
Abstract
Objective and design
Prostatic inflammation is the driving force in benign prostatic hyperplasia (BPH). This work investigated the potential modulatory effect of COX-2 inhibition on ADAM-17/EGFR/ERK1/2 axis.
Materials or subjects
Adult male Wistar rats were used.
Treatment
Celecoxib (10 and 20 mg/kg; i.p.) was injected i.p. daily for three weeks. Testosterone (TST) (3 mg/kg; s.c.) was used to induce BPH.
Methods
Prostatic inflammation and hyperplasia were assessed by organ weight and histopathology. Inflammatory mediators were measured using ELISA technique. Protein analysis was performed using western blotting and immunohistochemistry. Gene expression analysis was performed using qRT-PCR. Statistical analyses included one-way ANOVA and Tukey’s multiple comparison test.
Results
Testosterone-treated rats had a marked increase in COX-2, prostate weight, and index. Moreover, TST-induced COX-2 was inferred from cytoskeletal changes and was attributable to the overexpression of PGE2, NF-κB (p65), and IL-6. COX-2-derived PGE2 increased the activity of ADAM-17, TGF-α, and TNF-α. Consequently, EGFR–ERK1/2 pathway was over-activated, disrupting anti-apoptotic Bcl-2, cyclin D1, and pro-apoptotic Bax. Celecoxib reversed these effects.
Conclusion
COX-2 stimulates the ERK1/2 pathway via PGE2–ADAM-17-catalyzed shedding of TGF-α in testosterone-induced BPH. The results indicate a functional correlation between inflammation and hyperplasia in BPH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cyclooxygenase-2 (COX-2) is of major interest in several inflammatory disorders. COX-2 is an inducible cyclooxygenase isozyme that responds inappropriately to inflammatory stimuli. Unlike COX-2, COX-1 is normally expressed throughout the tissues and is required for optimal homeostasis. Generally, several factors may be implicated in the activation of COX-2, for example, nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB), tumor necrosis factor alpha (TNF-α), transforming growth factor alpha (TGF-α), and epidermal growth factor (EGF) (Morita 2002).
Benign prostatic hyperplasia (BPH) is a prevalent, aging-related chronic condition. BPH is seen as a clear-cut example of both inflammation and hyperplasia. It is characterized by the production of chemokines and cytokines, which promote inflammation. Prostaglandin E2 (PGE2) levels increase along with COX-2 levels, ultimately resulting in prostate cell proliferation (Chughtai et al. 2011). Despite the fact that COX-2 is highly expressed in BPH, it is not known how COX-2 causes prostatic enlargement (Robert et al. 2009). Testosterone (TST) is considered one of the most important risk factors for BPH. Besides being necessary to preserve sexual activity, the accumulated evidence indicates the inverse correlation of TST with age-related disorders in males, such as obesity, diabetes, metabolic syndrome, cardiovascular disorders, and cognitive impairment (Stanworth and Jones 2008). However, there are still some controversies surrounding the initiation and progression of BPH with high or low TST (Saad et al. 2011). Experimental and clinical studies have shown that the administration of anabolic steroid supplements stimulates the enlargement of the prostate in rats (Vargas et al. 2013), guinea pigs (Acosta et al. 2004), and even in bodybuilders (Kanayama et al. 2008, 2010).
Indeed, there is a well-identified interaction between COX-2 and epidermal growth factor receptor (EGFR). On the basis of efficacy, COX-2 inhibitors and EGFR inhibitors have reasonable therapeutic efficacy in non-malignant and malignant disorders (Dannenberg et al. 2005). Increased COX-2 transcription leads to increased synthesis of prostaglandins, especially PGE2, which can trans-activate EGFR signaling. PGE2 has been shown in several studies to increase cell proliferation through trans-activating EGFR signaling (Pai et al. 2002; Buchanan et al. 2003; Shao et al. 2003). Similar to other plasma membrane receptors, EGFRs commonly have an intracellular tyrosine kinase domain, a transmembrane segment, and an extracellular ligand-binding domain (Chen et al. 2016a). In the 1960s, the pivotal role of EGFR was well established in normal development and several disorders (Cohen 1962). Among all EGFR ligands, TGF-α and EGF have the greatest affinity. TGF-α binds to EGFR, resulting in the formation of homo-dimeric complexes, which are quickly followed by phosphorylation of the receptor. Typically, seven signaling molecules transmit the downstream effects of EGFR activation. The most significant pathway triggered by EGFR is ERK–MAPK; this pathway is known to be critical in cellular growth, differentiation, migration, and proliferation (Wee and Wang 2017).
Celecoxib (CXB) is a selective COX-2 inhibitor that has been demonstrated to help patients with BPH who experience nocturia (Falahatkar et al. 2008; Goodarzi et al. 2011). In addition, CXB is approved to be safe for several age-related inflammatory disorders (Goldenberg 1999; Dougados et al. 2001). In the current study, two daily doses of CXB were been selected (i.e., 10 mg/Kg and 20 mg/Kg) that were equivalent to those safe and well-tolerated in humans (Goodarzi et al. 2011; Nair et al. 2018). It is important to remember that CXB is preferred over other COX inhibitors owing to its selectivity for COX-2 and reduced side effects, particularly in elderly patients (Bushman and Jerde 2016). The present study employed CXB prophylactically to guarantee pre-inhibition of COX-2, which specifically impacts the relationship between COX-2 and the EGFR–ERK1/2 pathway in both the initiation and progression of testosterone-induced BPH in rats. Consequently, the main objective of the current work is to investigate the potential modulatory effect of COX-2 inhibition (via CXB) on ADAM-17/EGFR/ERK1/2 axis. Furthermore, this study re-evaluated CXB as a prophylactic approach for BPH and not only a symptomatic treatment for lower urinary tract symptoms induced by BPH.
Materials and methods
Materials
Celecoxib (CXB) was obtained from Pfizer Pharmaceuticals®, Cairo, Egypt, freshly dissolved in isotonic saline (Sun et al. 2013). Testosterone (TST) was obtained from Chemical Industries Development Co.® (CID) Giza, Egypt. It was reconstituted in olive oil to the required concentration (Abo-Youssef et al. 2020).
Animals
Eight-week-old male Wistar rats (150–200 g body weight) were kept in a controlled environment (22 °C ± 2 °C and constant humidity) under a 12 h light/dark cycle. A standard diet and water were freely available.
Ethics approval statement
The Institutional Animal Care and Use Committee of Beni-Suef University (BSU-IACUC-021–198) authorized the experiments prior to the study. Furthermore, the study protocols followed the National Institutes of Health’s recommendations for the care and use of laboratory animals (Garber 2011). The current animal study adhered to the ARRIVE guidelines (Percie du Sert et al. 2020).
Experimental design
Rats were randomly allocated to one of five groups; each contained six animals (Festing 2006). All animals were treated for three weeks. The treatment and groups were: the control group, administered isotonic saline (1 mL/kg/day; i.p.) plus 1 mL/kg/day olive oil subcutaneously; the CXB-20 group, was received celecoxib (CXB) (20 mg/kg/day; i.p.) (Sun et al. 2013; Nair et al. 2018) for 3 weeks plus 1 mL/kg/day olive oil subcutaneously; TST-induced BPH group, administered isotonic saline (1 mL/kg/day; i.p.) plus testosterone (TST) (3 mg/kg/day; s.c) for 2 weeks (Maggi et al. 1989; Pandita et al. 1998; Abo-Youssef et al. 2020); the CXB-10 plus TST group was received CXB (10 mg/kg/day; i.p.) (Sun et al. 2013; Nair et al. 2018) for 3 weeks, starting 1 week before TST administration (3 mg/kg/day; s.c.); the CXB–20 plus TST group was administered CXB (20 mg/kg/day; i.p.) (Sun et al. 2013; Nair et al. 2018) for three weeks, starting one week before TST administration (3 mg/kg/day; s.c.).
At the end of the experiment, the rats were weighed and then anesthetized by thiopental sodium (30 mg/kg; i.p) (Egyptian International Pharmaceutical Industries Company [EIPICO]®, Tenth of Ramadan City, Egypt.) (Fatehi-Hassanabad et al. 2005). Rats were then sacrificed by decapitated and prostate tissues were quickly dissected out, washed, then weighed. The prostate ventral lobes were divided into four sections. The first section was fixed in neutral buffered formalin (10%) for histological and immunohistochemical examinations. The second section was preserved in lysis buffer containing a protease inhibitor cocktail for western blotting analysis and stored at − 80 °C. The third section was stored in RNA later for use in quantitative real-time polymerase chain reaction analysis (qRT-PCR). The final section was kept at − 80 °C for use in further analysis.
Assessment of inflammation and hyperplasia in prostatic architecture
From each, the prostate was dissected and immediately weighed. The prostate index (PI) was determined by dividing the prostate weight (PW) by the overall body weight (Abo-Youssef et al. 2020).
Paraffin slices (4 µm) were produced from the fixed prostate samples (10% buffered formal saline) and then stained with hematoxylin and eosin. The tissue slides were then examined under a Leica microscope (Leica Microsystems GmbH®, Wetzlar, Germany). All light microscopic examinations and morphometric data utilized the Leica Application module for histological analysis, which was connected to a full HD microscopic imaging system (Leica Microsystems GmbH®, Germany) (Culling 1974). Each sample’s mean lining epithelial cell height was determined by scanning six non-overlapping fields from each ventral lobe tissue segment (Said et al. 2015).
Measurement of inflammatory mediators and metalloproteinase (ADAM-17 or TACE) activity in prostatic tissue using ELISA
In accordance with the manufacturer’s protocol, the assessment of COX-2 (Cusabio®, Houston, USA), NF-κB (Elabscience®, Houston, USA), PGE2 (Elabscience®, Houston, USA), IL-6 (Immuno-Biological Laboratories®, Minneapolis, USA), TNF-α (Cloud-Clone Corp.®, Texas, USA), ADAM-17 or TACE (Elabscience®, Houston, USA), and TGF-α (LifeSpan Biosciences®, Houston, USA) in prostatic tissue homogenates was performed using the corresponding rat ELISA kits.
Western blotting analysis of protein expression of p-EGFR and p-ERK1/2 in prostatic tissue
To determine the total protein content, the Bradford protein assay kit (Bio-Rad®, California, USA) was applied to each sample of the homogenized tissues (Bradford 1976). The protein levels of p-ERK1/2, T-ERK1/2, p-EGFR, and T-EGFR were assessed using western blotting. First, SDS lysis buffer was freshly prepared by adding the following components; 10 mM Tris, 100 mM NaCl, 25 mM ethylene-diamine tetra-acetic acid (EDTA), 25 mM ethylene glycol bis(2-aminoethyl) tetra-acetic acid (EGTA), 0.1% sodium dodecyl sulfate (SDS), 2% (v/v) Triton X-100 (pH 7.4), with 1:1000 protease inhibitor cocktail and phosphatase inhibitors (Elnagar et al. 2018; Abo-El Fetoh et al. 2020). Prostatic homogenates were then prepared in SDS buffer and subjected to SDS–polyacrylamide gel electrophoresis and western blotting analysis as described previously (Burnette 1981). To denature the proteins, 50 µg of total protein from each sample was mixed with an equivalent amount of 2 × electrophoresis sample buffer and heated at 95 °C for 10 min. After gel electrophoresis, the proteins were transferred onto polyvinylidene fluoride (PVDF) membranes (Bio-Rad®, California, USA) by semi-dry electro-blotting.
Primary incubation
Nonspecific binding to the PVDF membranes was initially blocked by incubation of the membrane in 5% non-fat dry milk in Tris-buffered saline containing 0.05% Tween (TBST) for 1 h. Then, the membrane was incubated with the primary antibodies against p-ERK1/2 (1:1000) (Cat# 8544–RRID: AB_11127856), T-ERK1/2 (1:2000) (Cat# 9102–RRID: AB_33074), p-EGFR (1:1000) (Cat# 2234–RRID: AB_331701), and T-EGFR (1:1000) (Cat# 2646–RRID:AB_2230881) (all from Cell Signaling Technology®, Massachusetts, USA) diluted in 1 × TBST buffer overnight at 4 °C. Four washes (each for 10 min) with 1 × TBST buffer were performed.
Secondary incubation
The PVDF membrane was then incubated with secondary antibody coupled to horseradish peroxidase (Cell Signaling Technology®, Massachusetts, USA, Cat# 7074–RRID: AB_2099233) diluted in 1 × TBST buffer (1:3000) for 30 min.
Detection
Enhanced chemiluminescence (ECL) solution (Perkin Elmer, Waltham®, Massachusetts, USA) was used to detect the signals with ChemiDoc imager (Bio-Rad®, California, USA) and the band intensity was measured using ImageLab® analysis software (Version 6.1.0 build 7, California, USA). T-ERK1/2 and T-EGFR were considered as loading control proteins, as discussed previously (Taylor et al. 2013; Abo-El Fetoh et al. 2020).
Detection of Cyclin D1 by immunohistochemistry
Prostatic tissue sections were dried, deparaffinized, and rehydrated before boiling in citrate buffer (pH 6.0) for 10 min. The tissue sections were gently washed for 2 h in TBS containing 5% bovine serum albumin (BSA). Anti-Cyclin D1 antibody (1:250) (Cell Signaling Technology®, Massachusetts, USA, Cat# 55506–RRID: AB_2827374) was incubated with the tissue sections overnight at 4 °C. After careful washing of the slides in TBS with 5% BSA, the slides were incubated with secondary antibody (Cell Signaling Technology®, Massachusetts, USA, Cat# 8114–RRID: AB_10544930), washed, and then treated with diaminobenzidine (DAB) for 15 min and washed in PBS. The slides were stained with hematoxylin, cleared in xylene, and dehydrated for microscopic examination (Kalyuzhny 2016). ImageLab® analysis software (Version 6.1.0 build 7, California, USA) was used to acquire the images and quantify the images. Six non-overlapping fields were randomly selected and scanned from each ventral lobe tissue section from each sample of immune-stained tissue sections for the determination of the mean percentage of immunohistochemical expression levels of cyclin D1 (Hsu et al. 2021).
Analysis of bax and Bcl-2 by real-time polymerase chain reaction (qRT-PCR)
qRT-PCR was used to measure the mRNA expression of pro-apoptotic Bax and anti-apoptotic Bcl-2. RExPrimer® was used to design primers (Piriyapongsa et al. 2009). The nucleotide sequences of the primers are shown in Table 1. Total RNA was extracted from prostatic tissues using TRIzol reagent Mini kit (Invitrogen®, Carlsbad, CA) in accordance with the manufacturer’s instructions. The mRNA expression of pro-apoptotic Bax and anti-apoptotic Bcl-2 was detected using a mixture of SYBR Green mastermix and Hot Start Taq RNA Polymerase (Jena Bioscience®, Jena, Germany). Based on the manufacturer’s guidelines, the following thermo-cycling conditions were applied in standard mode: 10 min at 95ºC, followed by 40 cycles of amplification (95ºC for 15 s, 57ºC for 45 s, and 72ºC for 45 s). ABI Prism® 7000 SDS Software was used to analyze the data. Finally, expression of each gene (i.e., number of copies) was calculated from the standard curve provided by each kit. The median of the RNA expression was calculated and used as a threshold to differentiate between the higher and lower expression within the factor groups. The mRNA expression was normalized to the expression of the housekeeping gene β-actin (Chen et al. 2016b).
Data and statistical analysis
Statistical analysis was conducted only for a group size of ≥ 5. Statistical significance was calculated using one-way ANOVA. Additionally, differences between groups were analyzed using Tukey’s multiple comparisons test. A probability (P) value of < 0.05 was considered to indicate statistical significance. All values are presented as the mean ± standard error of the mean (SEM). Data management and analysis were performed using GraphPad Prism® software for Windows (Version 9.2.0.332–San Diego, USA).
Results
The main hallmark of inflammation in testosterone (TST)-induced BPH is COX-2
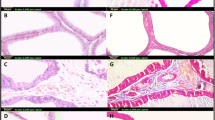
First, we confirmed the occurrence of BPH as a result of the subcutaneous injection of testosterone (TST). As shown in Table 2, the net PW was clearly increased in the TST-treated group (by 104%) above the control. Interestingly, PI was significantly raised in the TST-treated group (by 93%) above the control. In addition, we monitored the effect of TST-induced COX-2 on the cellular architecture of the prostate gland. The cross-sections and scoring analysis of TST-treated rats (Fig. 1c and Table 3) indicated massive congestion of the microvasculature (scoring + + + +) within prostate tissue. Furthermore, the presence of edema is apparent (scoring + +), with inflammatory cells in the perivascular compartment (scoring + +) and marked proliferation of the acinar lining epithelium (scoring + + + +). The abnormal histopathological changes were restored in the absence of COX-2, as shown in Fig. 1d and Fig. 1e. In addition, the quantitative analysis (Fig. 1f) revealed that the epithelium height was significantly increased by three-fold above the control. Other evidence presented in Fig. 2a shows that TST induced the highest level of COX-2 expression (by 81%) above the control. Compared with TST-treated rats, the pre-administration of 20 mg/kg/day celecoxib (CXB-20 plus TST) clearly attenuated COX-2 activity by 34%, whereas 10 mg/kg/day celecoxib (CXB-10 plus TST) showed only a small improvement.

TST-induced COX-2 clearly deteriorates the histological architecture of prostate in BPH. Histological examination of rat’s prostate: a Sections of control rats show normal histo-architecture of the ventral prostates. b Treatment with 20 mg of CXB alone showing unremarkable changes to control group. c Induction of BPH with TST exhibiting hypertrophy with increased epithelial thickness and intra-luminar projections and inflammatory infiltrate composed mainly of lymphocytes. d Treatment with 10 mg of CXB showing almost the same records as TST-induced group allover most of glandular acini with few scattered apparent intact acini in between. e Testosterone co-treated with 20 mg/kg CXB showing marked reduction in hypertrophy and hyperplasia. f Thickness of the basal epithelium cells showing significant reduction of cell membrane thickness with CXB-20–treated compared to TST-treated rats. The data are provided as mean ± SEM (n = 6). asignificant versus Control; bsignificant versus TST; csignificant versus CXB-10 + TST at P < 0.05. CXB Celecoxib; TST Testosterone. (Scale bar = 50 μm)

Summary of inflammatory markers induced by TST in BPH and the subsequent effect of celecoxib (CXB) on these markers: Evaluation of inflammatory markers using quantitative ELISA technique: Prostate content of COX-2 (ng/mg) a, NF-κB (p65) (ng/mg) b, PGE2 (Pg/mg) c and IL-6 (Pg/mg) d are significantly improved with co-administration of CXB. All inflammatory markers are further normalized with higher dose. The data are provided as mean ± SEM (n = 6). asignificant versus Control; bsignificant versus TST; csignificant versus CXB-10 + TST at P < 0.05. CXB Celecoxib, TST Testosterone; NF-κB (p65) Nuclear factor kappa-light-chain-enhancer of activated B cells, IL-6 Interleukin 6, COX-2 Cyclooxygenase enzyme isoform 2, PGE2 Prostaglandin E2
A positive correlation was found between COX-2 and PW, which was apparent the when COX-2 inhibitor (10 and 20 mg/kg/day CXB) was employed. As shown in Table 2, the concurrent administration of 20 mg/kg/day celecoxib (CXB-20 plus TST) efficiently decreased PW (by 37%) and PI (by 44%) compared with TST-treated rats. However, 10 mg/kg/day celecoxib (CXB-10 plus TST) caused a smaller improvement. In addition, the scoring values (Table 3) were alleviated by the COX-2 inhibitor. Interestingly, the inhibition of COX-2 (by 10 and 20 mg/kg/day CXB) exerted a significant reduction in the thickness of the cellular epithelium compared with TST-treated rats.
Testosterone (TST) activates the NF-κB inflammatory pathway in BPH
Next, we evaluated the involvement of the NF-κB inflammatory pathway in BPH. As shown in Fig. 2b, there was a marked difference in the activity of NF-κB in TST-treated rats compared with the control rats. Levels of NF-κB were significantly elevated in TST-treated rats (by 76%) compared with control rats. In addition, it has been well established that celecoxib (CXB) causes strong inhibition of NF-κB (Funakoshi-Tago et al. 2008). Compared with TST-treated rats, 20 mg/kg/day celecoxib (CXB-20 plus TST) reduced levels of NF-κB by 38%, whereas 10 mg/kg/day celecoxib (CXB-10 plus TST) resulted in only a minor improvement, indicating the dose-dependent effects of CXB.
COX-2-derived PGE2 is involved in testosterone (TST)-induced BPH
As illustrated in Fig. 2c, TST-induced rats had a significant increase in PGE2 activity (by 41%) compared with control rats. Compared with TST-induced rats, the absence of COX-2 in the 20 mg/kg/day celecoxib (CXB-20 plus TST) group clearly abolished PGE2 activity (by 27%), whereas 10 mg/kg/day celecoxib (CXB-10 plus TST) had a smaller effect.
Testosterone (TST)-induced COX-2 upregulates interleukin 6 in BPH
Interleukin 6 (IL-6) levels were clearly increased in TST-induced rats (by 1.5 times) compared with control rats (Fig. 2d). Compared with TST-treated rats, the concomitant administration of 20 mg/kg/day celecoxib (CXB-20 plus TST) strongly suppressed IL-6 levels (by 43%), whereas 10 mg/kg/day celecoxib (CXB-10 plus TST) caused a minor suppression.
Testosterone (TST)-induced COX-2 is critical for ERK1/2 phosphorylation in BPH
In this experiment, the mechanistic relevance between COX-2 and prostatic hyperplasia was considered. Several reports have considered the crosstalk between COX-2 and the EGFR–ERK1/2 pathway (Pai et al. 2002; Shao et al. 2003). Therefore, the potential effect of TST-induced COX-2 on the ERK1/2 pathway was examined. In line with high COX-2 expression (Fig. 2a), the results of the western blotting analysis in Fig. 3 revealed that TST-induced rats had a marked elevation in ERK1/2 phosphorylation (four times higher than the control rats).

TST-induced COX-2 is critical for phosphorylation of ERK1/2 in BPH. Assessment of phosphorylation of ERK1/2 using western blotting technique: Prostate content of p-ERK1/2/T-ERK1/2 is significantly improved with co-administration of CXB. It is apparent that inhibition of COX-2 is critical for decreasing phosphorylation of ERK1/2. The data are provided as mean ± SEM (n = 6). a significant versus Control; b significant versus TST; c significant versus CXB-10 + TST at P < 0.05. CXB Celecoxib, TST Testosterone, ERK1/2 Extracellular signal-regulated kinase 1 and 2
Furthermore, the significant inhibitory effects of COX-2 inhibitors support COX-2-mediated ERK1/2 activation (in the CXB-10 plus TST and CXB-20 plus TST groups). The quantitative analysis presented in Fig. 3 reveals an obvious reduction in ERK1/2 phosphorylation (by 66%) in 20 mg/kg/day celecoxib (CXB-20 plus TST), with only a minor effect in the 10 mg/kg/day celecoxib (CXB-10 plus TST) compared with TST-induced rats.
The metalloproteinase (ADAM-17 or TACE) is involved in COX-2-induced ERK1/2 phosphorylation in testosterone (TST)-treated animals
For this experiment, the upstream activators of ERK1/2 were assessed. Consequently, the functional involvement of COX-2 and ADAM-17 (TACE) (a member of the zinc metalloproteinase superfamily) has been investigated in the activation of the ERK1/2 pathway in TST-induced BPH. It is well known that COX-2-derived PGE2 induces ADAM-17 (TACE) activity (Yang and Chang 2018). To achieve this, the ADAM-17 (TACE) sandwich ELISA kit was utilized, which is the most sensitive tool for detection of ADAM-17 (TACE) activity (Trad et al. 2011; Yousef et al. 2020). It can be seen from Fig. 4a that the TST-treated rats have approximately 1.7 times more ADAM-17 (TACE) activity than control rats. As an additional proof, we identified the potential effect of high metalloproteinase activity of ADAM-17 (TACE) on its substrates (e.g., TNF-α and TGF-α). As shown in Fig. 4b and c, TST-treated rats had a marked increase in both TNF-α (by 66%) and TGF-α (by 1.2 times) compared with control rats.

TST-induced COX-2 triggers metalloproteinase activity of ADAM-17 (TACE) and its substrates in BPH. Evaluation of metalloproteinase activity of ADAM-17 and its substrates using quantitative ELISA technique: Prostate content of ADAM-17 or TACE (ng/mg) a, TNF-α (ng/mg) b and TGF-α (Pg/mg) c are significantly improved with co-administration of CXB. The data are provided as mean ± SEM (n = 6). asignificant versus Control; bsignificant versus TST; csignificant versus CXB-10 + TST at P < 0.05. CXB Celecoxib, TST Testosterone; ADAM-17 A disintegrin and metalloproteinase domain-17, TACE Tumor necrosis factor-alpha converting enzyme, TNF-α Tumor necrosis factor alpha, TGF-α Transforming growth factor alpha
Surprisingly, the pre-treatment with 20 mg/kg/day celecoxib (CXB-20 plus TST) significantly decreased ADAM-17 (TACE) activity (by 50%) compared with TST-treated rats. It is interesting that ADAM-17 (TACE) activity was restored to its normal ratio in the CXB-20 plus TST group; however, the improvement was smaller in the CXB-10 plus TST group. Moreover, the modulated metalloproteinase activity of ADAM-17 (TACE) is reflected on its substrates (e.g., TNF-α and TGF-α). As illustrated in Fig. 4b and c, rats in the CXB-20 plus TST group have significantly lower TNF-α (by 34%) and TGF-α (by 47%) activity compared with TST-treated rats.
COX-2-induced ERK1/2 phosphorylation is mediated via EGFR in testosterone (TST)-induced BPH
Transforming growth factor alpha (TGF-α) has long been recognized as one of the most effective EGFR ligands (Duffy et al. 2009; Schumacher and Rose-John 2021). There was a clear trend in of increasing TGF-α activity in TST-induced BPH. Therefore, we analyzed the potential impact of high TGF-α activity on EGFR. The western blotting analysis, presented in Fig. 5, shows that the phosphorylation of EGFR was strongly increased in TST-treated rats above control rats. Furthermore, the quantitative analysis in the lower panel of Fig. 5 shows a 2.5-fold increase in EGFR phosphorylation in TST-induced BPH.

TST-induced COX-2 activates ERK1/2 pathway via phosphorylation of EGFR in BPH. Assessment of phosphorylation of EGFR using western blotting technique: Prostate content of p-EGFR/T-EGFR is significantly improved with co-administration of CXB. It is apparent that inhibition of COX-2 leads to reduce phosphorylation of EGFR. The data are provided as mean ± SEM (n = 6). a significant versus Control; b significant versus TST; c significant versus CXB-10 + TST at P < 0.05. CXB Celecoxib, TST Testosterone, EGFR Epidermal growth factor receptor
Following the concomitant use of 20 mg/kg/day celecoxib (CXB-20 plus TST), a significant drop was observed in the phosphorylation of EGFR (Fig. 5). As shown in the lower panel of Fig. 5, rats in the CXB-20 plus TST group had decreased EGFR activity (by 80%) compared with TST-treated rats, with a less dramatic change in the CXB-10 plus TST group.
COX-2-induced prostatic hyperplasia is functionally correlated with the intracellular imbalance between proliferative and apoptotic markers in testosterone (TST)-induced rats
The question in the current experiment was to determine if there was an imbalance between proliferative and apoptotic signals in TST-induced BPH. As shown in Fig. 6, Cyclin D1 was highly expressed (in brown color) in TST-treated rats and quantified as approximately 11 times (Fig. 6f) higher than in control rats. In contrast, COX-2 inhibition restores the normal balance between proliferative and apoptotic signals. As illustrated in Fig. 6f, there was no increase in cyclin D1 expression in the CXB-20 plus TST group (79%) compared with TST-treated rats.

TST-induced cyclin D1 is abrogated with inhibition of COX-2. Immunohistochemical analysis of rat’s prostate (Scale bar = 50 μm): a Control sections demonstrate normal expression of cyclin D1. b Treatment with 10 mg of CXB alone showing almost the same normal expression of cyclin D1. c Induction of BPH with TST showing marked high expression of cyclin D1 (brown color). d Treatment with 10 mg of CXB showing almost the same expression of cyclin D1 (brown color) as TST-induced group. e Treatment with 20 mg of CXB showing significant improvement of cyclin D1 expression. f Quantitative analysis of cyclin D1 expression showing normalization of cyclin D1 expression with CXB-20–treated. The data are provided as mean ± SEM (n = 6). asignificant versus Control; bsignificant versus TST; csignificant versus CXB-10 + TST at P < 0.05. CXB Celecoxib, TST Testosterone
Further analysis was determined by qRT-PCR (Fig. 7a and Fig. 7b), which revealed the high mRNA expression of anti-apoptotic Bcl-2 (59% more than the control) and significantly lower mRNA expression of pro-apoptotic Bax (45% less than control) in TST-treated rats. Furthermore, mRNA expression of Bax was elevated in the CXB-20 plus TST (116%) compared with TST-treated rats (Fig. 7a). In addition, the mRNA expression of anti-apoptotic Bcl-2 was reduced (34%) in the CXB-20 plus TST compared with TST-treated rats (Fig. 7b).

TST-induced COX-2 disrupts the normal hemodynamic balance between apoptosis (pro-apoptotic Bax) and cell proliferation (anti-apoptotic Bcl-2). Assessment of proliferative versus apoptotic markers using quantitative PCR technique: Nuclear prostatic content of pro-apoptotic Bax a, and anti-apoptotic Bcl-2 b are significantly modulated after administration of CXB. All values are assigned as mRNA copies/ml (× 104). Together, pro-apoptotic Bax/anti-apoptotic Bcl-2 ratio c is significantly restored to its normal balance with CXB-20 treated rats. The data are provided as mean ± SEM (n = 6). asignificant versus Control; bsignificant versus TST; csignificant versus CXB-10 + TST at P < 0.05. CXB Celecoxib, TST Testosterone Bax Bcl-2 Associated X, Bcl-2 B-cell lymphoma 2
Collectively, the general trend in TST-induced BPH is for a lower ratio of pro-apoptotic Bax per anti-apoptotic Bcl-2 (65%) compared with the control group (Fig. 7c). On the other hand, COX-2 inhibition by 20 mg/kg/day celecoxib (CXB-20 plus TST), reversed the imbalance between proliferative and apoptotic signals by inducing a higher ratio of pro-apoptotic Bax per anti-apoptotic Bcl-2 compared with TST-induced BPH (Fig. 7c). The fewest changes were detected in the CXB-10 plus TST group.
Discussion
The BPH is the fourth most prevalent male diagnosis (Pizzorno et al. 2016). In general, the prevalence of BPH increases with age. There is an overall prevalence of 2%–25% with a further increase to 45% in older patients. From every 1000 persons, it is estimated that 9–41 patients are affected per year, with high treatment expenditure (USD 3–10 billion/year). Several factors, such as infectious agents, urine retrograde, metabolic syndrome, aging, and autoimmune disorders, may be involved in the initiation and progression of BPH nodules. All of these triggers participate in the activation of inflammatory signaling pathways, which ultimately results in prostatic enlargement (De Nunzio et al. 2020). In agreement with this concept, the current study evaluated the association between inflammation and hyperplasia in BPH. To this end, we selected the induction of BPH using testosterone (TST) in male rats, which is the most accepted experimental model of BPH in elderly patients (Altavilla et al. 2012; Abo-Youssef et al. 2020; Eid and Abdel-Naim 2020; Almukadi et al. 2021; Zhao et al. 2021).
Generally, TST is converted to dihydrotestosterone (DHT) via the 5-alpha-reductase enzyme. DHT is five times more active than TST. Consequently, DHT is able to induce prostatic hypertrophy by stimulating production of various growth factors (Madersbacher et al. 2019). As an additional pathway, TST is further bio-transformed into estradiol (the potent form of estrogen) through the action of CYP19/aromatase (Santen et al. 2009). Interestingly, the negative impact of estradiol on abnormal prostate enlargement was previously determined (Prins et al. 2006). As mentioned previously, BPH is well characterized with the presence of high levels of inflammatory mediators concurrently with prostatic hyperplasia (Abo-Youssef et al. 2020; Afify et al. 2020). However, the molecular mechanism through which prostatic inflammation induces hyperplasia has remained uncertain.
To prove this hypothesis, celecoxib (CXB) was employed prophylactically in the current study for three reasons. First, it is a standard COX-2 inhibitor (i.e., an inhibitor of inflammation) (Goldenberg 1999; Dougados et al. 2001). Second, the effectiveness of CXB (200 mg) as an anti-inflammatory in human BPH patients has also been shown in clinical trials (Falahatkar et al. 2008; Goodarzi et al. 2011). Third, CXB is proposed to also block the synthesis of IL-6 and prostaglandins, in particular prostaglandin E2 (PGE2), which is the main mediator in BPH (Anderson et al. 1996; Hinson et al. 1996). Consequently, CXB is the best choice for the validation of the current hypothesis. Based on the dose translation studies of Nair et al., the current preclinical study utilized 10 and 20 mg/Kg of CXB to best cover the human effective range experienced by Goodarzi et al. (Goodarzi et al. 2011; Nair et al. 2018). It is interesting to mention that COX-2 inhibitory action begins to weaken with daily doses above 50 mg/kg and entirely abates above 100 mg/kg (Niederberger et al. 2001). On safety, CXB has an FDA-boxed warning regarding possible adverse cardiovascular (C.V.S.) effects, notably heart attacks and strokes (Nissen et al. 2016; Barcella et al. 2019). In fact, there are some controversies about this issue. For instance, Barcella et al., discussed that even low doses of CXB have been associated with a significant C.V.S. risk (Barcella et al. 2019). However, Nissen et al., shown through clinical research that the substantial C.V.S risk only applies to doses more than 200 mg (Nissen et al. 2016). Interestingly, a similar dose of CXB was thoroughly examined and found to be safer for the entire G.I. tract than standard NSAIDs (Chan et al. 2010; García-Rayado et al. 2018). It is noteworthy that a benefit/risk ratio should be considered for prescribing CXB in patients at high C.V.S. risk (García-Rayado et al. 2018). Consistently, the present study was applied on rat equivalent dose (20 mg/Kg) to maximize the therapeutic efficacy with the least possible side effects.
Indeed, although TST normally declines with advancing age, intra-prostatic levels of DHT are maintained at normal levels. Elderly patients have a high incidence of coexisting clinical disorders that may further contribute to elevated levels of cytokines and immune responses in a “multi-hit” scenario. Accordingly, all of these factors may deviate the action of TST/DHT toward marked elevation of growth factors, including keratinocyte growth factor, EGF, insulin-like growth factors, and TGF-α. Ultimately, this pathway prompts the induction of stromal cell proliferation (Vignozzi et al. 2014). In addition, the same factors may be implicated in prostatic enlargement following abuse or long-administration of anabolic steroids in young athletes (Kanayama et al. 2008, 2010). Moreover, estrogen levels concurrently increase or remain constant. This provides strong support for the second scenario, which revealed the occurrence of BPH as a result of hormonal imbalance between androgens and estrogens in elderly patients. Regardless of TST level, BPH as a metabolic disorder may be essentially induced via TST, but it not crucial (Vignozzi et al. 2014).
The current study used a sequential process. Initially, the possible involvement of the NF-κB inflammatory pathway was explored in BPH. Indeed, NF-κB is a crucial component in a number of immunological and inflammatory processes. NF-κB is an inducible complex protein consisting of the p50 (NF-κB1) and p65 (RelA) subunits. When NF-κB is present in the cytoplasm, it is inactivated via sequestration with IκBα (the inhibitory protein). Gonzales et al. previously demonstrated that DHT binds to the intracellular androgen receptor (AR), resulting in the formation of a DHT–AR complex. It was also found that the DHT–AR complex increases nuclear translocation of NF-κB through the suppression of IκBα in cerebral blood vessels (Gonzales et al. 2009). Then, NF-κB binds to sequence-specific DNA binding sites and subsequently induces transcription of the gene encoding COX-2 (Liu et al. 2017). The results of the current study in BPH agree with these observations. Furthermore, several authors confirmed the inhibition of DHT-mediated NF-κB in the presence of anti-androgen therapy (Gonzales et al. 2009; Hsu et al. 2021). As CXB is a potent inhibitor of COX-2 (Goldenberg 1999) and NF-κB (Funakoshi-Tago et al. 2008), the current use of 10 and 20 mg/kg/day CXB effectively reduces the levels of COX-2 consistent with inhibition of NF-κB. Inflammation in BPH is therefore characterized primarily by COX-2. This observation is consistent with previous notation (Chughtai et al. 2011). It can therefore be assumed that PGE2 will also be induced.
For confirmatory purposes, the histopathological examination revealed severe deterioration of cellular architecture characterized by hallmarks of congestion, edema, and inflammation in BPH. The accumulated evidence agrees these results (Abo-Youssef et al. 2020; Afify et al. 2020). One possible explanation for this might be that inflammatory responses involve the translocation of phospholipase A2 from the cytosol to the nuclear membrane where the enzymatic hydrolysis of phospholipids takes place (Leslie 2004). Simultaneously, inflammation also induces transcription of the COX-2 and microsomal prostaglandin synthase E-1 (mPGE-1) genes. COX-2 acts on arachidonic acids (byproducts of phospholipids), producing prostaglandin H2, which is further converted to PGE2 (Morita 2002). The most clinically relevant findings are the strong inhibitory actions of CXB, which were clearly explored in histopathological examination. As further confirmation, the levels of PGE2 were markedly induced in the present study, concurrently with the high levels of COX-2. In contrast, the levels of both COX-2 and PGE2 are significantly suppressed in the presence of CXB. These results further support the idea that PGE2 is a principal mediator in inflammatory disorders (Park et al. 2006). Consequently, it was proposed that COX-2-derived PGE2 upregulates a positive feedback mechanism to induce expression of IL-6 in adjuvant arthritis (Anderson et al. 1996; Hinson et al. 1996). The current results mirror those of the previous studies that have examined the relationship between COX-2 and IL-6.
The next question in the current study was whether COX-2-derived PGE2 is essentially to the pathogenesis of BPH. The current findings corroborate the ideas of Pai et al., who suggested that PGE2 can induce gastric and intestinal hypertrophy. In addition, these results further support the involvement of PGE2 in colon cancer (Pai et al. 2002; Shao et al. 2003). Interestingly, the current results strengthen this idea by COX-2 inhibition. In the CXB-10 and CXB-20 groups, prostatic enlargement was markedly improved, and the CXB-20 group showed the best improvement.
The current results confirm the link between COX-2 and prostatic hypertrophy. Based on this, this study performed a thorough investigation of the mechanistic relevance of prostatic hypertrophy. Western blotting clearly showed the increase in ERK1/2 phosphorylation with COX-2 overexpression. The involvement of the ERK1/2 pathway in BPH is perhaps the most clinically significant finding in this study. Another finding was the marked inhibition of ERK1/2 phosphorylation in CXB-treated animals. In agreement with Ansari et al., COX-2-derived PGE2 induces cell proliferation, which may progress to tumor in keratinocytes (Ansari et al. 2008). To further prove the implication of ERK1/2 pathway in BPH, this study investigated upstream and downstream signals involved in this pathway.
There are several mechanisms for the induction of trophic action via COX-2-induced PGE2 (Yang and Chang 2018). One of them is via activation of metalloproteinase activity, which was the main focus of the current study. Several studies have demonstrated the role of ADAM-17 (TACE) as a cleaving enzyme in the activation of TNF-α and TGF-α (high-affinity EGFR ligands) (Duffy et al. 2009; Schumacher and Rose-John 2021). PGE2 overexpression causes ecto-domain shedding of TNF-α and TGF-α (the highest affinity EGFR ligand) via ADAM-17 (TACE). Active TGF-α binds to EGFR, causing it to dimerize and phosphorylate, activating the EGFR–ERK1/2 signaling cascade (Pai et al. 2002; Wee and Wang 2017). ERK1/2 activation plays an important role in inducing proliferative (e.g., cyclin D1 and anti-apoptotic Bcl-2) signals and inhibiting apoptotic signals (Bax) (Bonnefoy-Berard et al. 2004). Based on the findings of Abo-El Fetoh et al., phosphorylated ERK (p-ERK1/2) trans-locates from the cytosol to the nucleus (Abo-El Fetoh et al. 2020). p-ERK1/2 induces the expression of cell proliferation genes (i.e., cyclin D1 and anti-apoptotic Bcl-2) and abrogates the expression of pro-apoptotic Bax. Finally, the normal cell cycle is disrupted (Bonnefoy-Berard et al. 2004; Fu et al. 2019). At mRNA level, pro-apoptotic Bax, anti-apoptotic Bcl-2, and the Bax/Bcl-2 ratio were dysregulated compared with control samples. These results mirror those of the previous studies that have examined the dysregulation in pro-apoptotic Bax and anti-apoptotic Bcl-2 (Abdel-Naim et al. 2018; Abo-Youssef et al. 2020). Surprisingly, no differences were found when the protein levels of pro-apoptotic Bax and anti-apoptotic Bcl-2 were detected (Almukadi et al. 2021; D’Amico et al. 2021; Hsu et al. 2021). This implies that the normal hemodynamic balance of the prostate may be disrupted and further directed toward hyperplasia.
The activation of cAMP/protein kinase A pathway is considered another mechanism for PGE2-induced hypertrophy, and leads to an increase in the expression of amphiregulin (another EGFR ligand) (Shao et al. 2003). The activation of the intracellular Src-mediated pathway, the third mechanism, is independent of the release of extracellular EGFR ligand (Buchanan et al. 2003). Together, it is believed that all of these mechanisms activate the positive feedback loop to further increase gene expression of COX-2 and PGE2, leading to greater induction of the EGFR–ERK1/2 pathway (Dannenberg et al. 2005).
Conclusion
The present study uncovers the functional correlation between COX-2–PGE2 and ADAM-17 in BPH, which may be a future target for the management of BPH while preserving the current level of testosterone and mitigating interference from anti-androgen therapy. In summary, COX-2 induces the ERK1/2 pathway via PGE2–ADAM-17-catalyzed shedding of TGF-α in testosterone-induced BPH (Fig. 8). Furthermore, the current study suggests a novel treatment option (i.e., CXB) that is suitable not only for nocturia induced by BPH but also for prostatic hyperplasia.

Graphical diagram of functional correlation between inflammation and hyperplasia in TST-induced BPH. Cycloxygenase-2 (COX-2) upregulates EGFR–ERK1/2 signaling cascade via stimulation of PGE2-induced ADAM-17 and consequent shedding of TGF-α: The left panel (in red color) shows that TST-induced BPH stimulates the nuclear translocation of NF-κB [1–2] resulting in increasing gene transcription of COX-2 [3] and subsequent PGE2 [4]. PGE2 trans-activates EGFR via intracellular activation of ADAM-17 (TACE) [5] leading to shedding of TGF-α [6]. Consequently, active TGF-α phosphorylates EGFR and induce ERK1/2 phosphorylation [7–8]. Together, the nuclear translocation of p-ERK1/2 [9] induced prostatic hyperplasia via enhancing gene expression of cyclin D1 and anti-apoptotic Bcl-2 in consistent with downregulation of pro-apoptotic Bax expression. In the contrast, the panel in the right corner (in blue color) shows the potential effect of COX-2 inhibition in TST-induced BPH
Data availability
The data that support the findings of this study are included within the manuscript or the supplementary data.
Abbreviations
- COX-2:
-
Cyclooxygenase enzyme isoform 2
- PGE2:
-
Prostaglandin E2
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- IL-6:
-
Interleukin 6
- ADAM-17/TACE:
-
A disintegrin and metalloproteinase domain-17/Tumor necrosis factor-alpha converting enzyme
- TGF-α:
-
Transforming growth factor alpha
- TNF-α:
-
Tumor necrosis factor alpha
- EGFR:
-
Epidermal growth factor receptor
- ERK1/2:
-
Extracellular regulated kinase protein 1 and 2
- Bcl-2:
-
β-Cell lymphoma 2
- Bax:
-
Bcl-2-associated X protein
References
Abdel-Naim AB, Neamatallah T, Eid BG, Esmat A, Alamoudi AJ, Abd El-Aziz GS, Ashour OM (2018) 2-Methoxyestradiol attenuates testosterone-induced benign prostate hyperplasia in rats through inhibition of HIF-1 α /TGF- β /Smad2 Axis. Oxid Med Cell Longev. https://doi.org/10.1155/2018/4389484
Abo-El Fetoh ME, Helal GK, Saleh IG, Ewees M, Elshafey M, Elnagar MR, Akool E-S (2020) Cyclosporin A activates human hepatocellular carcinoma (HepG2 cells) proliferation: implication of EGFR-mediated ERK1/2 signaling pathway. Naunyn Schmiedebergs Arch Pharmacol 393:897–908. https://doi.org/10.1007/s00210-019-01798-w
Abo-Youssef AM, Afify H, Azouz AA, Abdel-Rahman HM, Abdel-Naim AB, Allam S (2020) Febuxostat attenuates testosterone-induced benign prostatic hyperplasia in rats via inhibiting JAK/STAT axis. Life Sci 260:118414. https://doi.org/10.1016/j.lfs.2020.118414
Acosta S, Dizeyi N, Feinstein R, Pierzynowski S, Abrahamsson PA (2004) Long-term testosterone stimulation induces hyperplasia in the guinea-pig prostate. Prostate Cancer Prostatic Dis 7:227–231. https://doi.org/10.1038/sj.pcan.4500744
Afify H, Abo-Youssef AM, Abdel-Rahman HM, Allam S, Azouz AA (2020) The modulatory effects of cinnamaldehyde on uric acid level and IL-6/JAK1/STAT3 signaling as a promising therapeutic strategy against benign prostatic hyperplasia. Toxicol Appl Pharmacol 402:115122. https://doi.org/10.1016/j.taap.2020.115122
Almukadi H, Eid BG, Shaik RA, Abdel-Naim AB, Esmat A (2021) Auraptene nanoparticles ameliorate testosterone-induced benign prostatic hyperplasia in rats: Emphasis on antioxidant, anti-inflammatory, proapoptotic and PPARs activation effects. Biomed Pharmacother 143:112199. https://doi.org/10.1016/j.biopha.2021.112199
Altavilla D, Minutoli L, Polito F, Irrera N, Arena S, Magno C et al (2012) Effects of flavocoxid, a dual inhibitor of COX and 5-lipoxygenase enzymes, on benign prostatic hyperplasia. Br J Pharmacol 167:95–108. https://doi.org/10.1111/j.1476-5381.2012.01969.x
Anderson GD, Hauser SD, Mcgarity KL, Bremer ME, Isakson PC, Gregory SA (1996) Selective inhibition of cyclooxygenase (COX)-2 reverses inflammation and expression of COX-2 and interleukin 6 in rat adjuvant arthritis. J Clin Investig 97:2672–2679. https://doi.org/10.1172/JCI118717
Ansari KM, Rundhaug JE, Fischer SM (2008) Multiple signaling pathways are responsible for prostaglandin E2-induced murine keratinocyte proliferation. Mol Cancer Res 6:1003–1016. https://doi.org/10.1158/1541-7786.MCR-07-2144
Barcella CA, Lamberts M, Mcgettigan P, Fosbøl EL, Lindhardsen J, Torp-Pedersen C et al (2019) Differences in cardiovascular safety with non-steroidal anti-inflammatory drug therapy—A nationwide study in patients with osteoarthritis. Basic Clin Pharmacol Toxicol 124:629–641. https://doi.org/10.1111/bcpt.13182
Bonnefoy-Berard N, Aouacheria A, Verschelde C, Quemeneur L, Marçais A, Marvel J (2004) Control of proliferation by Bcl-2 family members. Biochim Biophys Acta 1644:159–168. https://doi.org/10.1016/j.bbamcr.2003.10.014
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. https://doi.org/10.1016/0003-2697(76)90527-3
Buchanan FG, Wang D, Bargiacchi F, Dubois RN (2003) Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J Biol Chem 278:35451–35457. https://doi.org/10.1074/jbc.M302474200
Burnette WN (1981) “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem 112:195–203. https://doi.org/10.1016/0003-2697(81)90281-5
Bushman WA, Jerde TJ (2016) The role of prostate inflammation and fibrosis in lower urinary tract symptoms. Am J Physiol-Renal Physiol 311:F817–F821. https://doi.org/10.1152/ajprenal.00602.2015
Chan FKL, Lanas A, Scheiman J, Berger MF, Nguyen H, Goldstein JL (2010) Celecoxib versus omeprazole and diclofenac in patients with osteoarthritis and rheumatoid arthritis (CONDOR): a randomised trial. Lancet 376:173–179. https://doi.org/10.1016/S0140-6736(10)60673-3
Chen J, Zeng F, Forrester SJ, Eguchi S, Zhang M-Z, Harris RC (2016a) Expression and function of the epidermal growth factor receptor in physiology and disease. Physiol Rev 96:1025–1069. https://doi.org/10.1152/physrev.00030.2015
Chen W, Chen W, Zhu J, Chen N, Lu Y (2016b) Potent anti-inflammatory activity of tetramethylpyrazine is mediated through suppression of NF-k. Iran J Pharm Res. 15:197–204. https://pubmed.ncbi.nlm.nih.gov/27610159
Chughtai B, Lee R, Te A, Kaplan S (2011) Role of inflammation in benign prostatic hyperplasia. Reviews Urol 13:147–150. https://pubmed.ncbi.nlm.nih.gov/22110398/
Cohen S (1962) Isolation of a Mouse Submaxillary Gland Protein Accelerating Incisor Eruption and Eyelid Opening in the New-born Animal. J Biol Chem 237:1555–1562. https://doi.org/10.1016/S0021-9258(19)83739-0
Culling CFA (1974) Handbook of histopathological and histochemical techniques: including museum techniques. Butterworth-Heinemann. https://doi.org/10.1016/C2013-0-04011-X
D’amico R, Genovese T, Cordaro M, Siracusa R, Gugliandolo E, Peritore AF et al (2021) Palmitoylethanolamide/Baicalein Regulates the Androgen Receptor Signaling and NF-κB/Nrf2 Pathways in Benign Prostatic Hyperplasia. Antioxidants 10:1014. https://doi.org/10.3390/antiox10071014
Dannenberg AJ, Lippman SM, Mann JR, Subbaramaiah K, Dubois RN (2005) Cyclooxygenase-2 and Epidermal Growth Factor Receptor: Pharmacologic Targets for Chemoprevention. J Clin Oncol 23:254–266. https://doi.org/10.1200/jco.2005.09.112
De Nunzio C, Lombardo R, Cicione AM, Tubaro A (2020) Benign Prostatic Hyperplasia (BPH). In: Chapple CR, Steers WD, Evans CP (eds) Urologic principles and practice. Cham: Springer International Publishing. https://doi.org/10.1007/978-3-030-28599-9_20
Dougados M, Béhier J-M, Jolchine I, Calin A, Van Der Heijde D, Olivieri I et al (2001) Efficacy of celecoxib, a cyclooxygenase 2-specific inhibitor, in the treatment of ankylosing spondylitis: a six-week controlled study with comparison against placebo and against a conventional nonsteroidal antiinflammatory drug. Arthritis Rheum 44:180–185. https://doi.org/10.1002/1529-0131(200101)44:1%3c180::AID-ANR24%3e3.0.CO;2-K
Duffy MJ, Mckiernan E, Odonovan N, Mcgowan PM (2009) Role of ADAMs in cancer formation and progression. Clinical Cancer Res 15:1140–1144. https://doi.org/10.1158/1078-0432.Ccr-08-1585
Eid BG, Abdel-Naim AB (2020) Piceatannol attenuates testosterone-induced benign prostatic hyperplasia in rats by modulation of Nrf2/HO-1/NFκB Axis. Front Pharmacol. https://doi.org/10.3389/fphar.2020.614897
Elnagar MR, Walls AB, Helal GK, Hamada FM, Thomsen MS, Jensen AA (2018) Functional characterization of α7 nicotinic acetylcholine and NMDA receptor signaling in SH-SY5Y neuroblastoma cells in an ERK phosphorylation assay. Eur J Pharmacol 826:106–113. https://doi.org/10.1016/j.ejphar.2018.02.047
Falahatkar S, Mokhtari G, Pourreza F, Asgari SA, Kamran AN (2008) Celecoxib for treatment of nocturia caused by benign prostatic hyperplasia: a prospective, randomized, double-blind, placebo-controlled study. Urology 72:813–816. https://doi.org/10.1016/j.urology.2008.04.069
Fatehi-Hassanabad Z, Jafarzadeh M, Tarhini A, Fatehi M (2005) The antihypertensive and vasodilator effects of aqueous extract from berberis vulgaris fruit on hypertensive rats. Phytother Res 19:222–225. https://doi.org/10.1002/ptr.1661
Festing MFW (2006) Design and statistical methods in studies using animal models of development. ILAR J 47:5–14. https://doi.org/10.1093/ilar.47.1.5
Fu X, Feng Y, Shao B, Zhang Y (2019) Activation of the ERK/Creb/Bcl-2 pathway protects periodontal ligament stem cells against hydrogen peroxide-induced oxidative stress. Mol Med Rep 19:3649–3657. https://doi.org/10.3892/mmr.2019.10027
Funakoshi-Tago M, Shimizu T, Tago K, Nakamura M, Itoh H, Sonoda Y, Kasahara T (2008) Celecoxib potently inhibits TNFα-induced nuclear translocation and activation of NF-κB. Biochem Pharmacol 76:662–671. https://doi.org/10.1016/j.bcp.2008.06.015
Garber JC (2011) Guide for the Care and Use of Laboratory Animals. National Research Council - The National Academies Press, Washington, DC. https://doi.org/10.17226/12910
García-Rayado G, Navarro M, Lanas A (2018) NSAID induced gastrointestinal damage and designing GI-sparing NSAIDs. Expert Rev Clin Pharmacol 11:1031–1043. https://doi.org/10.1080/17512433.2018.1516143
Goldenberg MM (1999) Celecoxib, a selective cyclooxygenase-2 inhibitor for the treatment of rheumatoid arthritis and osteoarthritis. Clin Ther 21:1497–1513. https://doi.org/10.1016/S0149-2918(00)80005-3
Gonzales RJ, Duckles SP, Krause DN (2009) Dihydrotestosterone stimulates cerebrovascular inflammation through NFκB, modulating contractile function. J Cereb Blood Flow Metab 29:244–253. https://doi.org/10.1038/jcbfm.2008.115
Goodarzi D, Cyrus A, Khoddami Vishteh HR, Solhi H, Shirinkar M (2011) Effect of celecoxib on benign prostatic hyperplasia: results of a preliminary study. Urol Sci 22:147–150. https://doi.org/10.1016/j.urols.2011.07.001
Hinson RM, Williams JA, Shacter E (1996) Elevated interleukin 6 is induced by prostaglandin E2 in a murine model of inflammation: possible role of cyclooxygenase-2. Proc Natl Acad Sci 93:4885–4890. https://doi.org/10.1073/pnas.93.10.4885
Hsu CY, Lin YS, Weng WC, Panny L, Chen HL, Tung MC et al (2021) Phloretin ameliorates testosterone-induced benign prostatic hyperplasia in rats by regulating the inflammatory response. Oxi Stress Apoptos Life 11:743. https://doi.org/10.3390/life11080743
Kalyuzhny AE (2016) Immunohistochemistry. Nature. https://doi.org/10.1007/978-3-319-30893-7
Kanayama G, Hudson JI, Pope HG (2008) Long-term psychiatric and medical consequences of anabolic–androgenic steroid abuse: A looming public health concern? Drug Alcohol Depend 98:1–12. https://doi.org/10.1016/j.drugalcdep.2008.05.004
Kanayama G, Hudson JI, Pope HG (2010) Illicit anabolic–androgenic steroid use. Horm Behav 58:111–121. https://doi.org/10.1016/j.yhbeh.2009.09.006
Leslie CC (2004) Regulation of arachidonic acid availability for eicosanoid production. Biochem Cell Biol 82:1–17. https://doi.org/10.1139/o03-080
Liu T, Zhang L, Joo D, Sun S-C (2017) NF-κB signaling in inflammation. Signal Transduct Target Ther 2:17023. https://doi.org/10.1038/sigtrans.2017.23
Madersbacher S, Sampson N, Culig Z (2019) Pathophysiology of Benign Prostatic Hyperplasia and Benign Prostatic Enlargement: A Mini-Review. Gerontology 65:458–464. https://doi.org/10.1159/000496289
Maggi CA, Manzini S, Giuliani S, Meli A (1989) Infravesical outflow obstruction in rats: A comparison of two models. General Pharmacol Vascular System 20:345–349. https://doi.org/10.1016/0306-3623(89)90271-1
Morita I (2002) Distinct functions of COX-1 and COX-2. Prostagland Other Lipid Mediat 68–69:165–175. https://doi.org/10.1016/S0090-6980(02)00029-1
Nair A, Morsy MA, Jacob S (2018) Dose translation between laboratory animals and human in preclinical and clinical phases of drug development. Drug Dev Res 79:373–382. https://doi.org/10.1002/ddr.21461
Niederberger E, Tegeder I, Vetter G, Schmidtko A, Schmidt H, Euchenhofer C et al (2001) Celecoxib loses its anti-inflammatory efficacy at high doses through activation of NF-κB. FASEB J 15:1622–1624. https://doi.org/10.1096/fj.00-0716fje
Nissen SE, Yeomans ND, Solomon DH, Lüscher TF, Libby P, Husni ME et al (2016) Cardiovascular safety of celecoxib, naproxen, or ibuprofen for arthritis. N Engl J Med 375:2519–2529. https://doi.org/10.1056/NEJMc1702534
Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS (2002) Prostaglandin E2 transactivates EGF receptor: A novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med 8:289–293. https://doi.org/10.1038/nm0302-289
Pandita RK, Persson K, Hedlund P, Andersson K-E (1998) Testosterone-induced prostatic growth in the rat causes bladder overactivity unrelated to detrusor hypertrophy. Prostate 35:102–108. https://doi.org/10.1002/(SICI)1097-0045(19980501)35:2%3c102::AID-PROS3%3e3.0.CO;2-L
Park JY, Pillinger MH, Abramson SB (2006) Prostaglandin E2 synthesis and secretion: The role of PGE2 synthases. Clin Immunol 119:229–240. https://doi.org/10.1016/j.clim.2006.01.016
Percie Du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M et al (2020) The ARRIVE guidelines 20: updated guidelines for reporting animal research. PLoS Biol 18:3000410. https://doi.org/10.1371/journal.pbio.3000410
Piriyapongsa J, Ngamphiw C, Assawamakin A, Wangkumhang P, Suwannasri P, Ruangrit U et al (2009) RExPrimer: an integrated primer designing tool increases PCR effectiveness by avoiding 3’ SNP-in-primer and mis-priming from structural variation. BMC Geno 10:S4. https://doi.org/10.1186/1471-2164-10-S3-S4
Pizzorno JE, Murray MT, Joiner-Bey H (2016) 12 - Benign prostatic hyperplasia. In: Pizzorno J (ed) The Clinician’s Handbook of Natural Medicine (Third Edition). Edinburgh Churchill Livingstone
Prins GS, Huang L, Birch L, Pu Y (2006) The role of estrogens in normal and abnormal development of the prostate gland. Ann N Y Acad Sci 1089:1–13. https://doi.org/10.1196/annals.1386.009
Robert G, Descazeaud A, Nicolaïew N, Terry S, Sirab N, Vacherot F et al (2009) Inflammation in benign prostatic hyperplasia: A 282 patients’ immunohistochemical analysis. Prostate 69:1774–1780. https://doi.org/10.1002/pros.21027
Saad F, Yassin AA, Haider A, Gooren L (2011) Effects of testosterone on the lower urinary tract go beyond the prostate: New insights, new treatment options. Arab J Urol 9:147–152. https://doi.org/10.1016/j.aju.2011.06.003
Said MM, Hassan NS, Schlicht MJ, Bosland MC (2015) Flaxseed Suppressed Prostatic Epithelial Proliferation in a Rat Model of Benign Prostatic Hyperplasia. J Toxicol Environ Health A 78:453–465. https://doi.org/10.1080/15287394.2014.993779
Santen RJ, Brodie H, Simpson ER, Siiteri PK, Brodie A (2009) History of Aromatase: Saga of an Important Biological Mediator and Therapeutic Target. Endocr Rev 30:343–375. https://doi.org/10.1210/er.2008-0016
Schumacher N, Rose-John S (2021) ADAM17 orchestrates Interleukin-6, TNFα and signaling in inflammation and cancer. Mole Cell Res. https://doi.org/10.1016/j.bbamcr.2021.119141
Shao J, Lee SB, Guo H, Evers BM, Sheng H (2003) Prostaglandin E2 stimulates the growth of colon cancer cells via induction of amphiregulin. Can Res 63:5218–5223
Stanworth RD, Jones TH (2008) Testosterone for the aging male; current evidence and recommended practice. Clin Interv Aging 3:25–44. https://doi.org/10.2147/CIA.S190
Sun Y-H, Dong Y-L, Wang Y-T, Zhao G-L, Lu G-J, Yang J et al (2013) Synergistic analgesia of duloxetine and celecoxib in the mouse formalin test: a combination analysis. PLoS ONE 8:e76603. https://doi.org/10.1371/journal.pone.0076603
Taylor SC, Berkelman T, Yadav G, Hammond M (2013) A defined methodology for reliable quantification of western blot data. Mol Biotechnol 55:217–226. https://doi.org/10.1007/s12033-013-9672-6
Trad A, Hedemann N, Shomali M, Pawlak V, Grötzinger J, Lorenzen I (2011) Development of sandwich ELISA for detection and quantification of human and murine a disintegrin and metalloproteinase17. J Immunol Methods 371:91–96. https://doi.org/10.1016/j.jim.2011.06.015
Vargas RA, Oliveira LP, Frankenfeld S, Souza DB, Costa WS, Favorito LA, Sampaio FJ (2013) The prostate after administration of anabolic androgenic steroids: a morphometrical study in rats. Int Braz J Urol 39:675–682. https://doi.org/10.1590/S1677-5538.IBJU.2013.05.10
Vignozzi L, Rastrelli G, Corona G, Gacci M, Forti G, Maggi M (2014) Benign prostatic hyperplasia: a new metabolic disease? J Endocrinol Invest 37:313–322. https://doi.org/10.1007/s40618-014-0051-3
Wee P, Wang Z (2017) Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (basel). https://doi.org/10.3390/cancers9050052
Yang C-C, Chang K-W (2018) Eicosanoids and HB-EGF/EGFR in cancer. Cancer Metastasis Rev 37:385–395. https://doi.org/10.1007/s10555-018-9746-9
Yousef A, Saleh IG, Abd-Allah ARA, Elnagar MR, Akool E-S (2020) PEG-SOD attenuates the mitogenic ERK1/2 signaling cascade induced by cyclosporin A in the liver and kidney of albino mice. Chem Biol Interact 330:109245. https://doi.org/10.1016/j.cbi.2020.109245
Zhao Y, Zhang Y, Li Y, Yang M, Yuan J, Cao Y et al (2021) Yohimbine hydrochloride inhibits benign prostatic hyperplasia by downregulating steroid 5α-reductase type 2. Eur J Pharmacol 908:174334. https://doi.org/10.1016/j.ejphar.2021.174334
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). Open access agreement/funding (For Egypt) is established by Science, Technology & Innovation Funding Authority (STDF) in cooperation with Egyptian Knowledge Bank (EKB) under a Transformative Agreement plus fully OA agreement (January 2022). This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
MEA-ElF: Funding acquisition, Resources, Methodology, Investigation, Formal analysis, Writing—original draft. MMA-F: Conceptualization, Methodology, Data curation, Supervision, Writing—review & editing. WRM: Supervision, Methodology, Investigation, Formal analysis, Writing—review & editing. LAAR: Supervision, Writing–review. HA: Conceptualization, Resources, Methodology, Data curation, Validation, Formal analysis, Writing–original draft, Writing–review & editing.
Corresponding authors
Ethics declarations
Conflict of interests
The authors have no relevant financial or non-financial interests to disclose.
Ethics approval
The Institutional Animal Care and Use Committee of Beni-Suef University (BSU-IACUC-021–198) authorized the experiments prior to the study. Furthermore, the study's protocols followed the National Institutes of Health's recommendations for the care and use of laboratory animals. The current animal study is in adherence with the ARRIVE guidelines.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abo-El Fetoh, M.E., Abdel-Fattah, M.M., Mohamed, W.R. et al. Cyclooxygenase-2 activates EGFR–ERK1/2 pathway via PGE2-mediated ADAM-17 signaling in testosterone-induced benign prostatic hyperplasia. Inflammopharmacol 31, 499–516 (2023). https://doi.org/10.1007/s10787-022-01123-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-022-01123-7