
Abstract
The immune response plays a crucial role in preventing diseases, such as infections. There are two types of immune responses, specific and innate immunity, each of which consists of two components: cellular immunity and humoral immunity. Dysfunction in any immune system component increases the risk of developing certain diseases. Systemic lupus erythematosus (SLE), an autoimmune disease in the human body, develops an immune response against its own components. In these patients, due to underlying immune system disorders and receipt of immunosuppressive drugs, the susceptibility to infections is higher than in the general population and is the single largest cause of mortality in this group. COVID-19 infection, which first appeared in late 2019, has caused several concerns in patients with SLE. However, there is no strong proof of additional risk of developing COVID-19 in patients with SLE, and in some cases, studies have shown less severity of the disease in these individuals. This review paper discusses the immune disorders in SLE and COVID-19.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Coronavirus disease 2019 (COVID-19) is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a member of the Coronaviridae family of viruses, which is responsible for the pneumonia outbreak in late December 2019, later officially announced as a pandemic (Nokhostin et al. 2020; Cucinotta and Vanelli 2020). This virus can cause severe acute respiratory syndrome (SARS), a potentially life-threatening condition (Thienemann et al. 2020). Other manifestations of the disease include altered white blood cell (WBC) counts, impaired liver function tests (LFT), and increased inflammatory cytokines (Lei et al. 2020). Cytokines are one of the main components of the immune system, and their increased production causes impaired immune response called cytokine storm syndrome (CSS), seen in severe cases (Fajgenbaum et al. 2020). It has been shown that cytokine dysregulation can lead to more serious conditions such as autoimmune diseases (ADs), including systemic lupus erythematosus (SLE), in COVID-19 patients (Kyttaris 2019; Najafi et al. 2020). One suggested pathophysiology includes excessive cytokine production that can lead to a loss of tolerance to self-antigens (Franceschi et al. 2018; Hemminki et al. 2020; Patra et al. 2020).
ADs are caused by immune system response against self-antigens, which causes inflammation and damage to various tissues and organs of the body based on the nature of the disease (Bogoch et al. 2020). SLE is an AD that can involve the musculoskeletal, hematologic, integumentary, renal, and central nervous (CNS) systems (Kuhn et al. 2015). There is a close relationship between infections and mortality in SLE patients (Iliopoulos and Tsokos 1996). Approximately one-third of patients with SLE die due to infections, specifically respiratory infections (Rúa-Figueroa et al. 2017; Thomas et al. 2014). Multiple mechanisms expose SLE patients to infections, including alterations in immune responses, therapy with immunosuppressive drugs, and concomitant morbidities (Hou et al. 2018; Cohn 1991).
COVID-19 and ADs have many common disastrous outcomes, including kidney damage, which is accompanied by more severe clinical conditions in both diseases. Kidney damage in SLE patients is due to increased levels of interleukin-23 (IL)-23, interferon gamma (IFN-γ), and IL-17, which are produced by T helper 17 (Th17) cells (Zickert et al. 2015; Oke et al. 2017). Studies have revealed that the level of serum inflammatory cytokines in COVID-19 patients with better clinical conditions is lower than that in patients admitted to the intensive care unit (ICU) (Huang et al. 2020a; Xu et al. 2020a). This suggests that complications in COVID-19 patients may be due to an increase in the rate of cytokine production. On the other hand, similarities in COVID-19 and SLE pathophysiology may provide insight into new COVID-19 therapeutic approaches based on therapies currently used in SLE patients, which aim to suppress immune system over-activation and control cytokine output (Valencia et al. 2019). Therefore, this paper discusses the immune responses and immune system dysregulation in COVID-19 and the AD SLE.
The incidence of COVID-19 in autoimmune diseases
Despite the enhanced risk of COVID-19 in ADs, little evidence is available on the subject (Favalli et al. 2020), although some studies have proposed the possibility of an increased risk of developing COVID-19 in these patients (Cohn 1991; Sawalha et al. 2020; Gianfrancesco et al. 2020a; Horisberger et al. 2020). In a recent study by Mathian et al., among 17 SLE patients with COVID-19, 14 were admitted to the hospital, among which only two died due to severe respiratory infection (Mathian et al. 2019). In another study by Monti et al., among 320 patients treated with different antirheumatic agents, only four patients had confirmed diagnosis of COVID-19 (Monti et al. 2020). The data from the COVID-19 Global Rheumatology Alliance global registry as of March 1, 2022, showed that 1794 SLE patients had been diagnosed with COVID-19 (The Global Rheumatology Community’s Response to the Worldwide COVID-19 Pandemic 2022). A recent study considered the outcome of rheumatic disease patients who were under different therapies (Gianfrancesco et al. 2020b). Patients receiving prednisone at a dose greater than 10 mg/day were at higher risk for severe disease and ICU hospitalization. On the other hand, patients using anti-tumor necrosis factor alpha (TNF-α) agents had a decreased risk of admission. There was no change in the outcome of individuals treated with nonsteroidal anti-inflammatory drugs (NSAIDs) or hydroxychloroquine (HCQ) (Gianfrancesco et al. 2020b).
It is now well established that increased circulating levels of angiotensin-converting enzyme 2 (ACE2) and immunosuppressive therapies are potential mechanisms in favor of increased risk of COVID-19 in SLE patients (Sawalha et al. 2020). Paradoxically, the other aspect of immunosuppressant use in SLE patients is the decreased chance of severe lung injury in infected patients due to the anti-inflammatory effect of these agents (Horisberger et al. 2020). The ACE2 gene is located on the X chromosome, and DNA methylation plays a critical role in its expression (Jin et al. 2011). SLE patients have increased levels of ACE2 in their bloodstream resulting from decreased methylation of the ACE2 gene (Sawalha et al. 2020). Additionally, cell entry of COVID-19 viruses, the essential step in virus pathogenesis, is mediated by ACE2 (Hoffmann et al. 2020). Therefore, it is possible that the elevated levels of ACE2 in SLE patients, along with the increased levels of apolipoprotein E (ApoE) and TNF-α, might lead to a higher risk of COVID-19 viremia in these patients (Sawalha et al. 2020; Monti and Montecucco 2020).
Distinguishing SLE complications such as acute lupus pneumonitis from COVID-19 is not a straightforward diagnosis due to their clinical and laboratory similarities with severe cases of COVID-19 (Soloway et al. 2021; Kichloo et al. 2020). Soloway et al. reported a case of an SLE patient with acute lupus pneumonitis which masked her COVID-19 infection due to the consumption of a high dose of an immunosuppressive agent, including steroids, causing readmission for coronavirus pneumonitis (Soloway et al. 2021). In another study by Kichlo et al., a 22-year-old SLE patient with acute lupus pneumonitis, superinfected with COVID-19, was successfully managed with intravenous administration of a high dose of corticosteroids (Kichloo et al. 2020). These findings highlight the importance of considering COVID-19 in all SLE patients on high-dose steroid therapy who are admitted to hospitals, even with atypical findings for COVID-19.
In contrast to studies suggesting an increased likelihood of COVID-19 in SLE, some studies have raised the possibility of decreased COVID-19 risk in these patients. One issue considered by these studies is the drugs used in SLE treatment, which were initially thought to exacerbate or increase the risk of COVID-19 infection. However, over time, studies have revealed that not only are some of these drugs not harmful, but they can also be used to treat COVID-19 (Kokkotis et al. 2022; Brito et al. 2021; Guo et al. 2022; Wagner et al. 2021). It has been shown that an increased serum level of ACE2 is related to elevated TNF-α production (Channappanavar et al. 2016), the serum level of which is increased in COVID-19 patients (Huang et al. 2020a; Chen et al. 2020b). Thus it is not surprising that anti-TNF-α drugs which are used to treat SLE patients might have beneficial effects in the treatment of COVID-19 patients, as some studies have reported promising results (Kokkotis et al. 2022; Brito et al. 2021; Guo et al. 2022; Haga et al. 2008; Feldmann et al. 2020).
The incidence of autoimmune diseases in COVID-19
Different types of immune dysregulation are implicated in the main pathogenesis of both ADs and COVID-19, causing disturbed self-tolerance and autoimmune responses (Jakiela et al. 2018). As an example, autoantibodies which are the hallmark of ADs are also seen in COVID-19 patients (Huang et al. 2020b; Bastard et al. 2020). Some studies have also reported the incidence of new cases of ADs, including Guillain–Barré syndrome (GBS) (Alberti et al. 2020; Toscano et al. 2020; Virani et al. 2020; Coen et al. 2020; Scheidl et al. 2020; Caamaño and Beato 2020; Arnaud et al. 2020; Gutiérrez-Ortiz et al. 2020; Ottaviani et al. 2020; Dinkin et al. 2020), Kawasaki disease (KD) (Nathan et al. 2020; Toubiana et al. 2020; Jones et al. 2020; Riphagen et al. 2020; Verdoni et al. 2020; Rivera-Figueroa et al. 2020), SLE (Zamani et al. 2021), hemolytic anemia (Lazarian et al. 2020; Jacobs and Eichbaum 2021; AbouYabis and Bell 2021), and immune thrombocytopenic purpura (ITP) (Zulfiqar et al. 2020; Kuter 2021) after COVID-19 infection.
Autoimmunity in COVID-19
ADs result from loss of tolerance to self-antigens and aberrant activation of the immune system. Multiple factors, including environmental, genetic, and hormonal factors, contribute to the pathogenesis of these diseases (Wang et al. 2015; Moody et al. 2021). COVID-19 can mimic autoimmune and auto-inflammatory conditions by breaking the immunological tolerance, which is mediated by different mechanisms including bystander activation, molecular mimicry, and epitope spreading (Smatti et al. 2019; Rojas et al. 2018; Pacheco et al. 2019). Accordingly, it has been proposed that the possible mechanism of central respiratory depression in patients with COVID-19 is the similarity of the SARS-CoV-2 proteome to three human proteins, AIFM, DAB1, and SURF1, which are located in the respiratory pacemakers of the brainstem, also classified as pre-Bötzinger complex (Lucchese and Flöel 2020). Similarly, Kanduc et al. reported that the SARS-CoV-2 proteome contains several pentapeptides similar to pulmonary surfactants, revealing a possible mechanism behind the pulmonary damage in COVID-19 (Kanduc and Shoenfeld 2020). Moreover, after the clearance of the virus in pneumocytes, the inflammatory processes continue in the lung as a result of epitope cross-reaction between lung tissue and the SARS-CoV-2 virus (Huang et al. 2020b). Recent studies have reported strong cross-reactions between the spike protein of SARS-CoV-2 and various human tissue proteins such as transglutaminase 2, transglutaminase 3, ENA, thyroid peroxidase, mitochondria, myelin basic protein, α-myosin, collagen, claudin-5 + 6, and S100B, resulting in autoantibody production (Rodríguez et al. 2020; Vojdani and Kharrazian 2020). The other issue suggesting a similar mechanism behind immunological responses in SLE and COVID-19 is the same immunomodulatory agents used in the treatment of both SLE and COVID-19 patients (Luo et al. 2020; Ahn et al. 2019; Cantini et al. 2020).
The innate immune response in COVID-19 and SLE
The innate immune system serves as the primary barrier against viruses and uses pattern recognition receptors (PRRs) to induce immune responses (Diamond and Kanneganti 2022). This immune system consists of different immune cells, including neutrophils, macrophages, dendritic cells, monocytes, and especially innate lymphoid cells (ILCs) such as natural killer (NK) cells. PRRs recognize the pathogen-associated molecular patterns (PAMPs) which exist in all pathogens and the damage-associated molecular patterns (DAMPs), and are related to cell damage (Kanneganti 2020). The main PRR families consist of toll-like receptors (TLRs), retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), C-type lectin receptors, and absent in melanoma 2 (AIM2)-like receptors (Kanneganti 2020). Several types of PRRs, including TLRs, RLRs, and NLRs, are involved in the SARS-CoV-2 immune response (Diamond and Kanneganti 2022).
NK cells in COVID-19 and SLE
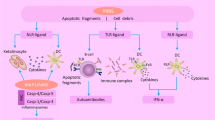
NK cells are one of the key elements of the innate immune system exhibiting cytotoxic activity, and play a critical role in the early response to viruses and proper coordination between innate and adaptive immune systems (Leem et al. 2021; Schuster et al. 2016). NK cell function is controlled by many activating and inhibiting receptors. Cell activation causes cytotoxicity and production of inflammatory cytokines, known as cytotoxic and inflammatory phenotypes, respectively (Schuster et al. 2016; Vivier et al. 2008; Lanier 2008; Kärre 2002). The inactivation of NK cells rests upon the "self" signals via the major histocompatibility complex class I (MHC-I) molecules expressed on all healthy cells, which leads to “self-tolerance” (Lanier 2008; Kärre 2002]. The inhibitory receptors that are expressed on NK cells and are specific for MHC-I include lectin-like CD94/NKG2A heterodimers and the killer cell immunoglobulin-like receptors (KIRs) (KLRG1 and TIGIT) (Lanier 2008; Long et al. 2013; Eeden et al. 2020). NK cells are also essential for modulating other immune components, especially T cells, and act in both direct and indirect (using antigen-presenting cells [APC] such as dendritic cells [DCs]) ways (Schuster et al. 2016; Lanier 2008). The direct interaction is mediated by cytokines, including IFN-γ as the activator agent and IL-10 as the inhibitory cytokine (Schuster et al. 2016; Lanier 2008). The cytotoxic elimination of activated T cells is the other direct way of regulating T cell function by NK cells. This is based on the increased activating ligands (NKG2D) and diminished inhibitory ligands (MHC-I) on activated T cells, making it possible for them to be recognized by NK cells (Lanier 2008; Kärre 2002). In addition, the decreased number of T cells, specifically T follicular helper (Tfh) cells, indirectly inhibits the B-cell-mediated immunity by NK cells (Zhang et al. 2013).
The total quantity of NK cells is reduced in SLE, and the existing NK cells show less cytotoxicity than normal NK cells (Park et al. 2009; Schleinitz et al. 2010 ). Sederberg et al. reported that there are several circulating autoantibodies against the multiple KIR receptors and the human leukocyte antigen (HLA) class I-binding receptors (NKG2A, NKG2C) in SLE patients, which disturb the self- and non-self-recognition by NK cells (Hagberg et al. 2015; Segerberg et al. 2019). Moreover, B- and T-cell-mediated altered immune response in ADs is thought to be the primary pathophysiology of these diseases. In terms of the altered adaptive immune response, these cells are impacted by the stimulation of NK cells and self-tolerance mechanisms (Gianchecchi et al. 2018). Similarly, researchers have demonstrated that COVID-19 patients have a decreased total number of NK and CD8+ T cells, which show a functionally exhausted phenotype (Zheng et al. 2020; Li et al. 2020; Wang et al. 2020). This is determined based on the enhanced expression of NKG2A, LAG3, PDCD1, and HAVCR2 as NK cell exhaustion indicators, and PD-1 and Tim-3 as T cell inhibitory factors (Zheng et al. 2020; Wilk et al. 2020). Increased expression of NKG2A in NK cells is accompanied by reduced expression of TNF-α, IFN-γ, and IL-2 and downregulation of granzyme B (Ndhlovu et al. 2012; Gleason et al. 2012). Tim-3 has a dual effect on NK cells. On the one hand, it increases the expression of IFN-γ in these cells, and on the other hand, its enhanced expression is accompanied by a significant drop in NK cell cytotoxicity (Ndhlovu et al. 2012; Gleason et al. 2012). Studies have suggested that lower expression of NKG2C may worsen COVID-19 outcome (Vietzen et al. 2020).
There are two types of conventional NK cells based on their surface molecules, including the CD56DIM/CD16POS/CD56BRIGHT/CD16NEG subsets. The first type has a cytotoxic phenotype and is predominantly found in peripheral blood (Vivier et al. 2008; Moretta et al. 2001), whereas the second type comprises cytokine-producing NK cells, usually located in the secondary lymphoid tissues and activated following interaction with IL-12, IL-15, and IL-18 cytokines (Vivier et al. 2008; Long et al. 2013; Moretta et al. 2001; Cooper et al. 2001). CD56DIM/CD16NEG (uCD56DIM) is the other member of the NK cell population, which is found in small numbers in healthy individuals and can be expanded in several disorders (Amand et al. 2017; Fan et al. 2008). A recent study demonstrated that the unconventional subset of NK cells (CD56DIM/CD16NEG) is increased in patients with COVID-19, regardless of the disease severity, and is associated with reduced NK cell cytotoxicity. Recovery to normal NK cell population was faster in patients with mild symptoms (Leem et al. 2021). In another study, Wilk et al. reported that the expression of multiple genes related to NK cell function and maturity is decreased in COVID-19 patients (Wilk et al. 2020), among which FCGR3A and FGFBP2 are specifically related to CD56DIM NK cells (Crinier et al. 2018). In a study by Leng et al., patients with COVID-19 were found to have elevated expression of CXCR3+, which is a marker of NK cells and is more common among the CD56BRIGHT NK cell subpopulation (Leng et al. 2020). Altogether, these findings suggest a shift toward more inflammatory NK cell traits in COVID-19 infection (Osman et al. 2020). Additionally, the balance between the CD56DIM and CD56BRIGHT NK cell population is disturbed in SLE patients, where the proportion of the CD56DIM subpopulation is decreased (Henriques et al. 2013; Spada et al. 2015; Liu et al. 2021). Interestingly, the CD56DIM NK cells present in SLE patients tend to produce elevated levels of IFN-γ, and the expression of activating receptors, including NKp44, NKp46, and CD69, is increased in these cells (Liu et al. 2021).
Cytokine storm
Cytokine storm is the excessive production of cytokines with inflammatory activity, present in many infectious and noninfectious conditions, resulting from inflammation (Tisoncik et al. 2012). It is believed that this phenomenon is a crucial component in COVID-19 pathogenesis and is related to its severe manifestations, such as acute respiratory distress syndrome (ARDS) (Huang et al. 2020a; Jiang et al. 2022; Hu et al. 2021). Indeed, many studies have demonstrated enhanced levels of cytokines of an inflammatory nature in COVID-19 patients, including IL-1β, TNF-α, IFN-γ, IL-2, IL-6, IL-10, granulocyte macrophage-colony stimulating factor (GM-CSF), inducible protein 10 (IP-10), and monocyte chemoattractant protein-1 (MCP-1) (Huang et al. 2020a; Ruan et al. 2020; Valle et al. 2020; Zhu et al. 2020; Chen et al.2020a).
Several mechanisms have been suggested as the principal mechanisms underlying cytokine storm progression in COVID-19, all common in the inadequate immune response against the virus (Jiang et al. 2022; Frisoni et al. 2021; Maiese et al. 2020; Blanco-Melo et al. 2020; Bastard et al. 2021; Wang et al. 2021; Lv et al. 2021; Domingo et al. 2020; Bourgonje et al. 2020; Zanza et al. 2021). Blanco-Melo et al. demonstrated that antiviral IFN production is impaired in COVID-19 patients (Blanco-Melo et al. 2020). Such impaired innate immune response makes replication much easier for the virus. Likewise, Lv and colleagues noted that the SARS-CoV-2 virus could survive in the M1 subpopulation of alveolar macrophages, facilitating the spread of infection throughout the lungs (Lv et al. 2021). Anti-type I IFN antibodies, along with the autoantibodies against other immune proteins, are another potential reason for decreased IFN response in SARS-CoV-2-infected patients (Bastard et al. 2021; Wang et al. 2021). Many studies have also shown increased levels of chemokines and cytokines after the SARS-CoV-2 virus enters the human body (Jiang et al. 2022; Blanco-Melo et al. 2020; Osman et al. 2020). As an example, increased function of the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome during COVID-19 results in increased production of IL-18 and IL-1β (Hadjadj et al. 2020; Schmidt and Lenz 2012; Zheng et al. 2014). After successful reproduction of SARS-CoV-2 in an environment lacking antiviral IFNs, accompanied by the increased levels of chemokines and cytokines, the exaggerated immune reaction to the virus causes a cytokine storm in the second stage of COVID-19 infection (Chowdhury et al. 2020). Based on the anti-inflammatory consequences of the ACE2 receptor (Sodhi et al. 2019; Nabah et al. 2004), Diamond et al. hypothesized a possible role of the renin–angiotensin–aldosterone system (RAAS) in the pathogenesis of increased cytokine storm in COVID-19. They also suggested that the decreased level of ACE2 receptor is responsible for the increased thrombo-inflammatory state in COVID-19 (Diamond 2020).
The role of cytokine storm in the pathogenesis of SLE is much more limited than in COVID-19. However, similar to COVID-19, there is supporting proof that shows an increased rate of cytokines such as TNF-α, B-lymphocyte stimulator (BLyS), type I IFNs, IL-6, IL-17, and IL-18 in SLE (Yap and Lai 2013). Furthermore, the use of cytokine inhibitory agents has been suggested as a therapeutic strategy in severe SLE patients and intriguingly, some studies have announced promising results (Huang et al. 2020a; Clark et al. 2013). In a recent study by Xu et al. on pristane-induced murine models of lupus, IL-38 levels were demonstrated to be increased in these mice. However, unexpectedly, they experienced a reduction in lupus development after using IL-38 suggesting the possible anti-inflammatory role of this cytokine in SLE (Xu et al. 2020b).
IFN responses in COVID-19 and SLE
The prominent role of type 1 interferon response in the pathogenesis of both rheumatoid and viral infections is undeniable (Rönnblom and Pascual 2008; Muskardin and Niewold 2018; Tariq et al. 2021; Vardhana and Wolchok 2020). However, many studies have reported decreased production of type I IFNs in COVID-19 (Blanco-Melo et al. 2020). Moreover, a recent study demonstrated that neutralizing autoantibodies against type I IFNs are seen in a minimum of 10% of patients with severe COVID-19 (Bastard et al. 2020). Accordingly, it can be theorized that high titers of type I IFN in patients with SLE may prevent the incidence of COVID-19 in these patients due to the more effective viral clearance.
Studies have reported the incidence of new cases of SLE after recombinant human IFN-α administration in patients with hematologic malignancies. These patients experience an improvement in their symptoms after the drug is ceased (Rönnblom et al. 1990; Niewold and Swedler 2005). Moreover, healthy relatives of SLE patients have been found to have high levels of type I IFN in their serum, suggesting a role of type I IFN genetic alterations in the immunopathology of SLE (Niewold et al. 2007; Kariuki et al. 2010). Furthermore, many SLE-associated disparities in the genes of the type I IFN pathway are accompanied by enhanced activity of the IFN-I pathway (Niewold 2011; Niewold et al. 2010; Bronson et al. 2012; Ghodke-Puranik and Niewold 2013). In addition, in some patients with SLE, elevated activity of type I IFN was reported during the year before they were diagnosed (Munroe et al. 2016).
Adaptive immunity (cell-mediated response)
As mentioned above, many studies have reported decreased number and exhausted phenotype (based on the excessive expression of PD-1 and Tim-3 as T cell exhaustion markers) of CD8+ T cells in COVID-19 patients (Zheng et al. 2020; Li et al. 2020; Wang et al. 2020). In contrast, based on elevated HLA-DR and CD38 as T cell activator markers, many other studies have demonstrated higher levels of T cell activation (Xu et al. 2020a; Diao et al. 2019; Wang et al. 2020; Qin et al. 2020). Furthermore, an elevated neutrophil-to-lymphocyte ratio (NLR) is found in COVID-19, indicating the inflammatory state of this disease (Qin et al. 2020). Taken together, these findings suggest significant changes in cell-based immunity against COVID-19, which are discussed below in detail.
T cells in COVID-19 and SLE
Although the main characteristic of SLE is the excessive production of autoantibodies by B cell lymphocytes, the role of T lymphocytes cannot be ignored. (Katsuyama et al. 2018; Mak and Kow 2014). Several functional changes in T cells are seen during SLE. For example, the expression of CD3ζ (CD247), a T cell surface protein, is decreased in SLE, leading to a shift in intracellular signaling to the Syk pathway (Katsuyama et al. 2018). The activation of the Syk pathway gives rise to vigorous calcium flux in T cells, leading to hyper-inflammation (Katsuyama et al. 2018). This process results in the upregulation of CD40 ligand (CD40L) in patients with active SLE.
CD40L is a co-stimulatory protein on T cells which helps them engage with other cells with CD40 molecules, such as B cells leading to activation of B cells (Mak and Kow 2014). As CD40 is present on many other cell types, including myocytes, platelets, and endothelial cells, which are robustly involved in positive inflammatory feedback, CD40L may cause several undesired side effects leading to inflammation (Goshua et al. 2020; Karnell et al. 2019).
The role of T lymphocytes in the occurrence of COVID-19 is much more of a double-edged blade. Although the protective role of T cells against the spike protein of the COVID-19 virus is established, the same cells could have uncontrolled proinflammatory responses (Toor et al. 2021). It has been shown that COVID-19 patients admitted to the ICU have higher levels of CD40L on their platelets and T lymphocytes, resulting in excessive inflammation, poor prognosis, and a hypercoagulable state (Goshua et al. 2020). This is concerning due to the hypercoagulable state of patients with active SLE that is already well established (Misra et al. 2020; Bowles et al. 2020).
Th17 cells in COVID-19 and SLE
Th17 cells are a member of CD4+ T lymphocytes that are capable of producing proinflammatory cytokines, including IL-17A, IL-17F, and IL-22, and are specified by the expression of the RAR-related orphan receptor gamma (RORγT) transcription factor (Yang et al. 2009; Littman and Rudensky 2010). This is why these cells are also known as an inflammatory Th subset (Yasuda et al. 2019). It has been shown that patients with SLE have increased numbers of Th17 cells and IL-17 cytokines in their bloodstream, and the severity of this immunological disorder is negatively associated with their clinical outcome (Shan et al. 2020). IL-17 is capable of inducing autoantibody synthesis in patients with lupus nephritis (LN) (Lee et al. 2019). Several studies have demonstrated increased levels of the mammalian target of rapamycin complex (mTORC] in SLE (Katsuyama et al. 2018). In a study by Singh and colleagues, development of a fulminant SLE was reported in a patient with a downregulated level of a negative regulator of mTORC1, which led to the patient’s death (Singh et al. 2013). mTORC is a nutrient sensor and is involved in the cell cycle management. mTOR has a primary role in the activation and polarization of naïve T cells (Katsuyama et al. 2018; Terrazzano et al. 2020; Zeng et al. 2013 Jul). Furthermore, a study demonstrated that SLE patients with LN have high rates of circulating Th17 cells and decreased Treg/Th17 ratio; however, no association with disease activity was found (Jakiela et al. 2018).
Another study by Abdel Galil and colleagues reported a direct association between IL-17 serum levels and proteinuria or anti-double-stranded DNA antibodies (anti-dsDNA ab) in patients with LN (Abdel Galil et al. 2015). As seen in SLE, many studies have shown increased activity of Th17 cells and elevated levels of IL-17 in COVID-19 (Biasi et al. 2020; Gadi et al. 2020). Another study established that the increased level of IL-17A is accompanied by increased damage to the lung tissue, leading to more severe disease in COVID-19 (Parackova et al. 2021). Furthermore, a high ratio of Th17 to Treg cells is correlated with poor prognosis in COVID-19 (Sadeghi et al. 2021).
Lymphopenia in SLE and COVID-19
Lymphopenia, with a predominant decrease in T lymphocytes, is an expected finding in SLE that has recently been detected in COVID-19 and used as a prognostic factor in these patients (Martin et al. 2017; Frater et al. 2020). Five main processes were found to be involved in the incidence of lymphopenia in SLE, including (1) the presence of immunoglobulin M (IgM) and/or IgG lymphocytotoxic antibodies against T-cell-rich antigens including CD4, CD45, MHC-I/II, glycophospholipids, and ribosomal P protein (Martin et al. 2017), (2) a decrease in the intracellular antioxidant glutathione, which causes increased apoptosis in T cells (Shah et al. 2012), (3) enhanced expression of Fas on naïve and memory T cells, which causes apoptosis (Bijl et al. 2001; Papo et al. 1998), (4) decreased expression of complement regulatory proteins, specifically CD55, CD59, and CD46, which expose T cells to complement-mediated cytolysis (Alegretti et al. 2012), and (5) decreased lymphopoiesis and increased T cell sequestration in secondary lymphoid organs and inflammatory sites due to the reduced number of CD34+ hematopoietic progenitors and enhanced level of IFN-γ which is associated with limited self-renewal of hematopoietic stem cells. This process is induced by transcription factor PU.1 hyper-expression (Spihlman et al. 2020). It has evident that SLE patients have elevated levels of IFN-γ (Oke et al. 2017; Munroe et al. 2016; Bengtsson et al. 2000; Rana et al. 2012).
Similar to the mechanisms implicated in SLE pathogenesis, multiple mechanisms are hypothesized to be involved in the development of lymphopenia in COVID-19. The first possible mechanism is the excessive production of inflammatory cytokines, which causes increased apoptosis in T cells. Studies have demonstrated an increased rate of T cell apoptosis mediated by type I IFN and IFN-γ, which are abundant in COVID-19 (Channappanavar et al. 2016; Perl et al. 2004). Epithelial cells infected with SARS-CoV-2 release TNF-α, which induces T cell apoptosis (Wang et al. 2018). Additional studies have proposed the role of other proinflammatory cytokines, including IFN-α, IL-1β, IL-6, IL-12, IL-18, and IL-33 (Fathi and Rezaei 2020; Okoye et al. 2017; Huang and Pranata 2020; Lin et al. 2020). In justification of high T cell apoptosis in COVID-19, one study demonstrated that the increased expression of the TP53 gene is a key factor in induction of apoptosis (Xiong et al. 2020). During COVID-19, T cell sequestration in the lungs, gastrointestinal tract, and lymphoid tissues is also involved in lymphopenia. A positive correlation is seen between lymphopenia and poor prognosis in patients with COVID-19 (Frater et al. 2020).
Adaptive immunity (humeral response)
Antibody role in COVID-19 and SLE
Anti-dsDNA autoantibodies are diagnostic hallmarks of SLE which lead to the deposition of the immune complexes throughout the body, specifically joints, blood vessels, and the renal system (Spihlman et al. 2020; Rekvig 2015). Different theories exist in terms of autoantibody production in SLE. Some studies have suggested the possible role of viruses, especially polyomavirus group viruses, while others have proposed the possible role of DNA-binding proteins through molecular mimicry (Rekvig 2015; Ahsan and Shah 2006). Moreover, increased apoptosis or insufficient clearance of apoptotic cells results in autoantigen–antibody complex production, which in turn stimulates IFN-α expression, leading to immune reactivity (Pan et al. 2020). Interestingly, a recent study reported elevated levels of autoantibodies such as antinuclear antibody (ANA) in about 34.5% of COVID-19 patients with severe infection (Vlachoyiannopoulos et al. 2020). Many studies have also shown positive anti-phospholipid antibodies (APL) consisting of anti-cardiolipin (aCL), lupus anticoagulant (LA), and anti-β2 glycoprotein (β2GPI) in COVID-19 infection, which may be accompanied by thrombotic events (Bowles et al. 2020; Zhang et al. 2020; Harzallah et al. 2020; Helms et al. 2020; Amin 2008; Zuo et al. 2020). However, a study reported no enhanced risk of increased major thromboembolic accidents in COVID-19 patients (Borghi et al. 2020). Surprisingly, another study showed that the APL antibodies in COVID-19 are not the same as those in SLE (Borghi et al. 2020).
IgM, IgG, and mucosal and systemic IgA are the most common types of SARS-CoV-2-specific antibodies in COVID-19 patients. Many of these antibodies are secreted in reaction to the S and N proteins of the virus (Guo et al. 2020; Lou et al. 2020; Whitman et al. 2020; Cervia et al. 2021; Jin et al. 2020; Xiang et al. 2020; Ma et al. 2020; Tang et al. 2021; Zhao et al. 2020; Long et al. 2020; Lynch et al. 2020; Yu et al. 2020). A significant association is seen between these antibodies and prognosis prediction in COVID-19 (Wang et al. 2021). The minimum positive rate of IgM and IgG antibodies reported in different studies was 84.6% and 80.8%, respectively (Lynch et al. 2020); however, the maximum positive rate was 100% for both antibodies (Guo et al. 2020; Lou et al. 2020; Whitman et al. 2020; Cervia et al. 2021; Jin et al. 2020; Xiang et al. 2020; Ma et al. 2020; Tang et al. 2021; Zhao et al. 2020; Long et al. 2020; Lynch et al. 2020; Yu et al. 2020). IgA antibody is less studied than the other two antibodies in COVID-19. In two different studies, the rate of positive IgA antibody in COVID-19 patients was reported to be 98.6% and 99.8% (Ma et al. 2020; Yu et al. 2020). The median seroconversion time for IgM antibody varied from 4 to 18 days in different studies (Guo et al. 2020; Lou et al. 2020; Ma et al. 2020; Tang et al. 2021; Zhao et al. 2020; Long et al. 2020; Lynch et al. 2020; Yu et al. 2020). This range for IgG antibody was 5 to 20 days (Guo et al. 2020; Lou et al. 2020; Ma et al. 2020; Tang et al. 2021; Zhao et al. 2020; Long et al. 2020; Lynch et al. 2020; Yu et al. 2020). The IgA seroconversion time was 4 to 13 days in different studies (Ma et al. 2020 Jul; Yu et al. 2020). The most commonly used methods for antibody measurement in clinical studies were enzyme-linked immunosorbent assay (ELISA), lateral flow immunoassay) (LFIA), chemiluminescent microparticle immunoassay (CMIA), and chemiluminescence immunoassay (CLIA) (Guo et al. 2020; Lou et al. 2020; Whitman et al. 2020; Cervia et al. 2021; Jin et al. 2020; Xiang et al. 2020; Ma et al. 2020; Tang et al. 2021; Zhao et al. 2020; Long et al. 2020; Lynch et al. 2020; Yu et al. 2020). Furthermore, many studies have shown a positive correlation between SARS-CoV-2-specific antibody levels and disease outcome in COVID-19 patients (Cervia et al. 2021; Ma et al. 2020; Tang et al. 2021; Lynch et al. 2020; Yu et al. 2020; Lin et al. 2020; Wang et al. 2020; Casadevall et al. 2020).
Conclusion
In general, it can be concluded that several immune disorders are involved in both COVID-19 and SLE diseases, which in some cases are common to both. These disorders affect almost all parts of the immune system, including the cellular and innate immune systems. Despite numerous studies in this regard, it seems that there are still many unanswered questions about the role of immunity and its disruption in the pathogenesis of both diseases, which necessitates the pursuit of further studies.
Data availability
Not applicable.
References
Abdel Galil SM, Ezzeldin N, El-Boshy ME (2015) The role of serum IL-17 and IL-6 as biomarkers of disease activity and predictors of remission in patients with lupus nephritis. Cytokine 76(2):280–7. https://doi.org/10.1016/j.cyto.2015.05.007
AbouYabis AN, Bell GT (2021) Hemolytic Anemia Complicating COVID-19 Infection. J Hematol 10(5): 221–227. https://doi.org/10.14740/jh906
Ahn D-G, Shin H-J, Kim M-H, Lee S, Kim H-S, Myoung J et al (2020) Current status of epidemiology, diagnosis, therapeutics, and vaccines for novel coronavirus disease 2019 (COVID-19). J Microbiol Biotechnol 30(3):313–324. https://doi.org/10.4014/jmb.2003.03011
Ahsan N, Shah KV (2006) Polyomaviruses and human diseases. Adv Exp Med Biol 577:1–18. https://doi.org/10.1007/0-387-32957-9_1
Alberti P, Beretta S, Piatti M, Karantzoulis A, Piatti ML, Santoro P et al (2020) Guillain-Barré syndrome related to COVID-19 infection. Neurol Neuroimmunol Neuroinflamm 7(4):e741. https://doi.org/10.1212/NXI.0000000000000741
Alegretti AP, Schneider L, Piccoli AK, Monticielo OA, Lora PS, Brenol JCT et al (2012) Diminished expression of complement regulatory proteins on peripheral blood cells from systemic lupus erythematosus patients. Clin Dev Immunol 2012:725684. https://doi.org/10.1155/2012/725684
Amand M, Iserentant G, Poli A, Sleiman M, Fievez V, Sanchez IP et al (2017) Human CD56(dim)CD16(dim) cells as an individualized natural killer cell subset. Front Immunol 8:699. https://doi.org/10.3389/fimmu.2017.00699
Amin NM (2008) Antiphospholipid syndromes in infectious diseases. Hematol Oncol Clin North Am 22(1):131–143. https://doi.org/10.1016/j.hoc.2007.10.001
Arnaud S, Budowski C, Tin SNW, Degos B (2020) Post SARS-CoV-2 Guillain-Barré syndrome. Clin Neurophysiol 131(7):1652–1654. https://doi.org/10.1016/j.clinph.2020.05.003
Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann H-H, Zhang Y et al (2020) Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370(6515):eabd4585. https://doi.org/10.1126/science.abd4585
Bastard P, Gervais A, Le Voyer T, Rosain J, Philippot Q, Manry J et al (2021) Autoantibodies neutralizing type I IFNs are present in~ 4% of uninfected individuals over 70 years old and account for~ 20% of COVID-19 deaths. Sci Immunol 6(62):eabl4340. https://doi.org/10.1126/sciimmunol.abl4340
Bengtsson AA, Sturfelt G, Truedsson L, Blomberg J, Alm G, Vallin H et al (2000) Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus 9(9):664–671. https://doi.org/10.1191/096120300674499064
Bijl M, Horst G, Limburg PC, Kallenberg CG (2001) Fas expression on peripheral blood lymphocytes in systemic lupus erythematosus (SLE): relation to lymphocyte activation and disease activity. Lupus 10(12):866–872. https://doi.org/10.1191/096120301701548517
Blanco-Melo D, Nilsson-Payant BE, Liu W-C, Uhl S, Hoagland D, Møller R, et al (2020) Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181(5):1036–1045.e9. https://doi.org/10.1016/j.cell.2020.04.026
Bogoch II, Watts A, Thomas-Bachli A, Huber C, Kraemer MUG, Khan K (2020) Pneumonia of unknown aetiology in Wuhan, China: potential for international spread via commercial air travel. J Travel Med 27(2):taaa008. https://doi.org/10.1093/jtm/taaa008
Borghi MO, Beltagy A, Garrafa E, Curreli D, Cecchini G, Bodio C et al (2020) Anti-phospholipid antibodies in COVID-19 are different from those detectable in the anti-phospholipid syndrome. Front Immunol 11: 584241. https://doi.org/10.3389/fimmu.2020.584241
Bourgonje AR, Abdulle AE, Timens W, Hillebrands J-L, Navis GJ, Gordijn SJ et al (2020) Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J Pathol 251(3):228–248. https://doi.org/10.1002/path.5471
Bowles L, Platton S, Yartey N, Dave M, Lee K, Hart DP et al (2020) Lupus anticoagulant and abnormal coagulation tests in patients with Covid-19. N Engl J Med 383(3):288–290. https://doi.org/10.1056/NEJMc2013656
Brito CA, Paiva JG, Pimentel FN, Guimarães RS, Moreira MR (2021) COVID-19 in patients with rheumatological diseases treated with anti-TNF. Ann Rheum Dis 80(5):e62 LP-e62. https://doi.org/10.1136/annrheumdis-2020-218171
Bronson PG, Chaivorapol C, Ortmann W, Behrens TW, Graham RR (2012) The genetics of type I interferon in systemic lupus erythematosus. Curr Opin Immunol 24(5):530–537. https://doi.org/10.1016/j.coi.2012.07.008
Caamaño DSJ, Beato RA (2020) Facial diplegia, a possible atypical variant of Guillain-Barré Syndrome as a rare neurological complication of SARS-CoV-2. J Clin Neurosci 77:230–232. https://doi.org/10.1016/j.jocn.2020.05.016
Cantini F, Niccoli L, Matarrese D, Nicastri E, Stobbione P, Goletti D (2020) Baricitinib therapy in COVID-19: a pilot study on safety and clinical impact. J Infect 81(2):318–356. https://doi.org/10.1016/j.jinf.2020.04.017
Casadevall A, Joyner MJ, Pirofski L-A (2020) SARS-CoV-2 viral load and antibody responses: the case for convalescent plasma therapy. J Clin Invest 130(10):5112–5114. https://doi.org/10.1172/JCI139760
Cervia C, Nilsson J, Zurbuchen Y, Valaperti A, Schreiner J, Wolfensberger A et al (2021) Systemic and mucosal antibody responses specific to SARS-CoV-2 during mild versus severe COVID-19. J Allergy Clin Immunol 147(2):545-557.e9. https://doi.org/10.1016/j.jaci.2020.10.040
Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK et al (2016) Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe 19(2):181–193. https://doi.org/10.1016/j.chom.2016.01.007
Chen L, Liu HG, Liu W, Liu J, Liu K, Shang J et al (2020a) Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia. Zhonghua Jiehe He Huxi Zazhi Chin J Tuberc Respir Dis 43:E005. https://doi.org/10.3760/cma.j.issn.1001-0939.2020.0005
Chen Z, Hu J, Zhang Z, Jiang S, Han S, Yan D, et al (2020b) Efficacy of hydroxychloroquine in patients with COVID-19: results of a randomized clinical trial. medRxiv. https://doi.org/10.1101/2020.03.22.20040758
Chowdhury MA, Hossain N, Kashem MA, Shahid MA, Alam A (2020) Immune response in COVID-19: a review. J Infect Public Health 13(11):1619–29. https://doi.org/10.1016/j.jiph.2020.07.001
Clark DN, Markham JL, Sloan CS, Poole BD (2013) Cytokine inhibition as a strategy for treating systemic lupus erythematosus. Clin Immunol 148(3):335–343. https://doi.org/10.1016/j.clim.2012.11.001
Coen M, Jeanson G, Almeida LAC, Hübers A, Stierlin F, Najjar I et al (2020) Guillain-Barré syndrome as a complication of SARS-CoV-2 infection. Brain Behav Immun 87:111-112. https://doi.org/10.1016/j.bbi.2020.04.074
Cohn LA (1991) The influence of corticosteroids on host defense mechanisms. J Vet Intern Med 5(2):95–104. https://doi.org/10.1111/j.1939-1676.1991.tb00939.x
Cooper MA, Fehniger TA, Caligiuri MA (2001) The biology of human natural killer-cell subsets. Trends Immunol 22(11):633–640. https://doi.org/10.1016/s1471-4906(01)02060-9
Crinier A, Milpied P, Escalière B, Piperoglou C, Galluso J, Balsamo A et al (2018) High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity 49(5):971-986.e5. https://doi.org/10.1016/j.immuni.2018.09.009
Cucinotta D, Vanelli M (2020) WHO Declares COVID-19 a pandemic. Acta Biomed 91(1):157–160. https://doi.org/10.23750/abm.v91i1.9397
De Biasi S, Meschiari M, Gibellini L, Bellinazzi C, Borella R, Fidanza L et al (2020) Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat Commun 11(1):3434. https://doi.org/10.1038/s41467-020-17292-4
Del Valle DM, Kim-Schulze S, Huang H-H, Beckmann ND, Nirenberg S, Wang B et al (2020) An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med 26(10):1636–1643. https://doi.org/10.1038/s41591-020-1051-9
Diamond B (2020) The renin–angiotensin system: an integrated view of lung disease and coagulopathy in COVID-19 and therapeutic implications. J Exp Med 217(8):e20201000. https://doi.org/10.1084/jem.20201000
Diamond MS, Kanneganti T-D (2022) Innate immunity: the first line of defense against SARS-CoV-2. Nat Immunol 23(2):165–176. https://doi.org/10.1038/s41590-021-01091-0
Diao B, Wang C, Tan Y, Chen X, Liu YY, Ning L et al (2020) Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol 11:827. https://doi.org/10.3389/fimmu.2020.00827
Dinkin M, Gao V, Kahan J, Bobker S, Simonetto M, Wechsler P et al (2020) COVID-19 presenting with ophthalmoparesis from cranial nerve palsy. Neurology 95(5):221–223. https://doi.org/10.1212/WNL.0000000000009700
Domingo P, Mur I, Pomar V, Corominas H, Casademont J, de Benito N (2020) The four horsemen of a viral apocalypse: the pathogenesis of SARS-CoV-2 infection (COVID-19). EBioMedicine 58:102887. https://doi.org/10.1016/j.ebiom.2020.102887
Fajgenbaum DC, June CH (2020) Cytokine storm. N Engl J Med 383(23):2255–2273. https://doi.org/10.1056/NEJMra2026131
Fan Y, Yang B, Wu C (2008) Phenotypically and functionally distinct subsets of natural killer cells in human PBMCs. Cell Biol Int 32(2):188–197. https://doi.org/10.1016/j.cellbi.2007.08.025
Fathi N, Rezaei N (2020) Lymphopenia in COVID-19: therapeutic opportunities. Cell Biol Int 44(9):1792–1797. https://doi.org/10.1002/cbin.11403
Favalli EG, Ingegnoli F, De Lucia O, Cincinelli G, Cimaz R, Caporali R (2020) COVID-19 infection and rheumatoid arthritis: Faraway, so close! Autoimmun Rev 19(5):102523. https://doi.org/10.1016/j.autrev.2020.102523
Feldmann M, Maini RN, Woody JN, Holgate ST, Winter G, Rowland M et al (2020) Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet 395(10234):1407–1409. https://doi.org/10.1016/S0140-6736(20)30858-8
Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A (2018) Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol 14(10):576–590. https://doi.org/10.1038/s41574-018-0059-4
Frater JL, Zini G, d’Onofrio G, Rogers HJ (2020) COVID-19 and the clinical hematology laboratory. Int J Lab Hematol 42(Suppl 1):11–18. https://doi.org/10.1111/ijlh.13229
Frisoni P, Neri M, D’Errico S, Alfieri L, Bonuccelli D, Cingolani M et al (2021) Cytokine storm and histopathological findings in 60 cases of COVID-19-related death: from viral load research to immunohistochemical quantification of major players IL-1β, IL-6, IL-15 and TNF-α. Forensic Sci Med Pathol 18(1):4–19. https://doi.org/10.1007/s12024-021-00414-9
Gadi N, Wu SC, Spihlman AP, Moulton VR (2020) What’s sex got to do with COVID-19? Gender-based differences in the host immune response to coronaviruses. Front Immunol 11:2147. https://doi.org/10.3389/fimmu.2020.02147
Ghodke-Puranik Y, Niewold TB (2013) Genetics of the type I interferon pathway in systemic lupus erythematosus. Int J Clin Rheumtol 8(6). https://doi.org/10.2217/ijr.13.58
Gianchecchi E, Delfino DV, Fierabracci A (2018) NK cells in autoimmune diseases: linking innate and adaptive immune responses. Autoimmun Rev 17(2):142–154. https://doi.org/10.1016/j.autrev.2017.11.018
Gianfrancesco M, Hyrich KL, Al-Adely S, Carmona L, Danila MI, Gossec L et al (2020b) Characteristics associated with hospitalisation for COVID-19 in people with rheumatic disease: data from the COVID-19 Global Rheumatology Alliance physician-reported registry. Ann Rheum Dis 79(7):859–866. https://doi.org/10.1136/annrheumdis-2020-217871
Gianfrancesco MA, Hyrich KL, Gossec L, Strangfeld A, Carmona L, Mateus EF et al (2020a) Rheumatic disease and COVID-19: initial data from the COVID-19 Global Rheumatology Alliance provider registries. Lancet Rheumatol 2(5):e250–3. https://doi.org/10.1016/S2665-9913(20)30095-3
Gleason MK, Lenvik TR, McCullar V, Felices M, O’Brien MS, Cooley SA et al (2012) Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood 119(13):3064–3072. https://doi.org/10.1182/blood-2011-06-360321
Goshua G, Pine AB, Meizlish ML, Chang C-H, Zhang H, Bahel P et al (2020) Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol 7(8):e575–e582. https://doi.org/10.1016/S2352-3026(20)30216-7
Guo L, Ren L, Yang S, Xiao M, Chang D, Yang F et al (2020) Profiling early humoral response to diagnose novel coronavirus disease (COVID-19). Clin Infect Dis an off Publ Infect Dis Soc Am 71(15):778–785. https://doi.org/10.1093/cid.ciaa310
Guo Y, Hu K, Li Y, Lu C, Ling K, Cai C et al (2022) Targeting TNF-α for COVID-19: recent advanced and controversies. Front Public Health 10:833967. https://doi.org/10.3389/fpubh.2022.833967
Gutiérrez-Ortiz C, Méndez-Guerrero A, Rodrigo-Rey S, San Pedro-Murillo E, Bermejo-Guerrero L, Gordo-Mañas R et al (2020) Miller Fisher syndrome and polyneuritis cranialis in COVID-19. Neurology 95(5):e601–e605. https://doi.org/10.1212/WNL.0000000000009619
Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N et al (2020) Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369(6504):718–724. https://doi.org/10.1126/science.abc6027
Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, Sata T et al (2008) Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc Natl Acad Sci U S A 105(22):7809–7814. https://doi.org/10.1073/pnas.0711241105
Hagberg N, Theorell J, Hjorton K, Spee P, Eloranta M-L, Bryceson YT et al (2015) Functional anti-CD94/NKG2A and anti-CD94/NKG2C autoantibodies in patients with systemic lupus erythematosus. Arthritis Rheumatol 67(4):1000–1011. https://doi.org/10.1002/art.38999
Harzallah I, Debliquis A, Drénou B (2020) Lupus anticoagulant is frequent in patients with Covid-19: response to reply. J Thromb Haemost. https://doi.org/10.1111/jth.14980
Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X et al (2020) High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med 46(6):1089–1098. https://doi.org/10.1007/s00134-020-06062-x
Hemminki K, Huang W, Sundquist J, Sundquist K, Ji J (2020) Autoimmune diseases and hematological malignancies: Exploring the underlying mechanisms from epidemiological evidence. Semin Cancer Biol 64:114–121. https://doi.org/10.1016/j.semcancer.2019.06.005
Henriques A, Teixeira L, Inês L, Carvalheiro T, Gonçalves A, Martinho A et al (2013) NK cells dysfunction in systemic lupus erythematosus: relation to disease activity. Clin Rheumatol 32(6):805–813. https://doi.org/10.1007/s10067-013-2176-8
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S et al (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181(2):271-280.e8. https://doi.org/10.1016/j.cell.2020.02.052
Horisberger A, Moi L, Ribi C, Comte D (2020) Impact of COVID-19 pandemic on SLE: beyond the risk of infection. Lupus Sci Med 7(1):e000408. https://doi.org/10.1136/lupus-2020-000408
Hou C, Jin O, Zhang X (2018) Clinical characteristics and risk factors of infections in patients with systemic lupus erythematosus. Clin Rheumatol 37(10):2699–2705. https://doi.org/10.1007/s10067-018-4198-8
Hu B, Huang S, Yin L (2021) The cytokine storm and COVID-19. J Med Virol 93(1):250–256. https://doi.org/10.1002/jmv.26232
Huang I, Pranata R (2020) Lymphopenia in severe coronavirus disease-2019 (COVID-19): systematic review and meta-analysis. J Intensive Care 8:36. https://doi.org/10.1186/s40560-020-00453-4
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al (2020a) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395(10223):497–506. https://doi.org/10.1016/S0140-6736(20)30183-5
Huang AT, Garcia-Carreras B, Hitchings MDT, Yang B, Katzelnick LC, Rattigan SM et al (2020b) A systematic review of antibody mediated immunity to coronaviruses: kinetics, correlates of protection, and association with severity. Nat Commun 11(1):4704. https://doi.org/10.1038/s41467-020-18450-4
Iliopoulos AG, Tsokos GC (1996) Immunopathogenesis and spectrum of infections in systemic lupus erythematosus. Semin Arthritis Rheum 25(5):318–336. https://doi.org/10.1016/s0049-0172(96)80018-7
Jacobs J, Eichbaum Q (2021) COVID-19 associated with severe autoimmune hemolytic anemia. Transfusion 61:635–640. https://doi.org/10.1111/trf.16226
Jakiela B, Kosałka J, Plutecka H, Bazan-Socha S, Sanak M, Musiał J (2018) Facilitated expansion of Th17 cells in lupus nephritis patients. Clin Exp Immunol 194(3):283–94. https://doi.org/10.1111/cei.13196
Jiang Y, Rubin L, Peng T, Liu L, Xing X, Lazarovici P et al (2022) Cytokine storm in COVID-19: from viral infection to immune responses, diagnosis and therapy. Int J Biol Sci 18(2):459–472. https://doi.org/10.7150/ijbs.59272
Jin B, Li Y, Robertson KD (2011) DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer 2(6):607–617. https://doi.org/10.1177/1947601910393957
Jin Y, Wang M, Zuo Z, Fan C, Ye F, Cai Z et al (2020) Diagnostic value and dynamic variance of serum antibody in coronavirus disease 2019. Int J Infect Dis IJID off Publ Int Soc Infect Dis 94:49–52. https://doi.org/10.1016/j.ijid.2020.03.065
Jones VG, Mills M, Suarez D, Hogan CA, Yeh D, Segal JB et al (2020) COVID-19 and Kawasaki disease: novel virus and novel case. Hosp Pediatr 10(6):537–540. https://doi.org/10.1542/hpeds.2020-0123
Kanduc D, Shoenfeld Y (2020) On the molecular determinants of the SARS-CoV-2 attack. Clin Immunol 215:108426. https://doi.org/10.1016/j.clim.2020.108426
Kanneganti T-D (2020) Intracellular innate immune receptors: life inside the cell. Immunol Rev 297(1):5–12. https://doi.org/10.1111/imr.12912
Kariuki SN, Franek BS, Kumar AA, Arrington J, Mikolaitis RA, Utset TO et al (2010) Trait-stratified genome-wide association study identifies novel and diverse genetic associations with serologic and cytokine phenotypes in systemic lupus erythematosus. Arthritis Res Ther 12(4):1–12. https://doi.org/10.1186/ar3101.aar6584
Karnell JL, Albulescu M, Drabic S, Wang L, Moate R, Baca M et al (2019) A CD40L-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci Transl Med 11(489):eaar6584. https://doi.org/10.1126/scitranslmed.aar6584
Kärre K (2002) NK cells, MHC class I molecules and the missing self. Scand J Immunol 55(3):221–228. https://doi.org/10.1046/j.1365-3083.2002.01053.x
Katsuyama T, Tsokos GC, Moulton VR (2018) Aberrant T Cell Signaling and Subsets in Systemic Lupus Erythematosus. Front Immunol 9:1088. https://doi.org/10.3389/fimmu.2018.01088
Kichloo A, Aljadah M, Albosta M, Wani F, Singh J, Solanki S (2020) COVID-19 and acute lupus pneumonitis: diagnostic and treatment dilemma. J Investig Med High Impact Case Rep 8:232470962093343. https://doi.org/10.1177/2324709620933438
Kokkotis G, Kitsou K, Xynogalas I, Spoulou V, Magiorkinis G, Trontzas I et al (2022) Systematic review with meta-analysis: COVID-19 outcomes in patients receiving anti-TNF treatments. Aliment Pharmacol Ther 55:154–167. https://doi.org/10.1111/apt.16717
Kuhn A, Bonsmann G, Anders H-J, Herzer P, Tenbrock K, Schneider M (2015) The diagnosis and treatment of systemic lupus erythematosus. Dtsch Arztebl Int 112(25):423–432. https://doi.org/10.3238/arztebl.2015.0423
Kuter DJ (2021) Exacerbation of immune thrombocytopenia following COVID-19 vaccination. Br J Haematol 195(3):365–370. https://doi.org/10.1111/bjh.17645
Kyttaris VC (2019) Targeting cytokines to treat autoimmunity. Clin Immunol 206:108251. https://doi.org/10.1016/j.clim.2019.108251
Lanier LL (2008) Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 9(5):495–502. https://doi.org/10.1038/ni1581
Lazarian G, Quinquenel A, Bellal M, Siavellis J, Jacquy C, Re D et al (2020) Autoimmune haemolytic anaemia associated with COVID-19 infection. Br J Haematol 190(1):29–31. https://doi.org/10.1111/bjh.16794
Lee SSHS-Y, Lee SSHS-Y, Seo H-B, Ryu J-G, Jung K, Choi JW et al (2019) Inhibition of IL-17 ameliorates systemic lupus erythematosus in Roquinsan/san mice through regulating the balance of TFH cells, GC B cells, Treg and Breg. Sci Rep 9(1):5227. https://doi.org/10.1038/s41598-019-41534-1
Leem G, Cheon S, Lee H, Choi SJ, Jeong S, Kim E-S, et al (2021) Abnormality in the NK-cell population is prolonged in severe COVID-19 patients. J Allergy Clin Immunol 148(4):996–1006.e18. https://doi.org/10.1016/j.jaci.2021.07.022
Lei J, Li J, Li X, Qi X (2020) CT imaging of the 2019 novel coronavirus (2019-nCoV) pneumonia. Radiology 295(1):18. https://doi.org/10.1148/radiol.2020200236
Leng Z, Zhu R, Hou W, Feng Y, Yang Y, Han Q et al (2020) Transplantation of ACE2(-) mesenchymal stem cells improves the outcome of patients with COVID-19 pneumonia. Aging Dis 11(2):216–228. https://doi.org/10.14336/AD.2020.0228
Li D, Chen Y, Liu H, Jia Y, Li F, Wang W et al (2020) Immune dysfunction leads to mortality and organ injury in patients with COVID-19 in China: insights from ERS-COVID-19 study. Signal Transduct Target Ther 5:62. https://doi.org/10.1038/s41392-020-0163-5
Lin L, Lu L, Cao W, Li T (2020) Hypothesis for potential pathogenesis of SARS-CoV-2 infection-a review of immune changes in patients with viral pneumonia. Emerg Microbes Infect 9(1):727–732. https://doi.org/10.1080/22221751.2020.1746199
Lin Y, Wu C, Wang X, Liu S, Zhao K, Kemper T et al (2020) Glucosamine promotes hepatitis B virus replication through its dual effects in suppressing autophagic degradation and inhibiting MTORC1 signaling. Autophagy 16(3):548–561. https://doi.org/10.1080/15548627.2019.1632104
Littman DR, Rudensky AY (2010) Th17 and regulatory T cells in mediating and restraining inflammation. Cell 140(6):845–58. https://doi.org/10.1016/j.cell.2010.02.021
Liu M, Liu J, Zhang X, Xiao Y, Jiang G, Huang X (2021) Activation status of CD56dim natural killer cells is associated with disease activity of patients with systemic lupus erythematosus. Clin Rheumatol 40(3):1103–1112. https://doi.org/10.1007/s10067-020-05306-x
Long EO, Kim HS, Liu D, Peterson ME, Rajagopalan S (2013) Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol 31:227–258. https://doi.org/10.1146/annurev-immunol-020711-075005
Long Q-X, Liu B-Z, Deng H-J, Wu G-C, Deng K, Chen Y-K et al (2020) Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat Med 26(6):845–848. https://doi.org/10.1038/s41591-020-0897-1
Lou B, Li T-D, Zheng S-F, Su Y-Y, Li Z-Y, Liu W et al (2020) Serology characteristics of SARS-CoV-2 infection after exposure and post-symptom onset. Eur Respir 56(2):2000763. https://doi.org/10.1183/13993003.00763-2020
Lucchese G, Flöel A (2020) Molecular mimicry between SARS-CoV-2 and respiratory pacemaker neurons. Autoimmun Rev 19(7):102556. https://doi.org/10.1016/j.autrev.2020.102556
Luo P, Liu Y, Qiu L, Liu X, Liu D, Li J (2020) Tocilizumab treatment in COVID-19: a single center experience. J Med Virol 92(7):814–818. https://doi.org/10.1002/jmv.25801
Lv J, Wang Z, Qu Y, Zhu H, Zhu Q, Tong W et al (2021) Distinct uptake, amplification, and release of SARS-CoV-2 by M1 and M2 alveolar macrophages. Cell Discov 7(1):1–12. https://doi.org/10.1038/s41421-021-00258-1
Lynch KL, Whitman JD, Lacanienta NP, Beckerdite EW, Kastner SA, Shy BR (2020) Magnitude and kinetics of anti-SARS-CoV-2 antibody responses and their relationship to disease. Clin Infect Dis 72:301–308. https://doi.org/10.1093/cid/ciaa979
Ma H, Zeng W, He H, Zhao D, Jiang D, Zhou P et al (2020) Serum IgA, IgM, and IgG responses in COVID-19. Cell Mol Immunol 17(7):773–775. https://doi.org/10.1038/s41423-020-0474-z
Maiese A, Passaro G, Matteis ADE, Fazio V, Raffaele LR, Di PM (2020) Thromboinflammatory response in SARS-CoV-2 sepsis. Med Leg J 88(2):78–80. https://doi.org/10.1177/0025817220926915
Mak A, Kow NY (2014) The Pathology of T Cells in Systemic Lupus Erythematosus. J Immunol Res 2014:1–8. https://doi.org/10.1155/2014/419029
Martin M, Guffroy A, Argemi X, Martin T (2017) Systemic lupus erythematosus and lymphopenia: clinical and pathophysiological features. La Rev Med Interne 38(9):603–613. https://doi.org/10.1016/j.revmed.2017.01.005
Mathian A, Mahevas M, Rohmer J, Roumier M, Cohen-Aubart F, Amador-Borrero B et al (2020) Clinical course of coronavirus disease 2019 (COVID-19) in a series of 17 patients with systemic lupus erythematosus under long-term treatment with hydroxychloroquine. Ann Rheum Dis 79(6):837–839. https://doi.org/10.1136/annrheumdis-2020-217566
Misra DP, Agarwal V, Gasparyan AY, Zimba O (2020) Rheumatologists’ perspective on coronavirus disease 19 (COVID-19) and potential therapeutic targets. Clin Rheumatol 39(7):2055–2062. https://doi.org/10.1007/s10067-020-05073-9
Monti S, Montecucco C (2020) Can hydroxychloroquine protect patients with rheumatic diseases from COVID-19? Response to: “does hydroxychloroquine prevent the transmission of COVID-19?” by Heldwein and Calado and “SLE, hydroxychloroquine and no SLE patients with COVID-19: a comment.” Ann Rheum Dis 79:e62. https://doi.org/10.1136/annrheumdis-2020-217524
Monti S, Balduzzi S, Delvino P, Bellis E, Quadrelli VS, Montecucco C (2020) Clinical course of COVID-19 in a series of patients with chronic arthritis treated with immunosuppressive targeted therapies. Ann Rheum Dis 79(5):667–668. https://doi.org/10.1136/annrheumdis-2020-217424
Moody R, Wilson K, Flanagan KL, Jaworowski A, Plebanski M (2021) Adaptive immunity and the risk of autoreactivity in COVID-19. Int J Mol Sci 22(16):8965. https://doi.org/10.3390/ijms22168965
Moretta A, Bottino C, Vitale M, Pende D, Cantoni C, Mingari MC et al (2001) Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol 19:197–223. https://doi.org/10.1146/annurev.immunol.19.1.197
Munroe ME, Lu R, Zhao YD, Fife DA, Robertson JM, Guthridge JM et al (2016) Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann Rheum Dis 75(11):2014–2021. https://doi.org/10.1136/annrheumdis-2015-208140
Muskardin TLW, Niewold TB (2018) Type I interferon in rheumatic diseases. Nat Rev Rheumatol 14(4):214–228. https://doi.org/10.1038/nrrheum.2018.31
Nabah YNA, Mateo T, Estellés R, Mata M, Zagorski J, Sarau H et al (2004) Angiotensin II induces neutrophil accumulation in vivo through generation and release of CXC chemokines. Circulation 110(23):3581–3586. https://doi.org/10.1161/01.CIR.0000148824.93600.F3
Najafi S, Rajaei E, Moallemian R, Nokhostin F (2020) The potential similarities of COVID-19 and autoimmune disease pathogenesis and therapeutic options: new insights approach. Clin Rheumatol 39(11):3223–3235. https://doi.org/10.1007/s10067-020-05376-x
Nathan N, Prevost B, Corvol H (2020) Atypical presentation of COVID-19 in young infants. Lancet 395(10235):1481. https://doi.org/10.1016/S0140-6736(20)30980-6
Ndhlovu LC, Lopez-Vergès S, Barbour JD, Jones RB, Jha AR, Long BR et al (2012) Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 119(16):3734–3743. https://doi.org/10.1182/blood-2011-11-392951
Niewold TB (2011) Interferon alpha as a primary pathogenic factor in human lupus. J Interf Cytokine Res off J Int Soc Interf Cytokine Res 31(12):887–892. https://doi.org/10.1089/jir.2011.0071
Niewold TB, Swedler WI (2005) Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin Rheumatol 24(2):178–181. https://doi.org/10.1007/s10067-004-1024-2
Niewold TB, Hua J, Lehman TJA, Harley JB, Crow M (2007) High serum IFN-α activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun 8(6):492–502. https://doi.org/10.1038/sj.gene.6364408
Niewold TB, Clark DN, Salloum R, Poole BD (2010) Interferon alpha in systemic lupus erythematosus. J Biomed Biotechnol 2010:948364. https://doi.org/10.1155/2010/948364
Nokhostin F, Dargahi MalAmir M, Tutunchi S, Rezaeeyan H (2020) Evaluation of prognostic/diagnostic value of hematological markers in the detection of inflammation in coronavirus disease: a review study. J Adv Med Biomed Res 28(128):171–174. https://doi.org/10.30699/jambs.28.128.171
Oke V, Brauner S, Larsson A, Gustafsson J, Zickert A, Gunnarsson I et al (2017) IFN-λ1 with Th17 axis cytokines and IFN-α define different subsets in systemic lupus erythematosus (SLE). Arthritis Res Ther 19(1):139. https://doi.org/10.1186/s13075-017-1344-7
Okoye IS, Houghton M, Tyrrell L, Barakat K, Elahi S (2017) Coinhibitory receptor expression and immune checkpoint blockade: maintaining a balance in CD8(+) T cell responses to chronic viral infections and cancer. Front Immunol 8:1215. https://doi.org/10.3389/fimmu.2017.01215
Osman MS, van Eeden C, CohenTervaert JW (2020) Fatal COVID-19 infections: is NK cell dysfunction a link with autoimmune HLH? Autoimmun Rev 19:102561. https://doi.org/10.1016/j.autrev.2020.102561
Osman M, Faridi RM, Sligl W, Shabani-Rad M-T, Dharmani-Khan P, Parker A et al (2020) Impaired natural killer cell counts and cytolytic activity in patients with severe COVID-19. Blood Adv 4(20):5035–5039. https://doi.org/10.1182/bloodadvances.2020002650
Ottaviani D, Boso F, Tranquillini E, Gapeni I, Pedrotti G, Cozzio S et al (2020) Early Guillain-Barré syndrome in coronavirus disease 2019 (COVID-19): a case report from an Italian COVID-hospital. Neurol Sci 41(6):1351–1354. https://doi.org/10.1007/s10072-020-04449-8
Pacheco Y, Acosta-Ampudia Y, Monsalve DM, Chang C, Gershwin ME, Anaya J-M (2019) Bystander activation and autoimmunity. J Autoimmun 103:102301. https://doi.org/10.1016/j.jaut.2019.06.012
Pan L, Lu M-P, Wang J-H, Xu M, Yang S-R (2020) Immunological pathogenesis and treatment of systemic lupus erythematosus. World J Pediatr 16(1):19–30. https://doi.org/10.1007/s12519-019-00229-3
Papo T, Parizot C, Ortova M, Piette JC, Frances C, Debre P et al (1998) Apoptosis and expression of soluble Fas mRNA in systemic lupus erythematosus. Lupus 7(7):455–461. https://doi.org/10.1191/096120398678920460
Parackova Z, Bloomfield M, Klocperk A, Sediva A (2021) Neutrophils mediate Th17 promotion in COVID-19 patients. J Leukoc Biol 109(1):73–76. https://doi.org/10.1002/JLB.4COVCRA0820-481RRR
Park Y-W, Kee S-J, Cho Y-N, Lee E-H, Lee H-Y, Kim E-M et al (2009) Impaired differentiation and cytotoxicity of natural killer cells in systemic lupus erythematosus. Arthritis Rheum 60(6):1753–1763. https://doi.org/10.1002/art.24556
Patra MC, Shah M, Choi S (2020) Toll-like receptor-induced cytokines as immunotherapeutic targets in cancers and autoimmune diseases. Semin Cancer Biol 64:61–82. https://doi.org/10.1016/j.semcancer.2019.05.002
Perl A, Gergely PJ, Nagy G, Koncz A, Banki K (2004) Mitochondrial hyperpolarization: a checkpoint of T-cell life, death and autoimmunity. Trends Immunol 25(7):360–367. https://doi.org/10.1016/j.it.2004.05.001
Qin C, Zhou L, Hu Z, Zhang S, Yang S, Tao Y et al (2020) Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in Wuhan, China. Clin Infect Dis an off Publ Infect Dis Soc Am 71(15):762–768. https://doi.org/10.1093/cid/ciaa248
Rana A, Minz RW, Aggarwal R, Anand S, Pasricha N, Singh S (2012) Gene expression of cytokines (TNF-α, IFN-γ), serum profiles of IL-17 and IL-23 in paediatric systemic lupus erythematosus. Lupus 21(10):1105–1112. https://doi.org/10.1177/0961203312451200
Rekvig OP (2015) Anti-dsDNA antibodies as a classification criterion and a diagnostic marker for systemic lupus erythematosus: critical remarks. Clin Exp Immunol 179(1):5–10. https://doi.org/10.1111/cei.12296
Riphagen S, Gomez X, Gonzalez-Martinez C, Wilkinson N, Theocharis P (2020) Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 395(10237):1607–1608. https://doi.org/10.1016/S0140-6736(20)31094-1
Rivera-Figueroa EI, Santos R, Simpson S, Garg P (2020) Incomplete Kawasaki disease in a child with Covid-19. Indian Pediatr 57(7):680–681. https://doi.org/10.1007/s13312-020-1900-0
Rodríguez Y, Novelli L, Rojas M, De Santis M, Acosta-Ampudia Y, Monsalve DM et al (2020) Autoinflammatory and autoimmune conditions at the crossroad of COVID-19. J Autoimmun 114:102506. https://doi.org/10.1016/j.jaut.2020.102506
Rojas M, Restrepo-Jiménez P, Monsalve DM, Pacheco Y, Acosta-Ampudia Y, Ramírez-Santana C et al (2018) Molecular mimicry and autoimmunity. J Autoimmun 95:100–123. https://doi.org/10.1016/j.jaut.2018.10.012
Rönnblom L, Pascual V (2008) The innate immune system in SLE: type I interferons and dendritic cells. Lupus 17(5):394–399. https://doi.org/10.1177/0961203308090020
Rönnblom LE, Alm GV, Oberg KE (1990) Possible induction of systemic lupus erythematosus by interferon-alpha treatment in a patient with a malignant carcinoid tumour. J Intern Med 227(3):207–210. https://doi.org/10.1111/j.1365-2796.1990.tb00144.x
Rúa-Figueroa Í, López-Longo J, Galindo-Izquierdo M, Calvo-Alén J, Del Campo V, Olivé-Marqués A et al (2017) Incidence, associated factors and clinical impact of severe infections in a large, multicentric cohort of patients with systemic lupus erythematosus. Semin Arthritis Rheum 47(1):38–45. https://doi.org/10.1016/j.semarthrit.2017.01.010
Ruan Q, Yang K, Wang W, Jiang L, Song J (2020) Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan China. Intensive Care Med 46:846–848. https://doi.org/10.1007/s00134-020-05991-x
Sadeghi A, Tahmasebi S, Mahmood A, Kuznetsova M, Valizadeh H, Taghizadieh A et al (2021) Th17 and Treg cells function in SARS-CoV2 patients compared with healthy controls. J Cell Physiol 236(4):2829–2839. https://doi.org/10.1002/jcp.30047
Sawalha AH, Zhao M, Coit P, Lu Q (2020) Epigenetic dysregulation of ACE2 and interferon-regulated genes might suggest increased COVID-19 susceptibility and severity in lupus patients. medRxiv 2020.03.30.20047852. https://doi.org/10.1016/j.clim.2020.108410
Scheidl E, Canseco DD, Hadji-Naumov A, Bereznai B (2020) Guillain-Barr é syndrome during SARS-CoV-2 pandemic: a case report and review of recent literature. J Peripher Nerv Syst 25(2):204–207. https://doi.org/10.1111/jns.12382
Schleinitz N, Vély F, Harlé J-R, Vivier E (2010) Natural killer cells in human autoimmune diseases. Immunology 131(4):451–458. https://doi.org/10.1111/j.1365-2567.2010.03360x
Schmidt RL, Lenz LL (2012) Distinct licensing of IL-18 and IL-1β secretion in response to NLRP3 inflammasome activation. PLoS ONE 7(9):e45186. https://doi.org/10.1371/journal.pone.0045186
Schuster IS, Coudert JD, Andoniou CE, Degli-Esposti MA (2016) “Natural Regulators”: NK cells as modulators of T cell immunity. Front Immunol 7:235. https://doi.org/10.3389/fimmu.2016.00235
Segerberg F, Lundtoft C, Reid S, Hjorton K, Leonard D, Nordmark G et al (2019) Autoantibodies to killer cell immunoglobulin-like receptors in patients with systemic lupus erythematosus induce natural killer cell hyporesponsiveness. Front Immunol 10:2164. https://doi.org/10.3389/fimmu.2019.02164
Shah D, Sah S, Nath SK (2013) Interaction between glutathione and apoptosis in systemic lupus erythematosus. Autoimmun Rev 12(7):741–751. https://doi.org/10.1016/j.autrev.2012.12.007
Shan J, Jin H, Xu Y (2020) T cell metabolism: a new perspective on Th17/treg cell imbalance in systemic lupus erythematosus. Front Immunol 11:1027. https://doi.org/10.3389/fimmu.2020.01027
Singh N, Birkenbach M, Caza T, Perl A, Cohen PL (2013) Tuberous sclerosis and fulminant lupus in a young woman. J Clin Rheumatol Pract Rep Rheum Musculoskelet Dis 19(3):134. https://doi.org/10.1097/RHU.0b013e318289c033
Smatti MK, Cyprian FS, Nasrallah GK, Al Thani AA, Almishal RO, Yassine HM (2019) Viruses and autoimmunity: a review on the potential interaction and molecular mechanisms. Viruses 11(8):762. https://doi.org/10.3390/v11080762
Sodhi CP, Nguyen J, Yamaguchi Y, Werts AD, Lu P, Ladd MR, et al (2019) A dynamic variation of pulmonary ACE2 is required to modulate neutrophilic inflammation in response to pseudomonas aeruginosa lung infection in mice. J Immunol 203(11):3000 LP–3012. https://doi.org/10.4049/jimmunol.1900579
Soloway S, DePace NL, Soloway AM, Colombo J (2021) Lupus pneumonitis therapy masks coronavirus (COVID-19). Case Rep Rheumatol 2021:6645780. https://doi.org/10.1155/2021/6645780
Spada R, Rojas JM, Barber DF (2015) Recent findings on the role of natural killer cells in the pathogenesis of systemic lupus erythematosus. J Leukoc Biol 98(4):479–487. https://doi.org/10.1189/jlb.4RU0315-081RR
Spihlman AP, Gadi N, Wu SC, Moulton VR (2020) COVID-19 and systemic lupus erythematosus: focus on immune response and therapeutics. Front Immunol 11:589474. https://doi.org/10.3389/fimmu.2020.589474
Tang J, Ravichandran S, Lee Y, Grubbs G, Coyle EM, Klenow L et al (2021) Antibody affinity maturation and plasma IgA associate with clinical outcome in hospitalized COVID-19 patients. Nat Commun 12(1):1221. https://doi.org/10.1038/s41467-021-21463-2
Tariq S, Van Eeden C, Tervaert JWC, Osman MS (2021) COVID-19, rheumatic diseases and immune dysregulation—a perspective. Clin Rheumatol 40(2):433–442. https://doi.org/10.1007/s10067-020-05529-y
Terrazzano G, Rubino V, Palatucci AT, Giovazzino A, Carriero F, Ruggiero G (2020) An open question: is it rational to inhibit the mTor-dependent pathway as COVID-19 therapy? Front Pharmacol 11:856. https://doi.org/10.3389/fphar.2020.00856
The global rheumatology community’s response to the worldwide Covid-19 pandemic. 2022. Available from: https://rheum-covid.org/updates/combined-data.html [Accessed on 1 Mar 2022 ]
Thienemann F, Pinto F, Grobbee DE, Boehm M, Bazargani N, Ge J et al (2020) World heart federation briefing on prevention: coronavirus disease 2019 (COVID-19) in low-income countries. Glob Heart 15(1):31. https://doi.org/10.5334/gh.778
Thomas G, Mancini J, Jourde-Chiche N, Sarlon G, Amoura Z, Harlé J-R et al (2014) Mortality associated with systemic lupus erythematosus in France assessed by multiple-cause-of-death analysis. Arthritis Rheumatol 66(9):2503–2511. https://doi.org/10.1002/art.38731
Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG (2012) Into the eye of the cytokine storm. Microbiol Mol Biol Rev 76(1):16–32. https://doi.org/10.1111/imm.13262
Toor SM, Saleh R, Sasidharan Nair V, Taha RZ, Elkord E (2021) T-cell responses and therapies against SARS-CoV-2 infection. Immunology 162(1):30–43. https://doi.org/10.1111/imm.13262
Toscano G, Palmerini F, Ravaglia S, Ruiz L, Invernizzi P, Cuzzoni MG et al (2020) Guillain-Barré syndrome associated with SARS-CoV-2. N Engl J Med 382(26):2574–2576. https://doi.org/10.1056/NEJMc2009191
Toubiana J, Poirault C, Corsia A, Bajolle F, Fourgeaud J, Angoulvant F et al (2020) Kawasaki-like multisystem inflammatory syndrome in children during the covid-19 pandemic in Paris, France: prospective observational study. BMJ 369:m2094. https://doi.org/10.1136/bmj.m209
Valencia JC, Egbukichi N, Erwin-Cohen RA (2019) Autoimmunity and cancer, the paradox comorbidities challenging therapy in the context of preexisting autoimmunity. J Interf Cytokine Res 39(1):72–84. https://doi.org/10.1089/jir.2018.0060
van Eeden C, Khan L, Osman MS, Cohen Tervaert JW (2020) Natural killer cell dysfunction and its role in COVID-19. Int J Mol Sci 21(17):6351. https://doi.org/10.3390/ijms21176351
Vardhana SA, Wolchok JD (2020) The many faces of the anti-COVID immune response. J Exp Med 217(6): e20200678. https://doi.org/10.1084/jem.20200678
Verdoni L, Mazza A, Gervasoni A, Martelli L, Ruggeri M, Ciuffreda M et al (2020) An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet 395(10239):1771–1778. https://doi.org/10.1016/S0140-6736(20)31103-X 13 May 2020
Vietzen H, Zoufaly A, Traugott M, Aberle J, Aberle S, Puchhammer-Stöckl E. (2020) REPRINT: NK cell receptor NKG2C deletion and HLA-Evariants are risk factors for severe COVID-19. Research Square 2020. https://doi.org/10.21203/rs.3.rs-34505/v1
Virani A, Rabold E, Hanson T, Haag A, Elrufay R, Cheema T et al (2020) Guillain-Barré syndrome associated with SARS-CoV-2 infection. Idcases 20:e00771. https://doi.org/10.1016/j.idcr.2020.e00771
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S (2008) Functions of natural killer cells. Nat Immunol 9(5):503–510. https://doi.org/10.1038/ni1582
Vlachoyiannopoulos PG, Magira E, Alexopoulos H, Jahaj E, Theophilopoulou K, Kotanidou A et al (2020) Autoantibodies related to systemic autoimmune rheumatic diseases in severely ill patients with COVID-19. Ann Rheum Dis 79:1661–1663. https://doi.org/10.1136/annrheumdis-2020-218009
Vojdani A, Kharrazian D (2020) Potential antigenic cross-reactivity between SARS-CoV-2 and human tissue with a possible link to an increase in autoimmune diseases. Clin Immunol 217:108480. https://doi.org/10.1016/j.clim.2020.108480
Wagner C, Griesel M, Mikolajewska A, Mueller A, Nothacker M, Kley K et al (2021) Systemic corticosteroids for the treatment of COVID-19. Cochrane Database Syst Rev 8(8):CD014963. https://doi.org/10.1002/14651858.CD014963
Wang L, Wang F-S, Gershwin ME (2015) Human autoimmune diseases: a comprehensive update. J Intern Med 278(4):369–395. https://doi.org/10.1111/joim.12395
Wang M, Zhang C, Tian T, Zhang T, Wang R, Han F et al (2018) Increased regulatory T cells in peripheral blood of acute myeloid leukemia patients rely on tumor necrosis factor (TNF)-α-TNF receptor-2 pathway. Front Immunol 9:1274. https://doi.org/10.3389/fimmu.2018.01274
Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J et al (2020) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan. China JAMA 323(11):1061–1069. https://doi.org/10.1001/jama.2020.1585
Wang Y, Zhang L, Sang L, Ye F, Ruan S, Zhong B et al (2020) Kinetics of viral load and antibody response in relation to COVID-19 severity. J Clin Invest 130(10):5235–5244. https://doi.org/10.1172/JCI138759
Wang F, Nie J, Wang H, Zhao Q, Xiong Y, Deng L et al (2020) Characteristics of peripheral lymphocyte subset alteration in COVID-19 pneumonia. J Infect Dis 221(11):1762–1769. https://doi.org/10.1093/infdis/jiaa150
Wang EY, Mao T, Klein J, Dai Y, Huck JD, Jaycox JR et al (2021) Diverse functional autoantibodies in patients with COVID-19. Nature 595(7866):283–288. https://doi.org/10.1038/s41586-021-03631-y
Wang Y, Li J, Li H, Lei P, Shen G, Yang C (2021) Persistence of SARS-CoV-2-specific antibodies in COVID-19 patients. Int Immunopharmacol 90:107271. https://doi.org/10.1016/j.intimp.2020.107271
Whitman JD, Hiatt J, Mowery CT, Shy BR, Yu R, Yamamoto TN et al (2020) Evaluation of SARS-CoV-2 serology assays reveals a range of test performance. Nat Biotechnol 38(10):1174–1183. https://doi.org/10.1038/s41587-020-0659-0
Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martínez-Colón GJ, McKechnie JL, et al (2020) A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med 26(7):1070–6. https://doi.org/10.1038/s41591-020-0944-y
Xiang F, Wang X, He X, Peng Z, Yang B, Zhang J et al (2020) Antibody detection and dynamic characteristics in patients with coronavirus disease 2019. Clin Infect Dis an off Publ Infect Dis Soc Am 71(8):1930–1934. https://doi.org/10.1093/cid/ciaa461
Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A et al (2020) Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect 9(1):761–770. https://doi.org/10.1080/22221751.2020.1747363
Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C et al (2020a) Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med 8:420–422. https://doi.org/10.1016/S2213-2600(20)30076-X
Xu W-D, Su L-C, Liu X-Y, Wang J-M, Yuan Z-C, Qin Z et al (2020b) IL-38: A novel cytokine in systemic lupus erythematosus pathogenesis. J Cell Mol Med 24(21):12379–12389. https://doi.org/10.1111/jcmm.15737
Yang J, Chu Y, Yang X, Gao D, Zhu L, Yang X et al (2009) Th17 and natural treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum 60(5):1472–1483. https://doi.org/10.1002/art.24499
Yap DYH, Lai KN (2013) The role of cytokines in the pathogenesis of systemic lupus erythematosus—from bench to bedside. Nephrology 18(4):243–255. https://doi.org/10.1111/nep.12047
Yasuda K, Takeuchi Y, Hirota K (2019) The pathogenicity of Th17 cells in autoimmune diseases. Semin Immunopathol 41(3):283–297. https://doi.org/10.1007/s00281-019-00733-8
Yu H-Q, Sun B-Q, Fang Z-F, Zhao J-C, Liu X-Y, Li Y-M et al (2020) Distinct features of SARS-CoV-2-specific IgA response in COVID-19 patients. Eur Respir J 56(2):2001526. https://doi.org/10.1183/13993003.01526-2020
Zamani B, Moeini Taba S-M, Shayestehpour M (2021) Systemic lupus erythematosus manifestation following COVID-19: a case report. J Med Case Rep 15(1):29. https://doi.org/10.1186/s13256-020-02582-8
Zanza C, Tassi MF, Romenskaya T, Piccolella F, Abenavoli L, Franceschi F et al (2021) Lock, stock and barrel: Role of renin-angiotensin-aldosterone system in coronavirus disease 2019. Cells 10(7):1752. https://doi.org/10.3390/cells10071752
Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H (2013) mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 499(7459):485–490. https://doi.org/10.1038/nature12297
Zhang X, Ing S, Fraser A, Chen M, Khan O, Zakem J et al (2013) Follicular helper T cells: new insights into mechanisms of autoimmune diseases. Ochsner J 13(1):131–139. https://www.ochsnerjournal.org/content/13/1/131
Zhang Y, Xiao M, Zhang SS, Xia P, Cao W, Jiang W et al (2020) Coagulopathy and antiphospholipid antibodies in patients with covid-19. N Engl J Med 382(17):e38. https://doi.org/10.1056/NEJMc2007575
Zhao J, Yuan Q, Wang H, Liu W, Liao X, Su Y et al (2020) Antibody responses to SARS-CoV-2 in patients with novel coronavirus disease 2019. Clin Infect Dis an off Publ Infect Dis Soc Am 71(16):2027–2034. https://doi.org/10.1093/cid/ciaa344
Zheng Y, Sun L, Jiang T, Zhang D, He D, Nie H (2014) TNFα promotes Th17 cell differentiation through IL-6 and IL-1β produced by monocytes in rheumatoid arthritis. J Immunol Res 2014:385352. https://doi.org/10.1155/2014/385352
Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D et al (2020) Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol 17:533–535. https://doi.org/10.1038/s41423-020-0402-2
Zhu Z, Cai T, Fan L, Lou K, Hua X, Huang Z et al (2020) Clinical value of immune-inflammatory parameters to assess the severity of coronavirus disease 2019. Int J Infect Dis 95:332–339. https://doi.org/10.1016/j.ijid.2020.04.041
Zickert A, Amoudruz P, Sundström Y, Rönnelid J, Malmström V, Gunnarsson I (2015) IL-17 and IL-23 in lupus nephritis - association to histopathology and response to treatment. BMC Immunol 16(1):7. https://doi.org/10.1186/s12865-015-0070-7
Zulfiqar A-A, Lorenzo-Villalba N, Hassler P, Andrès E (2020) Immune thrombocytopenic purpura in a patient with Covid-19. N Engl J Med 382(18):e43. https://doi.org/10.1056/NEJMc2010472
Zuo Y, Estes SK, Ali RA, Gandhi AA, Yalavarthi S, Shi H et al (2020) Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci Transl Med 12(570):eabd3876. https://doi.org/10.1126/scitranslmed.abd3876
Funding
None.
Author information
Authors and Affiliations
Contributions
SAA and FF: study design/final approval of the version to be published and agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. SSH, SMH, FF, and SAA: data analysis, drafting and revision of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hejazian, S., Hejazian, S., Farnood, F. et al. Dysregulation of immunity in COVID-19 and SLE. Inflammopharmacol 30, 1517–1531 (2022). https://doi.org/10.1007/s10787-022-01047-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-022-01047-2