. Hypercalcemia was defined as a calcium level > 11 mg/dL, and hypercalciuria as a urinary calcium excretion > 320 mg in a 24-h urine specimen.
Serum lysozyme levels were reported in the hospital files of 49 patients. However, this marker is a poorly reliable feature of sarcoidosis, as increases have been detected in 25–26% of patients receiving corticosteroids (CS) at the time of diagnosis [25, 26]. Therefore, serum lysozyme data were not considered further in our study.
All patients underwent a comprehensive ophthalmologic examination, including complete biomicroscopic assessment, intraocular pressure (IOP), visual acuity, pupillary reaction, ocular motility, visual field testing, ophthalmoscopy with fluorescein or indocyanine green angiography, fundus autofluorescence imaging, and optical coherence tomography. Additional tests were performed as requested, both at the time of diagnosis and at variable intervals during follow-up.
Categorical data are reported as the number and percentage. The data of independent groups were compared using Fisher’s exact test or a Chi-squared as appropriate. Mean and standard deviation were used to assess patient age by group; the data were compared using a t-test for independent samples. Sex distributions in the groups were compared using a Chi-squared test. A p value < 0.05 was considered significant for all tests. All of the statistical analyses were performed using the R-software, V.3.1.1 (https://www.r-project.org).
Diagnostic procedures were carried out in accordance with the ethical standards of the University of Bari Medical School and conformed with the tenets of the 1964 Helsinki Declaration and its later amendments.
Results
The results of the diagnostic procedures carried out in our patients with ocular sarcoidosis are summarized in Table 1. Serum levels of ACE were increased in the large majority, although the specificity and sensitivity of a high ACE level for the diagnosis of sarcoidosis are only 90% and 41%, respectively [27]. sIL-2R levels, measured in the serum samples of only 11 out of the 115 patients with SS, were elevated in 8 of them (72.7%), including 3 patients with ocular sarcoidosis. Although sIL2-receptor determinations are more reliable (94% specificity and 98% sensitivity) than the serum ACE level in detecting SS [24], the small number of sera examined in our study prevented an adequate analysis.
Hypercalcemia was detected at diagnosis in 5 (3 males and 2 females) out of the 33 patients tested (15%), and hypercalciuria in 11 (5 males and 6 females) of the 27 patients tested (41%), with the highest levels of urinary calcium occurring in 3 patients with severe active sarcoidosis. The administration of CS induced a prompt reversal of the metabolic defect (data not shown).
Pulmonary manifestations seen on chest X-rays and chest CT scan were the most common clinical features, with the findings ranging in severity from isolated bilateral hilar lymphadenopathy (stage 1, Fig. 1a–c) to pulmonary nodules scattered in both lung fields with a perilymphatic distribution and ground-glass parenchymal opacities. Further disease progression resulted in fibrosis, mostly localized in the hilar and peri-hilar regions but also visible in the upper lobes (stage 4, Fig. 1d, e). In selected patients, 18F-FDG/PET was employed to locate the most suitable and accessible biopsy site (Fig. 1f) [22].

Radiographic patterns seen in the thorax of patients with systemic (including ophthalmic) sarcoidosis. a Bilateral hilar lymphadenopathy (BHL) with polycyclic outlines. b In another patient, BHL is associated with mediastinal enlargement along the right para-tracheal boundary (arrow). c Axial contrast computed tomography (CT) image shows typical BHL (arrows). Lymph nodes with a density lower than that of the vasa are disposed around the bronchi and pulmonary arteries. d Micronodules a few millimeters in diameter tend to form granulomatous conglomerates that surround the broncho-vascular peduncles. Lung involvement extends along the broncho-vascular axis, from the hila toward the periphery. Note the diffuse distribution in both lung fields of lung nodules with a typical peri-lymphatic distribution and thickening of the pleural surfaces. e Advanced-stage pulmonary sarcoidosis. Fibrosis is typically localized in the hilar and peri-hilar districts and in the upper lobes, with large cystic spaces. The hila are stretched upwards. f 18F-FDG-PET/CT shows hypermetabolism in many organs and a clear prevalence in both lungs
Typical non-necrotizing epithelioid granulomas were seen in lung, lymph node or skin biopsies (Fig. 2a–i). The granulomas, consisting of epithelioid cells, giant cells and lymphocytes, exhibited a lymphatic pattern that surrounded the bronchovascular structures; angioinvasion was seen in some cases as well.

Typical histological features of sarcoidosis. a–c Pulmonary sarcoidosis. a Non-necrotizing granulomas are distributed along broncho-vascular bundles (hematoxylin–eosin [HE], × 20). b Several non-confluent granulomas with tightly packed epithelioid cells and Langhans multinucleated giant cells, surrounded by a sparse rim of lymphocytes (HE, × 200). c A well-defined granuloma adjacent to the vessel wall (HE, × 400). D–F: Lymph node sarcoidosis. d The nodal architecture is completely replaced by densely packed granulomas (HE, × 20). e and f Non-necrotizing granulomas consisting of epithelioid histiocytic cells (more clearly recognizable in f, and lymphocytes (HE, × 400). g–i: Cutaneous sarcoidosis. g Dense, non-necrotizing granulomas and inflammatory cells in the dermis are accompanied by epidermal hyperkeratosis (HE, × 200). h The same image as in G but at higher magnification shows that the granulomas are uniform in size and shape (HE, × 400). i The granulomatous infiltrate can occasionally extend from the dermis to the subcutaneous adipose tissue (HE, × 400)
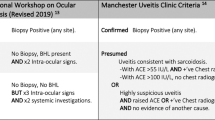
Ocular manifestations were detected in 33 of our 115 patients with SS (29%). The involvement was bilateral in 29 patients (88%) and the presenting feature in 13 of these patients (39%). The diagnostic procedures carried out in the 33 patients are summarized in Table 1. The First International Workshop on Ocular Sarcoidosis for the Standardization of Uveitis Nomenclature (SUN) published diagnostic criteria [28], which were later revised in a consensus workshop [29]. Based on these criteria, definite biopsy-supported ocular sarcoidosis was diagnosed in 32 of the 33 patients (97%). In the remaining patient, in whom no biopsy was performed, the diagnosis of ocular sarcoidosis was supported by an ophthalmologic examination indicating posterior uveitis, chest X-ray and CT scans showing bilateral hilar lymphadenopathy, parenchymal nodules and ground glass opacities, and BAL fluid with CD4 lymphocytosis of 18% and a CD4/CD8 ratio of 4:1.
Among the variable patterns of ocular sarcoidosis, eyelid and orbital involvement as well as tarsal and bulbar granulomas were uncommon findings. By contrast, anterior chronic granulomatous uveitis was the most frequent ophthalmologic manifestation and was diagnosed in 12 patients (36%), with bilateral involvement in 11 of them. Ocular pain, redness, photophobia, and lachrymation of variable severity were present in 9 patients, whereas the remaining 3 patients had a mild, almost silent uveitis. The ophthalmologic examination of the 12 patients revealed stellate keratic precipitates of the large mutton-fat-type (n = 8 patients, Fig. 3a, b), Koeppe nodules on the pupillary border (n = 3 patients) and Busacca nodules of the stroma (n = 1 patient, Fig. 3c). Posterior synechiae, glaucoma, and cataract as complications were observed in 4, 2, and 3 patients, respectively.

Sarcoidosis-related uveitis. a Slit-lamp photograph from a patient with granulomatous anterior uveitis due to sarcoidosis shows large mutton-fat keratic precipitates. b In another patient, smaller granulomatous keratic precipitates are seen. c Multiple Busacca nodules are clearly recognizable in this slit-lamp photograph from a patient with sarcoid anterior uveitis. d Fundus photograph from a patient with sarcoid intermediate uveitis shows snowball opacities. e Sarcoid retinal vasculitis with candle wax drippings
Anterior uveitis was also diagnosed in a 34-year-old male patient with a clinically distinct phenotype of sarcoidosis called Löfgren syndrome, which is characterized by acute-onset and recurrent erythema nodosum, acute iridocyclitis, bilateral hilar adenopathy and migratory polyarthritis [30]. In this patient, steroid-induced remission was achieved in just over one year. In a 41-year-old female patient who complained of dry eyes and mouth, anterior uveitis was associated with low-grade fever, bilateral parotid gland enlargement, and right facial nerve palsy. She was diagnosed with uveoparotid fever (Heerfordt syndrome) [31]. A therapeutic daily regimen of 0.5 mg prednisone/kg body weight with progressive tapering resulted in complete remission within 3 months and no further evidence of the disease over the next year.
Intermediate uveitis was detected in 3 patients (9.1%), in whom blurred vision and worsening floaters were common complaints. Slit lamp examination in the first patient, who had been diagnosed with pars planitis, showed several inflammatory cells suspended in the vitreous. In the other two patients, a higher number of cellular aggregates commonly termed “snowballs” (Fig. 3d) were identified in the vitreous, which were associated with vascular leakage on fluorescein angiography. Cataract and macular edema were observed as late complications in all three patients.
Posterior uveitis was diagnosed in 7 patients (21%) who complained of worsening vision, visual field disturbances, and floaters but who had no evidence of central nervous system involvement. On funduscopic examination, four patients had the typical fundus findings known as “candle-wax drippings” (Fig. 3e), that result from segmental retinal periphlebitis with perivenous exudation. Capillary obstruction with vitreous hemorrhage was detected in 2 patients and multifocal chorioretinitis with choroidal granulomas was observed in the seventh patient.
Finally, panuveitis with inflammation of all uveal components was recorded in 9 patients (27%). The clinical presentation included a variable combination of symptoms including redness and photophobia, ocular pain, lachrymation, blurring of vision, floaters, reduction of visual acuity, and photopsia. Fluorescein angiography revealed multiple chorioretinal peripheral lesions in 4 patients and multiple choroidal granulomas in the posterior pole associated with peripheral chorioretinal scars in 3 patients. The remaining 2 patients had only mild ocular complaints at the time of the first ophthalmological examination, despite macular edema, faint vitreous hemorrhage, and increased intraocular pressure (IOP).
Table 2 compares the basic demographic data, laboratory signs, disease phenotype [32], number of organs involved and the medical treatment of patients with (group A) and without (group B) ocular manifestations at diagnosis. Mean age was significantly higher in group B than in group A patients. Apart from ocular disease, which was the distinguishing feature in all group A patients but by definition absent in group B patients, the lungs and thoracic lymph nodes were the most frequently involved sites in both groups. The frequency of skin, peripheral lymph node, liver, spleen, heart and kidney involvement did not significantly differ between the two groups. There was also no significant difference in the number of group A and group B patients in whom from 1 to ≥ 4 organs were determined to be involved at the time of diagnosis.
The mean duration of follow-up was 48.8 ± 19.2 months (range, 13–84). A later appearance of ocular disease was recorded in 5 group B patients during follow-up: 2 patients developed posterior uveitis 15 and 18 months after the diagnosis whereas in the other 3 patients panuveitis was detected approximately 24, 31 and 36 months after the initial diagnosis. In all five patients, the ocular manifestations were diagnosed 6 weeks to 4 months after the discontinuation of CS treatment (3 patients) or during tapering to a maintenance dose (2 patients). Seven group A patients who had lung involvement at the time of diagnosis experienced pulmonary exacerbations within 1–2 years after the initial diagnosis. Three additional group A patients developed liver and heart involvement within 2 years of diagnosis, while still receiving low doses of CS.
Treatment
Given the persistent mystery regarding the etiology of sarcoidosis, its first-line treatment continues to be based on non-specific agents. Topical CS eyedrops, mostly prednisolone acetate 1% with variable frequency of administration (usually four times daily), were used to treat our patients with anterior uveitis, with the exception of two patients who instead received rimexolone 1% eyedrops. A mild increase in the IOP but no development of glaucoma was observed in three patients. CS eyedrops were combined with cycloplegics to relieve ciliary muscle spasm and to prevent the formation of posterior synechiae.
Of the three patients with intermediate uveitis, one was treated with sub-Tenon’s injections of triamcinolone acetonide, whereas the other two patients received a dexamethasone intravitreal implant (Ozurdex®, Allergan Pharmaceuticals) that was replaced every 5–6 months. The same type of intravitreal implant was employed in 3 and 4 patients with posterior uveitis and panuveitis, respectively. An increased IOP was a common complication and resulted in glaucoma and cataract in 3 patients.
Twenty-three patients, treated or not with topical CS, were administered systemic (invariably oral) CS. The initial dose of 0.5–1 mg/kg/day was gradually tapered according to the clinical course while maintaining the best balance between efficacy and side-effects (Fig. 4). The decision to switch from topical CS eyedrops or intravitreal CS implants to the systemic administration of CS, with or without steroid-sparing agents, was based on both the persistent severity of the uveitic disease and the concomitant occurrence of extra-ocular manifestations, especially respiratory tract involvement. Following the initiation of systemic treatment, none of the patients required additional Ozurdex implants.

Choroidal sarcoidosis and the response to corticosteroids. A fundus photograph of the left eye of a 65-year-old man with systemic sarcoidosis shows multiple choroidal nodules. The patient was started on a daily regimen of 1 mg oral prednisone/kg body weight. Serial fundus photographs after the initiation of treatment reveal a gradual reduction in the size of the nodules with scarring
Although CS remains the first-line treatment in the large majority of sarcoidosis patients, clear evidence of its long-term benefits from randomized controlled studies is lacking [33]. In addition, CS-related complications such as hypertension, hyperglycemia, Cushing-like syndrome, and osteoporosis, alone or in combination, develop in the majority of patients.
A wide variety of steroid-sparing agents, including azathioprine, methotrexate (MTX), leflunomide, and mycophenolate mofetil, are commonly used as second-line therapy in patients with refractory or relapsing SS and in those who suffer serious adverse effects from CS [33,34,35]. In our series, MTX was the most commonly employed immunosuppressive agent and, in combination with CS, was administered at a dose of 25 mg weekly to the 4 and 6 patients with posterior uveitis and panuveitis, respectively. In these same patients, in step with progressive tapering of the CS to a daily maintenance dose of 0.1–0.2 mg/kg and the concomitant reduction of MTX to 15 mg weekly, a low-dose of the antimetabolite azathioprine (50 mg daily) was added to the therapeutic regimen. Overall, satisfactory results were achieved in all patients in terms of both efficacy and safety. None of the patients were treated with mycophenolate mofetil, whose efficacy in a comparative randomized clinical trial was not higher than that of MTX; on the contrary, treatment success was 22% higher with MTX [36].
Three patients with posterior uveitis and 3 with panuveitis received a combination of gradually tapered CS (from 0.5 to 0.1 mg/kg daily) plus adalimumab (40 mg subcutaneously every 2 weeks for 12 weeks). These patients had concomitant lung involvement (pulmonary infiltration with or without lymphadenopathy [37], with a decreased diffusing lung capacity for carbon monoxide (DLCO), and had experienced frequent relapses of their ocular manifestations during stepwise reductions in the doses of CS and MTX. In 5 of the 6 patients treated with tapered CS and adalimumab, uveitic flares did not occur throughout the 12-month period of post-treatment follow-up. In all 5 patients, mean levels of C-reactive protein and ACE significantly decreased and DLCO improved, albeit to a variable extent. In the sixth patient, panuveitis worsened by the end of CS plus adalimumab treatment and a full-dose regimen of 1 mg CS/kg daily combined with 25 mg weekly of MTX was therefore resumed. All six patients administered adalimumab reported side effects of injection-site allergic reaction, fatigue, and malaise.
The overall visual outcome in both groups was satisfactory, although complete follow-up data were available only for the first 2 years and were fragmentary thereafter. Of the 62 eyes affected by sarcoid uveitis at diagnosis, the best corrected visual acuity (BCVA) was 20/20 or better in 24 eyes (38.7%), not worse than 20/50 in 36 eyes (58.1%) and reduced to counting fingers in the remaining two eyes. After one year, 29 eyes (46.8%) had a VA of 20/20 or better; three eyes lost 2 lines of VA and the VA of the two eyes with vision reduced to counting fingers remained unchanged. According to our last reliable VA data, recorded 2 years from the time of diagnosis, there was a further improvement in 37 (59.7%) of the eyes, with a VA of 20/20 or better. However, there was no recovery in the eyes that had lost 2 lines of VA and in those with counting fingers vision. Neither bilateral severe visual impairment nor blindness occurred in any of our patients.
Ocular surgery became inevitable in 16 patients (48%) and consisted of cataract extraction in 7 patients, trabeculectomy in 5 patients, and retinal pigment epithelial detachment (RPED) in 4 patients. One of the RPED patients had anterior granulomatous uveitis; the detachment was ascribed to a choriocapillary inflammatory process. The other 3 patients had panuveitis. In one of these patients, a rapid reduction in the size of the choroidal granulomas following CS treatment may have favored a tear in the RPE whereas in the other 2 patients budding during choroidal neovascularization through Bruch’s membrane was hypothesized as the underlying mechanism of serous RPED.
One patient with posterior uveitis became legally blind in her right eye after developing cystoid macular edema.
Discussion
The diagnosis of ocular sarcoidosis in our cohort was firmly established on the basis of typical ophthalmological features, radiologic findings and histological evidence of non-caseating epithelioid granulomas in biopsy specimens from ocular and/or extra-ocular tissue. An increased proportion of CD4 + helper T-cells with an elevated CD4/CD8 ratio in BAL fluid, detected in a high proportion of our patients (Table 1), has a specificity of ~ 95% for sarcoidosis, although the sensitivity is slightly below 60% [21]. In these cases, the diagnosis is further supported by the additional detection of high serum ACE levels [38] and by the presence of dysregulated calcium homeostasis. Hypercalcemia was detected in 15% of our patients at diagnosis. It has been reported with a similarly low prevalence (5–11%) in other studies [25,26,27], although it may be missed in those with subacute sarcoidosis, given the frequently undulating course of this form of the disease. Hypercalciuria, shown to be three times more frequent than hypercalcemia in SS [26], was determined in 41% of our patients.
Both literature reports and our own experience consistently evidence an association of chronic SS with a wide spectrum of ocular inflammatory manifestations, detected in 29% of our patients and occurring as the presenting feature in over one-third of them. Any segment of the eye and its adnexa may be affected, but uveitis is the most frequently diagnosed condition [11,12,13, 39]. The prevalence of uveitis as the initial presenting complaint of SS ranges from 20–30% [14, 16] to almost 80% [11], depending on the population studied and the length of ophthalmic follow-up. Conversely, 30–60% [13] and up to 80% [12, 15, 16] of patients with SS may develop ophthalmic disease at some point during the course of the disease.
In our cohort, the anatomical types of intraocular inflammation were, in descending order of frequency, anterior uveitis, panuveitis, and posterior and intermediate uveitis. At variance from studies in which the majority of patients were Black, posterior and anterior uveitis is the most common localization of sarcoid uveitis in Caucasian patients (reviewed in [12]). Depending on disease severity and the timing of therapy initiation, sarcoid uveitis remains a potentially sight-threatening condition that can cause permanent visual deterioration of variable degree [14, 40]. In our patients, a reliable estimation of the impact of sarcoid uveitis on visual function was restricted to the first 2 years of follow-up, when, compared with the BCVA at diagnosis, a VA of 20/20 or better was recorded in the large majority of the affected eyes. However, no improvement was found in the eyes that had lost two lines of VA one year after diagnosis and in those with counting fingers vision. Irreversible visual loss is most frequently associated with posterior uveitis complicated by cystoid macular edema, which occurred in one of our patients, but ocular hypertension and cataract formation may also lead to visual loss, albeit with a lower frequency [13, 14].
Pulmonary involvement is the most common extraocular manifestation of SS, occurring in > 90% of patients, with consequent respiratory symptoms and abnormal lung function [5, 10, 41]. The comparative analysis of our patients with (group A) and without (group B) ocular manifestations at diagnosis showed that patients in group A were significantly younger and had significantly less pulmonary involvement than those in group B (Table 2), suggesting that ocular sarcoidosis has its own specificity. However, the measured values of ACE, calcemia and calciuria were not significantly different between the two groups. In addition, the drugs used in treatment were fairly variable across the two groups and the outcomes only partially comparable. One possible explanation for the lower prevalence of lung involvement in group A patients is that the occurrence of eye symptoms compels the patient to seek a prompt ophthalmological examination, which usually results in an earlier diagnosis. With the initiation of a suitable therapy, involvement of the pulmonary parenchyma and mediastinal lymph nodes may be delayed or even prevented. Alternatively, considering that all our patients were ethnically homogeneous Caucasians, the differences in the prevalence patterns of the individual organ manifestations may reflect environmental factors [1, 42].
Although the etiology of sarcoidosis is unknown, a better understanding of the pathogenetic mechanisms underlying its onset is clearly important for the development of a more rational and effective therapeutic approach. The basic mechanism that triggers and maintains the inflammatory state is an interaction between lymphocytes and macrophages within the granulomatous areas, with the compartmentalization of T-cells expressing the helper (Th) phenotype [43]. Immunologically, sarcoidosis is characterized by an exaggerated Th type 1 (Th1) immune response to a so far unknown antigen(s) (Fig. 5). The interaction between antigens and innate immune receptors, such as Toll-like receptor 2, induces the activation of macrophages which then become highly efficient antigen-presenting cells able to interact with T cells. Activated macrophages also release pro-inflammatory cytokines, including tumor necrosis factor (TNF)-α, a central mediator in the initiation and amplification of the granulomatous response, as well as interleukin (IL)-12, IL-18 and IL-6, which modulate the type of T cell response [44, 45]. The levels of chemokines and chemokine receptors associated with a Th1 response are also increased [46] and, together with pro-inflammatory cytokines, orchestrate granuloma formation.

Hypothetical pathogenetic pathway of sarcoidosis. Antigens interact with genetic, epigenetic and environmental factors to cause the following: a TLR2-mediated macrophage activation with the production of pro-inflammatory cytokines, such as TNF-α, IL-12, IL-18 and IL-6; b antigen presentation by MHC class II molecules on macrophages to CD4+ T cells; activation and differentiation of CD4+ T cells into Th1 and Th17.1 effector cells that produce IFN-γ and IL-2, and Th17 cells that produce IL-17; c impaired regulatory T cell response; d up-regulation of the mTORC1 pathway; e up-regulation of SAA followed by SAA aggregation within the granuloma and amplification of the Th1 immune response. Abbreviations. TLR: toll-like receptor; MHC: major histocompatibility complex; IL: interleukin; TNF: tumor necrosis factor; IFN: interferon; mTORC1: mechanistic target of rapamycin complex 1; SAA: serum amyloid A
Topical, regional, and systemic CS remain the mainstay of therapy for chronic ocular sarcoidosis. Steroid-sparing immunosuppressive agents, which in our cohort consisted of MTX and azathioprine, may be required in patients who are or become dependent on, unresponsive to or poorly tolerant of CS treatment. However, for patients with SS not controlled by first- and second-line therapies, the obvious therapeutic choice is a combination of CS and a biologic agent. This was the case in 6 patients of our cohort, who were treated with CS plus adalimumab. All but one of these patients remained free of uveitic flares for one year post-treatment. Successful use of the chimeric antibody infliximab and the fully humanized monoclonal antibody adalimumab in the treatment of severe and refractory chronic sarcoidosis has been reported and both have become established third-line agents [47,48,49]. These antibodies target TNF-α, which, as mentioned above (Fig. 5), plays a central role in the initiation and amplification of the inflammatory response. In addition to TNF-α inhibitors, a steadily growing list of other biologic agents, such as lymphocyte inhibitors and specific receptor antagonists, are being tested in patients with non-infectious uveitis, including sarcoidosis, both in observational case series and in non-randomized off-label studies [16, 50].
The caution shown by experienced clinicians in recommending systemic biologic agents reflects the high cost of these drugs, the scarcity of randomized clinical trials confirming their efficacy and long-term safety data. Although a significant proportion of patients with systemic autoimmune diseases have been effectively treated with licensed biologic agents as second- or third-line therapies targeting a specific molecular component of the immune system, the paradoxical induction by these drugs of organ-specific or systemic autoimmune processes, including sarcoidosis, has been reported. Perez-Alvarez et al. [51] documented over 1500 cases involving these unexpected events and thus clearly demonstrated that biologics, in spite of their undoubted utility, may be a double-edged sword [52].
Among the strengths of our study are: (1) the diagnosis of systemic sarcoidosis supported by biopsies of involved organs in the large majority of patients; (2) the homogeneous collection of data, made possible through a carefully planned collaboration between an eye-care clinic and a clinical immunology tertiary center of the same university hospital; (3) the length of the patients’ follow-up, which exceeded 4 years in the majority, (n = 71, 62%) and (4) clinical and ophthalmological assessments made by the same internists and the same ophthalmologists to avoid or reduce the risk of variability.
The potential shortcomings must be noted as well, including: (1) the retrospective, observational nature of the study; (2) the relatively small number of patients, compared to multicenter, population-based cohort studies [10, 42, 53,54,55],(3) clinical and biochemical data that were only partially available for 5 of our patients with ocular manifestations and (4) the fact that an ophthalmologic examination was not performed in the first 17 patients diagnosed with systemic sarcoidosis, in whom the occurrence of subclinical or mildly symptomatic ocular manifestations could not, therefore, be excluded.
Patient education and scheduled periodic controls are recommended to achieve a timely diagnosis, to taper or conversely upgrade treatment as required, and to limit—to the extent possible—extraocular damage. A multidisciplinary collaboration among different healthcare providers, including the ophthalmologist, internist, immunologist and rheumatologist, will likely result in a more precise diagnostic workup and a more effective therapeutic strategy for all patients with sarcoidosis. Two important clinical challenges that remain to be met are the recognition of patients at risk of developing progressive ocular and multi-organ disease, and the decision when to stop treatment. Their resolution awaits the results of longitudinal studies with congruous numbers of patients.
References
Evans M, Sharma O, LaBree L, Smith RE, Rao NA (2007) Differences in clinical findings between Caucasians and African Americans with biopsy-proven sarcoidosis. Ophthalmology 114(2):325–333
Coulon C, Kodjikian L, Rochepeau C, Perard L, Jardel S, Burillon C et al (2019) Ethnicity and association with ocular, systemic manifestations and prognosis in 194 patients with sarcoid uveitis. Graefes Arch Clin Exp Ophthalmol 257(11):2495–2503
Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J (2014) Sarcoidosis. Lancet 383(9923):1155–1167
Mañá J, Rubio-Rivas M, Villalba N, Marcoval J, Iriarte A, Molina-Molina M et al (2017) Multidisciplinary approach and long-term follow-up in a series of 640 consecutive patients with sarcoidosis: cohort study of a 40-year clinical experience at a tertiary referral center in Barcelona, Spain. Medicine (Baltimore) 96(29):e7595
Chen ES, Moller DR (2011) Sarcoidosis—scientific progress and clinical challenges. Nat Rev Rheumatol 7(8):457–467
Atmaca LS, Atmaca-Sönmez P, Idil A, Kumbasar OO, Celik G (2009) Ocular involvement in sarcoidosis. Ocul Immunol Inflamm 17(2):91–94
Judson MA, Boan AD, Lackland DT (2012) The clinical course of sarcoidosis: presentation, diagnosis, and treatment in a large white and black cohort in the United States. Sarcoidosis Vasc Diffuse Lung Dis 29(2):119–127
Jabs DA, Johns CJ (1986) Ocular involvement in chronic sarcoidosis. Am J Ophthalmol 102(3):297–301
Ohara K, Okubo A, Sasaki H, Kamata K (1992) Intraocular manifestations of systemic sarcoidosis. Jpn J Ophthalmol 36(4):452–457
Baughman RP, Teirstein AS, Judson MA, Rossman MD, Yeager H Jr, Bresnitz EA et al (2001) Case control etiologic study of sarcoidosis (ACCESS) research group. clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med 164:1885–1889
Ma SP, Rogers SL, Hall AJ, Hodgson L, Brennan J, Stawell RJ, Lim LL (2019) Sarcoidosis-related uveitis: clinical presentation, disease course, and rates of systemic disease progression after uveitis diagnosis. Am J Ophthalmol 198:30–36
Rothova A (2000) Ocular involvement in sarcoidosis. Br J Ophthalmol 84(1):110–116
Jamilloux Y, Kodjikian L, Broussolle C, Sève P (2014) Sarcoidosis and uveitis. Autoimmun Rev 13(8):840–849
Pasadhika S, Rosenbaum JT (2015) Ocular sarcoidosis. Clin Chest Med 36(4):669–683
Yang SJ, Salek S, Rosenbaum JT (2017) Ocular sarcoidosis: new diagnostic modalities and treatment. Curr Opin Pulm Med 23(5):458–467
Matsou A, Tsaousis KT (2018) Management of chronic ocular sarcoidosis: challenges and solutions. Clin Ophthalmol 12:519–532
Barisani-Asenbauer T, Maca SM, Mejdoubi L, Emminger W, Machold K, Auer H (2012) Uveitis—a rare disease often associated with systemic diseases and infections—a systematic review of 2619 patients. Orphanet J Rare Dis 7:57
Llorenç V, Mesquida M, Sainz de la Maza M, Keller J, Molins B, Espinosa G et al (2015) Epidemiology of uveitis in a Western urban multiethnic population. the challenge of globalization. Acta Ophthalmol 93(6):561–567
Bodaghi B, Cassoux N, Wechsler B, Hannouche D, Fardeau C, Papo T et al (2001) Chronic severe uveitis: etiology and visual outcome in 927 patients from a single center. Medicine (Baltimore) 80(4):263–270
Kitamei H, Kitaichi N, Namba K, Kotake S, Goda C, Kitamura M et al (2009) Clinical features of intraocular inflammation in Hokkaido. Japan Acta Ophthalmol 87(4):424–428
Costabel U, Bonella F, Ohshimo S, Guzman J (2010) Diagnostic modalities in sarcoidosis: BAL, EBUS, and PET. Semin Respir Crit Care Med 31(4):404–408
Sobic-Saranovic D, Grozdic I, Videnovic-Ivanov J, Vucinic-Mihailovic V, Artiko V, Saranovic D et al (2012) The utility of 18F-FDG PET/CT for diagnosis and adjustment of therapy in patients with active chronic sarcoidosis. J Nucl Med 53(10):1543–1549
Rubini G, Cappabianca S, Altini C, Notaristefano A, Fanelli M, Stabile Ianora AA et al (2014) Current clinical use of 18FDG-PET/CT in patients with thoracic and systemic sarcoidosis. Radiol Med 119(1):64–74
Gundlach E, Hoffmann MM, Prasse A, Heinzelmann S, Ness T (2016) Interleukin-2 receptor and angiotensin-converting enzyme as markers for ocular sarcoidosis. PLoS ONE 11(1):e0147258
Pascual RS, Gee JB, Finch SC (1973) Usefulness of serum lysozyme measurement in diagnosis and evaluation of sarcoidosis. N Engl J Med 289(20):1074–1076
Thi Hong Nguyen C, Kambe N, Kishimoto I, Ueda-Hayakawa I, Okamoto H (2017) Serum soluble interleukin-2 receptor level is more sensitive than angiotensin-converting enzyme or lysozyme for diagnosis of sarcoidosis and may be a marker of multiple organ involvement. J Dermatol 44(7):789–797
Ungprasert P, Carmona EM, Crowson CS, Matteson EL (2016) Diagnostic utility of angiotensin-converting enzyme in sarcoidosis: a population-based study. Lung 194(1):91–95
Herbort CP, Rao NA, Mochizuki M (2009) Members of Scientific Committee of First International Workshop on Ocular Sarcoidosis. International criteria for the diagnosis of ocular sarcoidosis: results of the first International Workshop on Ocular Sarcoidosis (IWOS). Ocul Immunol Inflamm 17(3):160–169
Mochizuki M, Smith JR, Takase H, Kaburaki T, Acharya NR, Rao NA (2019) Feb 23 International Workshop on Ocular Sarcoidosis Study Group. Revised criteria of International Workshop on Ocular Sarcoidosis (IWOS) for the diagnosis of ocular sarcoidosis. Br J Ophthalmol pii: bjophthalmol-2018-313356
Brown F, Tanner LS. Lofgren Syndrome. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2019 Jan https://www.ncbinlm.nih.gov/books/NBK482315
Dua A, Manadan A (2013) Images in clinical medicine. Heerfordt's syndrome, or uveoparotid fever. N Engl J Med 369(5):458
Judson MA, Baughman RP, Teirstein AS, Terrin ML, Yeager H Jr (1999) Defining organ involvement in sarcoidosis: the ACCESS proposed instrument. ACCESS research group. a case control etiologic study of sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 16(1):75–86
James WE, Baughman R (2018) Treatment of sarcoidosis: grading the evidence. Expert Rev Clin Pharmacol 11(7):677–687
Müller-Quernheim J, Kienast K, Held M, Pfeifer S, Costabel U (1999) Treatment of chronic sarcoidosis with an azathioprine/prednisolone regimen. Eur Respir J 14(5):1117–1122
Baughman RP, Winget DB, Lower EE (2000) Methotrexate is steroid sparing in acute sarcoidosis: results of a double blind, randomized trial. Sarcoidosis Vasc Diffuse Lung Dis 17(1):60–66
Rathinam SR, Babu M, Thundikandy R, Kanakath A, Nardone N, Esterberg E et al (2014) A randomized clinical trial comparing methotrexate and mycophenolate mofetil for noninfectious uveitis. Ophthalmology 121(10):1863–1870
Baughman RP, Shipley R, Desai S, Drent M, Judson MA, Costabel U et al (2009) Sarcoidosis investigators. changes in chest roentgenogram of sarcoidosis patients during a clinical trial of infliximab therapy: comparison of different methods of evaluation. Chest 136(2):526–535
Baughman RP, Lower EE, du Bois RM (2003) Sarcoidosis. Lancet 361(9363):1111–1118
Ungprasert P, Tooley AA, Crowson CS, Matteson EL, Smith WM (2019) Clinical characteristics of ocular sarcoidosis: a population-based study 1976–2013. Ocul Immunol Inflamm 27(3):389–395
Abad S, Meyssonier V, Allali J, Gouya H, Giraudet AL, Monnet D et al (2004) Association of peripheral multifocal choroiditis with sarcoidosis: a study of thirty-seven patients. Arthritis Rheum 51(6):974–982
Prasse A (2016) The diagnosis, differential diagnosis, and treatment of sarcoidosis. Dtsch Arztebl Int 113(33–34):565–574
Schupp JC, Freitag-Wolf S, Bargagli E, Mihailović-Vučinić V, Rottoli P, Grubanovic A et al (2018) Phenotypes of organ involvement in sarcoidosis. Eur Respir J 51(1):1700991
Angi MR, Forattini F, Chilosi M, Cipriani A, De Caro G, Semenzato G (1990) Immunopathology of ocular sarcoidosis. Int Ophthalmol 14(1):1–11
Ziegenhagen MW, Rothe ME, Zissel G, Müller-Quernheim J (2002) Exaggerated TNFalpha release of alveolar macrophages in corticosteroid resistant sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 19(3):185–190
Zissel G, Prasse A, Müller-Quernheim J (2010) Immunologic response of sarcoidosis. Semin Respir Crit Care Med 31(4):390–403
Zissel G, Prasse A, Müller-Quernheim J (2007) Sarcoidosis–immunopathogenetic concepts. Semin Respir Crit Care Med 28(1):3–14
Erckens RJ, Mostard RL, Wijnen PA, Schouten JS, Drent M (2012) Adalimumab successful in sarcoidosis patients with refractory chronic non-infectious uveitis. Graefes Arch Clin Exp Ophthalmol 250(5):713–720
Saketkoo LA, Baughman RP (2016) Biologic therapies in the treatment of sarcoidosis. Expert Rev Clin Immunol 12(8):817–825
Adler BL, Wang CJ, Bui TL, Schilperoort HM, Armstrong AW (2019) Anti-tumor necrosis factor agents in sarcoidosis: a systematic review of efficacy and safety. Semin Arthritis Rheum 48(6):1093–1104
Hassan M, Karkhur S, Bae JH, Halim MS, Ormaechea MS, Onghanseng N et al (2019) New therapies in development for the management of non-infectious uveitis: a review. Clin Exp Ophthalmol 47(3):396–417
Perez-Alvarez R, Pérez-de-Lis M, Ramos-Casals M (2013) Biologics-induced autoimmune diseases. Curr Opin Rheumatol 25(1):56–64
Ramos-Casals M, Perez-Alvarez R, Diaz-Lagares C, Cuadrado M-J, Khamashta MA (2010) Autoimmune diseases induced by biological agents: a double-edged sword? Autoimmun Rev 9(3):188–193
Heiligenhaus A, Wefelmeyer D, Wefelmeyer E, Rösel M, Schrenk M (2011) The eye as a common site for the early clinical manifestation of sarcoidosis. Ophthalmic Res 46(1):9–12
Birnbaum AD, French DD, Mirsaeidi M, Wehrli S (2015) Sarcoidosis in the national veteran population: association of ocular inflammation and mortality. Ophthalmology 122(5):934–938
Acharya NR, Browne EN, Rao N, Mochizuki M (2018) Distinguishing features of ocular sarcoidosis in an international cohort of uveitis patients. Ophthalmology 125(1):119–126
Acknowledgements
Open access funding provided by Università degli Studi di Bari Aldo Moro within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
RD, GA, and FD conceived and designed the study, had full access to all the data in the study, and take responsibility for the integrity of the data and the accuracy of the analysis. RD and FD wrote the manuscript. JB and TTK revised the manuscript for important intellectual content and supplied additional images of sarcoid uveitis. PL, AM and DS retrieved and collected the data, and helped in the preparation of the figures. FAZ provided helpful insight into all histopathological diagnoses. All authors reviewed the manuscript, approved the draft submission, and accept responsibility for all aspects of this study.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dammacco, R., Biswas, J., Kivelä, T.T. et al. Ocular sarcoidosis: clinical experience and recent pathogenetic and therapeutic advancements. Int Ophthalmol 40, 3453–3467 (2020). https://doi.org/10.1007/s10792-020-01531-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10792-020-01531-0