
Abstract
Hepatic ischemia–reperfusion injury (HIRI) is the main reason for organ failure following liver surgery; however, its underlying causes are complex, and include oxidative stress, sterile inflammatory, and mitochondrial damage. Unfortunately, treatments for HIRI are based on supportive therapy, and no specific drugs or methods are currently available. Chlorogenic acid (CGA) is a dietary polyphenol with a wide range of pharmacological effects and it has a protective effect on HIRI; however, its specific mechanism remains unclear. In this study, we investigated that CGA pretreatment exerts protective effects against HIRI and the potential underlying mechanisms. We found that CGA pretreatment reduced ALT, AST, MDA, TNF-α, and IL-1β levels following HIRI, improved SOD and GSH levels, and alleviated pathological liver tissue damage, with the highest CGA dose (100 mg/kg.d) exerted the strongest effect. In addition, we showed that CGA pretreatment significantly decreased the levels of reactive oxygen species following HIRI, inhibited HMGB1 release by decreasing IRF-1 expression, inhibited the expression of HMGB1, TLR-4, MyD88, P-IκB-α, NF-κB P65, and P-P65, and promoted IκB-α degradation. Thus, CGA appears to inhibit oxidative stress and inflammatory responses during HIRI. Furthermore, we found that CGA pretreatment reduced hepatocyte apoptosis following HIRI, alleviated mitochondrial damage, promoted BCL-2 expression, inhibited Bax upregulation, and inhibited cytochrome C release to prevent caspase activation, thereby reducing the expression of the caspase-independent pathway components, ENDOG and AIF. Together, our findings suggest that CGA can protect against HIRI by inhibiting oxidative stress, the HMGB1/TLR-4/NF-κB signaling pathway–mediated inflammatory responses, and mitochondria-mediated apoptosis. Thus, CGA appears to be a promising therapeutic approach for treating HIRI.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Hepatic ischemia–reperfusion injury (HIRI) occurs during liver transplantation, liver resection, and other liver surgeries, and is an important cause of early transplantation failure, tissue injury, organ rejection, and even liver failure [1]. The complex pathological process of HIRI is not well studied, and include oxidative stress, aseptic inflammatory, and apoptosis [2]. However, no pharmacological approaches have been approved for the prevention or treatment of HIRI [3], so it remains a significant challenge for clinicians.
Although multiple studies have investigated the potential mechanisms of HIRI, they currently remain unclear. Reactive oxygen species (ROS) is one of the most prominent factors of HIRI causing liver injury and is mainly produced by Kupffer cells and mitochondria during ischemia–reperfusion (I/R) [4]. Glycogen depletion and ATP deficiency during ischemia does not pose a risk to the liver; however, the acute and massive ROS release, as well as inflammation that occur during reperfusion can cause extensive hepatocellular injury and necrosis [4, 5]. Studies have shown that the scavenging of excessive ROS will be beneficial to mitigate the sterile inflammatory response and apoptosis [6, 7]. Therefore, excessive ROS production is one of the earliest and most important factors that contribute toward liver damage during HIRI.
Numerous studies have shown that ROS is associated with the early production of endogenous damage-associated molecular patterns (DAMPs); high mobility group box 1 (HMGB1), one of the most studied DAMPs, mediates the complex aseptic inflammatory response [8]. HMGB1 normally binds to nuclear DNA in its deacetylated form; however, during I/R, ROS promote the expression of IRF-1, an early response transcription factor that can enhance histone acetyl transferase activity and promote the acetylation of HMGB1. Acetylated (Ac)-HMGB1 then dissociates from nuclear DNA and enters the cytoplasm, where intracellular Ca2+ can promote the loading of HMGB1 into lysosomes for extracellular secretion [9,10,11]. Ac-HMGB1 mainly recognizes and binds to the Toll-like receptor 4 (TLR-4) receptor which then activates nuclear factor-κB (NF-κB) to mediate sterile inflammatory responses [12]. In addition, accumulating evidence has suggested that both inflammatory responses and I/R injury are attenuated in the absence of TLR-4 [13]. Therefore, HMGB1/TLR-4 pathway blockade could be a therapeutic target for treating HIRI.
Recent evidence has indicated that apoptosis is the main mode of I/R-induced hepatocyte death and that mitochondrial function exerts a key role in HIRI development [14, 15]. Animal studies have also shown that infusion with active mitochondria before reperfusion can significantly reduce HIRI [16] and that inhibiting apoptosis can reduce the degree of IRI in the liver and heart [17]. ROS-induced oxidative stress plays a key role in cell apoptosis and promotes mitochondrial damage and apoptosis signaling pathways during HIRI, especially in the early stages [18]. Excessive ROS accumulation in the liver after I/R can lead to mitochondrial crest loss, decreased mitochondrial membrane potential, and disrupted the balance of pro-apoptotic (Bad, Bax, and Bid) and anti-apoptotic (BCL-2 and BCL-XL), enhanced mitochondrial permeability transition pore opening, causing cytochrome C (Cyt-c) spillover, and initiation of endogenous apoptosis [19, 20]. Several drugs have recently been reported to reduce ROS induction by HIRI, thereby reducing hepatocyte apoptosis [21,22,23]. Consequently, ROS links and integrates the complex inflammatory response and mitochondrial damage-mediated apoptosis, and provides a new avenue for its prevention and treatment.
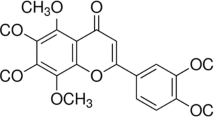
Traditional Chinese medicines have been used for many years and have a wide range of application, low toxicity, and few side effects. However, there has been no breakthrough in the protective effect of drug pretreatment for HIRI, thus it is important to develop herbal medicines or monomers with protective effects against HIRI. Chlorogenic acid (CGA), also known as 5-O-caffeoylquinic acid (Fig. S1), is widely found in fruits, vegetables, and spices, and is one of the most abundant and powerful polyphenol compounds in the human diet [24]. It has many important effects such as antioxidant, antibacterial, hepatoprotective, cardioprotective, and anti-inflammatory properties [25]. CGA ameliorates alcohol-induced liver injury by scavenging ROS and inhibits CCl4-induced liver fibrosis in rats, potentially by being related to TLR-4/MyD88/NF-κB signaling pathway inhibition [26, 27]. Furthermore, CGA has demonstrated protective effects against oxidative stress, inflammation, and apoptosis in methotrexate-induced rat liver injury [28]. Since CGA can protect against liver injury via mechanisms related to ROS removal, the inhibition of inflammation, and apoptosis, we aimed to investigate the protective effects of CGA against HIRI as well as its potential underlying mechanisms to identify potential new targets for the prevention and treatment of HIRI.
MATERIALS AND METHODS
Animals
Forty male Sprague-Dawley rats (160–200 g; SPF grade) were purchased (Tianqin, Changsha, China) and housed in the animal center (Zunyi Medical University, Zunyi, China) under an ambient temperature of 21—27 °C at 50% humidity. All rat experiments were approved by the Laboratory Animal Welfare & Ethics Committee of Zunyi Medical University (approval no: KLLY(A)-2020-029).
Animal Grouping and Pretreatment
CGA was obtained from Huamike Biological Co., Ltd (Beijing, China; purity > 98%). The rats were randomly assigned to five groups: the sham-operated group (Sham, n = 8), I/R + low-dose CGA group (I/R + CGA-L, n = 8), I/R + medium-dose CGA group (I/R + CGA-M, n = 8), I/R + high-dose CGA group (I/R + CGA-H, n = 8), and the ischemia–reperfusion group (I/R, n = 8). The I/R + CGA-L, I/R + CGA-M, and I/R + CGA-H groups were administered 20 mL/kg.d CGA by gavage at concentrations of 25, 50, and 100 mg/kg.d for 10 days, respectively, as reported by previous studies [29, 30]. The Sham and I/R groups were administered 20 mL/kg.d normal saline by gavage for 10 days.
Rat HIRI Model
The rat HIRI model was established as described previously [31]. The rats were anesthetized by intraperitoneal injection of 1% pentobarbital (50 mg/kg, obtained from the Affiliated Hospital of Zunyi Medical University), and all groups, except for the Sham group, were dissected at the hepatoduodenal ligament, then, non-invasive vascular clips were used to block the vessels and bile ducts leading to the middle and left lobes of the liver, thus causing 70% of the liver ischemia. Thereafter, the ischemic liver lobes changed color from bright red (Figure S2A) to grayish white (Fig. S2B, C). The vascular clips were released after 1 h and the liver was reperfused for 4 h (I/R treatment times were based on the method of Gao W et al. [32]), and the color gradually changed from gray to bright red (Fig. S2D), indicating a successful I/R model. In the Sham group, the hepatoduodenal ligament was dissected but hepatic blood flow was not blocked.
Cell Culture and Transfection
Normal human hepatocytes (HL7702, L02) were obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai, China), thawed, and cultured in RPMI-1640 medium containing 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum at 37 °C in an incubator with 5% CO2. Some of the cells were inoculated in six-well plates (1 × 105 cells/well) and cultured in complete RPMI-1640 medium. Upon reaching 50% confluence, L02 cells were transfected for 48 h with TLR-4 overexpression plasmids (NMID: NM_138554.1; HANBIO, Shanghai, China; MOI = 20) and empty plasmids using lentivirus; the transfection efficiency was increased using polybrene (6 μg/mL).
Cell Proliferation Assay
The effects of CGA on L02 cells cytotoxicity and proliferation were detected using the Cell Counting Kit-8 assay kit (CCK-8; Dojindo, Beijing, China), according to the manufacturer’s instructions. The L02 cells were cultured overnight in 96-well plates (5 × 103 cells/well). The following day, the cells were washed and replaced with medium containing different CGA concentrations and incubation was continued for 48 h. Thereafter, 10 μL of CCK8 reagent was added to each well and incubated at 37 °C for 2 h. Lastly, the absorbance at 450 nm was detected by an absorbance reader (Molecular Devices, CA, USA).
Cell Grouping and Pretreatment
The cells were divided into 5 groups upon successful transfection of the TLR-4 overexpression plasmid: a normal L02 cell group (NC), a TLR-4 overexpression + CGA group (OE+CGA), a TLR-4 overexpression group (OE), a hypoxia/reoxygenation + CGA group (H/R+CGA), and a hypoxia/reoxygenation (H/R) group. The NC, OE and H/R groups were given complete medium, while the OE+CGA and H/R+CGA groups were given a mixture of CGA and complete medium, and they were incubated for 48 h at 37 °C with 5% CO2. On the third day, the H/R+CGA and H/R groups underwent hypoxia preadaptation for 6 h in a three-gas hypoxia incubator (Thermo, MA, USA) with N2 (94%), O2 (1%), and CO2 (5%) in sugar-free Dulbecco’s modified Eagle medium. Then the cells were replaced with complete RPMI-1640 medium and cultured in a 5% CO2 incubator at 37 ℃ for a further 12 h. The remaining 3 groups were simultaneously replaced with sugar-free Dulbecco’s modified Eagle medium and cultured in a 37 °C incubator with 5% CO2 for 6 h. Thereafter, they were replaced with complete medium, and incubation was continued for 12 h. Hypoxia (6 h) and reoxygenation (12 h) treatment times were based on the method of Huang et al. [2].
Sample Collection
After liver reperfusion, the inferior vena cava of each rat was dissected and exposed and 4–6 ml blood was extracted. The serum was collected after blood coagulation, centrifuged for 10 min at 3000 rpm, and stored in a refrigerator at −80 °C for liver function analysis. Left liver lobe samples were taken uniformly from all rats and each sample was divided into several parts. The first part (0.5 cm × 0.5 cm) was stored in 10% formalin and sent to the Department of Pathology, Affiliated Hospital of Zunyi Medical University, where it was embedded in paraffin and cut into 5 μm-thick sections for TUNEL and hematoxylin-eosin (H&E) staining. The second part (0.5 cm × 0.5 cm) was cut into 10 μm-thick frozen sections for dihydroethidium (DHE) staining. The third part (0.1 g) was added to 1 mL of 0.9% saline, pulverized three times using a Xinyi-96 high-flux tissue grinder (Ningbo, China; 1500 rpm, 20 s). The ground liver tissue was allowed to stand 30 min and centrifuged for 10 min at 13,300 rpm, after which the supernatant was collected to obtain a 10% liver homogenate. The remaining part was stored at −80°C until analyzed.
Determination of Liver Function and Detection of Oxidative Stress and Inflammatory Markers
The serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were analyzed by an AU5800 biochemical analyzer (Beckman Coulter, CA, USA). Superoxide dismutase (SOD), malondialdehyde (MDA) and glutathione (GSH) levels in liver homogenates, which are both oxidative stress markers, were measured using SOD, MDA and GSH kits (Njjcbio, Nanjing, China) according to the manufacturer’s instructions. The liver homogenate levels of the inflammatory markers HMGB1, interleukin-1β (IL-1β), and tumor necrosis factor alpha (TNF-α) were detected by enzyme-linked immunosorbent assay kits (LIANKE, Hangzhou, China) according to the manufacturer’s instructions.
Histopathological Analysis
To evaluate liver tissue damage, paraffin sections were made and H&E staining was performed. The stained pathology sections were dried and examined by two experienced pathologists using a light microscope (Leica, Germany). According to Suzuki’s [33] criteria, histopathological scores (0–4) were given for each field: 0 = Normal hepatocytes; 1 = individual cellular edema, vacuolization, or single cellular necrosis; 2 = little cellular edema, vacuolization, or necrosis ≤ 30% ; 3 = moderate cellular edema, vacuolization, or necrosis ≤ 60%; and 4 = severe edema, vacuolization and necrosis > 60%.
Real-Time Quantitative PCR (RT-qPCR) Assays
Total RNA in liver specimens or cells were extracted using RNAiso Plus (TAKARA, Tokyo, Japan), quantified, and reverse-transcribed into cDNA. Target mRNA was amplified using standard two-step PCR and a reaction volume of 15 µL (including 7.5 μl TB Green Premix Ex Taq II, 1 μl specific forward primer, 1 μl specific reverse primer, 3 μl cDNA, and 2.5 μl sterile water). Thereafter, the 2−ΔΔCq method was used to measure the gene relative expression levels and β-actin was used for normalization. Primer sequences for the target genes are shown in Table S1.
Western Blotting Assays
The total protein was extracted from the collected cells or liver tissue samples using a lysis solution (RIPA buffer; Solarbio, Beijing, China), including protease inhibitors and phosphatase inhibitors, and then quantified using a BCA protein assay kit (Solarbio), according to the manufacturer’s instructions. Total proteins were separated by sodium dodecyl sulfate-polyacrylamide gels (8–15%) and transferred to polyvinylidene fluoride membranes. Non-specific binding was blocked using 5% non-fat milk (phosphorylated protein using 5% fetal bovine serum) for at least 1 h under room temperature. Next, the membranes and diluted primary antibodies were incubated overnight at 4 °C. The following day, the membranes were incubated with secondary antibodies (Proteintech, Wuhan, China; 1:2000) for at least 1 h at room temperature. The membranes were exposed to super sensitive ECL luminescence reagent (Meilunbio, Dalian, China) with an imaging system (Bio-Rad, CA, USA). The protein bands were analyzed, and their grayscale values were counted using Image Lab software (v.5.2; Bio-Rad). Details of the primary antibodies, including their source and working dilutions, are presented in Table S2.
Immunoprecipitation (IP) Assays
The total protein was extracted by the same method as mentioned in the “Western Blotting Assays” section. Then, 1mg of the protein sample was mixed with 1 µg goat normal IgG and 20 µL of Protein A+G Agarose (Beyotime, Shanghai, China) and shaken slowly for 1.5 h at 4 °C. Thereafter, the supernatant was collected after centrifugation for 5 min at 2500 rpm to remove nonspecific binding; simultaneously, the goat normal IgG was used as a negative control. 1.5 µg of HMGB1 primary antibodies were added to each supernatant at 4 °C and shaken slowly overnight. The next day, another 40 µL Protein A+G Agarose was added, incubated for 2 h at 4 °C and centrifuged instantaneously at 13,300 rpm. After five washes with PBS, the supernatant was removed, and the sample was analyzed using western blotting.
Fluorescence Analysis
ROS in the frozen liver sections was labeled with DHE (Beyotime), which produces red fluorescence. Apoptotic hepatocytes were detected using a mitochondrial membrane potential and apoptosis detection kit (Beyotime), according to the manufacturer’s instructions. Live cells with intact mitochondrial membrane potential were labeled with red fluorescence using MitoTracker Red CMXRos and apoptotic cells were labeled with green fluorescence using Annexin V- Fluorescein isothiocyanate (FITC). Nuclei were stained with blue fluorescence using 4,6-diamidino-2-phenylindole (DAPI) or Hoechst 33342. All steps were performed according to the instructions provided by the supplier. Next, the stained sections or cells were photographed under a fluorescent microscope (Olympus, Tokyo, Japan) and analyzed using Image-Pro Plus software (v.6.0; Bio-Rad).
TUNEL Assays
A one-step TUNEL assay kit with FITC (Beyotime) was used to detect apoptosis in liver tissue. The slides were dewaxed and washed three times with PBS, incubated with proteinase K at 37 ℃ for 25 min, and then incubated with TUNEL reaction solution for 1 h in the dark. Next, 1 drop of anti-fluorescence quencher containing DAPI was added, and after 10 min the coverslip was covered and observed under a fluorescence microscope (Olympus). The fluorescence pictures were analyzed using Image-Pro Plus software (v.6.0; Bio-Rad).
Statistical Analysis
All results were shown as means ± standard error (SEM) and were analyzed using SPSS (v.22) or GraphPad Prism software (v.8). One-way ANOVA was used for the comparison of data between groups conforming to normal distribution. Statistical significance was defined as p < 0.05. Since the histopathological grading data were not normally distributed, nonparametric comparisons were performed using the Kruskal–Wallis test. The level of significance for two-way comparisons between groups was defined as p < 0.005.
RESULTS
CGA improved liver function, oxidative stress, inflammation levels, and liver histopathology injury in rats
To demonstrate whether different concentrations of CGA pretreatment had a protective effect on HIRI and to obtain the optimal CGA concentration, we examined the levels of liver function, oxidative stress markers, inflammation factors, and the degree of liver histopathological damage in rats (Fig. 1). First, we assessed the effect of CGA on liver function by measuring its serum biomarker levels (Fig. 1A). The serum levels of ALT and AST were significantly increased after I/R injury compared with that of the Sham group (p < 0.01). In addition, compared with that of the I/R group, the serum levels of AST were significantly decreased after pretreatment with all doses of CGA; however, only medium and high doses of CGA pretreatment could decrease the ALT levels (p < 0.05). Therefore, CGA pretreatment could reverse liver function injury after HIRI and the most significant decrease was observed in the I/R+CGA-H group (p < 0.05).

CGA pretreatment ameliorated liver function, oxidative stress, inflammation levels and the degree of liver histopathological damage in rats.
To examine whether CGA affected oxidative stress in the liver, we measured liver homogenate levels of MDA, SOD and GSH (Fig. 1B). After HIRI, MDA levels increased, and SOD, GSH levels decreased significantly compared with that of the Sham group (p < 0.05). However, after CGA pretreatment, MDA levels decreased and SOD, GSH levels significantly increased compared to that of the I/R group (p < 0.05). This indicates that CGA pretreatment could reduce oxidative stress in the liver and this effect was most significant in the I/R + CGA-H group (p < 0.01).
Next, we investigated the effect of CGA on liver inflammation by measuring the liver homogenate levels of HMGB1, TNF-α, and IL-1β (Fig. 1C). The levels of HMGB1, TNF-α, and IL-1β were significantly increased after HIRI compared to that of the Sham group (p < 0.05). In particular, compared to that of the I/R group, HMGB1 levels were significantly decreased after all doses of CGA pretreatment (p < 0.05); however, only low and high doses of CGA pretreatment resulted in a significant decrease in IL-1β levels (p < 0.01), while only the medium and high doses of CGA pretreatment resulted in a significant decrease in TNF-α levels (p < 0.01). Therefore, CGA pretreatment could significantly decrease aseptic inflammation caused by HIRI and the most significant decrease was observed in the I/R + CGA-H group (p < 0.01).
Later, to investigate the histopathological effect of CGA on the liver, we performed H&E staining on liver tissues (Fig. 1D). The results revealed that hepatocytes had a normal pathological structure in the Sham group (Fig. 1D: a, f); however, those in the I/R group displayed hepatocyte edema, disorganized hepatic cord structure, vacuolation, lamellar necrosis, hemorrhage, and inflammatory cell infiltration (Fig. 1D: e, j). In contrast, hepatocellular lesions were reduced in the CGA groups (Fig. 1D: b–d, g–i). Kruskal–Wallis analyses of the histopathological scores between the different groups revealed that the low and medium CGA doses and I/R groups scored higher than the Sham group and that only the I/R + CGA-H group had a lower score compared to that of the I/R group (p < 0.005; Fig. 1E).
Since the highest CGA dose (100 mg/kg.d) exerted the strongest effect, it was used for subsequent experiments.
CGA Reduced Liver ROS Levels in Rats
To examine the effect of CGA on cellular ROS levels in rat liver tissue, we stained frozen liver sections from each group with a DHE fluorescent probe and DAPI (Fig. 2A, B). Interestingly, compared to that of the Sham group, DHE fluorescence intensity significantly increased in rats subjected to HIRI (p < 0.05) and decreased after CGA pretreatment, compared to that in the I/R group (p < 0.01). The results show that CGA could inhibit ROS production during HIRI in rats, which may in turn reduce oxidative stress-induced liver injury.

CGA pretreatment reduced ROS levels in rats.
CGA Reduced the Active Secretion of HMGB1 In Vivo
The expression of IRF-1 protein in the liver tissue of rats in each group was detected by western blotting, and Ac-HMGB1 expression in the liver tissue of rats in each group was detected by IP combined with western blotting. The results showed that IRF-1 and Ac-HMGB1 expression levels in the liver tissue of the I/R+CGA and I/R groups were increased compared with that of the Sham group (p < 0.05). However, the expression levels of IRF-1 and Ac-HMGB1 in the I/R+CGA group were significantly decreased compared with that of the I/R group (p < 0.01, Fig. 3A, B). The results suggest that CGA can effectively inhibit the active secretion of HMGB1 during HIRI in rats.

CGA attenuates the expression levels of inflammatory pathway related proteins in rats.
CGA Decreased the Levels of HMGB1/TLR-4/NF-κB Signaling Pathway Components In Vivo
RT-qPCR analysis revealed that the mRNA levels of HMGB1 and TLR-4 in the I/R+CGA group were significantly lower than those in the I/R group (p < 0.01), among which the TLR-4 mRNA levels were not significantly different from that in the Sham group (p > 0.05, Fig. 3C). The results suggest that CGA could inhibit the expression of HMGB1 and its downstream inflammatory pathway receptor, TLR-4, at the transcriptional level during HIRI in rats.
Next, to further determine the mechanism underlying the observed effects of CGA in vivo, we examined the expression of the TLR-4/NF-κB signaling pathway components in the liver tissues of each group (Fig. 3D). Compared with that of the Sham group, the protein expression of TLR-4, MyD88, P65, P-P65, P-IκB-α, IL-1β, and TNF-α was significantly increased after HIRI (p < 0.05), whereas the protein expression of IκB-α decreased. Moreover, the expression of TLR-4, MyD88, P65, P-P65, P-IκB-α, IL-1β, and TNF-α was significantly decreased after CGA pretreatment (p < 0.01) and the expression of IκB-α was significantly increased compared to that of the I/R group (p < 0.01). The results show that CGA could inhibit the NF-κB signaling pathway mediated by TLR-4, and thus reduce the sterile inflammatory response during HIRI in rats.
CGA Attenuated Apoptosis in Rat Liver Tissue
First, we examined the effect of CGA on apoptosis by performing TUNEL staining on liver tissues (Fig. 4A). The results showed that apoptotic cells were significantly increased after I/R, compared to that of the Sham group, but were significantly reduced after CGA pretreatment (p < 0.01). We observed apoptotic cells after HIRI; however, it is unknown whether this is due to a mitochondria-mediated apoptotic pathway? Therefore, we further examined mitochondrial apoptosis pathway-related proteins to prove our hypothesis (Fig. 4B). Compared to that of the Sham group, BCL-2 expression significantly decreased after HIRI (p < 0.01), and its expression significantly increased after CGA pretreatment compared to that of the I/R group (p < 0.05). Conversely, Bax, Cyt-c, cleaved-caspase9, cleaved-caspase3, ENDOG, and AIF expression significantly increased after I/R injury compared to that of the Sham group (p < 0.01); however, their expression significantly decreased after CGA pretreatment compared to that of the I/R group (p < 0.05). The results indicate that CGA significantly attenuated the occurrence of hepatocyte apoptosis during HIRI in rats by inhibiting both the mitochondria-mediated caspase-dependent apoptotic and caspase-independent apoptotic pathways. It remains to be further explored in in vitro experiments whether the underlying cause is related to the reduction of hepatocyte mitochondrial damage by CGA.

CGA pretreatment reduced hepatocyte apoptosis and the expression levels of mitochondria-mediated apoptosis pathway related proteins in rats.
CGA Decreased the Levels of HMGB1/TLR-4/NF-κB Signaling Pathway Components In Vitro
L02 cell lines overexpressing stable TLR-4 were screened using lentiviral transfection, and GFP expression was observed under a fluorescence microscope (Fig. 5A). RT-qPCR and western blotting were used to detect TLR-4 expression in the normal L02 cell (NC), empty plasmid transfection (EMT), and TLR-4 overexpression (OE) groups. The results showed that TLR-4 mRNA and protein levels were significantly upregulated in the OE group compared to the NC and EMT groups (p < 0.01), while the results in the EMT and NC groups were not significantly different (P > 0.05, Fig. 5B, C), indicating that the transfection was successful and the empty plasmid had no effect on the results of this experiment.

CGA decreased the levels of TLR-4/NF-κB signaling pathway components in vitro.
CCK8 results showed no toxic effect of 12.5–200 µM CGA on L02 cells, while 12.5–25 µM CGA promoted their proliferation (Fig. 5D). To determine the optimal CGA concentration, cells were divided into NC, EMT, OE, and OE + CGA (12.5, 50, or 200 µM) groups according to the CCK-8 assay results. The OE + CGA groups were administered 12.5, 50, and 200 µM CGA and cultured with TLR-4 overexpressing cells for 48 h. The protein expression of TLR-4 was detected by western blotting, revealing that the TLR-4 expression was significantly lower in the 50 and 200 µM CGA groups than in the OE group (p < 0.01). TLR-4 expression was particularly significant in the CGA 200 μM group (p < 0.01, Fig. 5E); therefore, CGA 200 µM was selected for subsequent experiments.
Next, we measured the protein expression of the TLR-4/NF-κB signaling pathway components in L02 cells (Fig. 5F). Compared to the protein levels in the NC group, TLR-4, MyD88, P65, P-P65 and P-IκB-α protein levels were significantly increased in the OE and H/R groups (p < 0.01), whereas the IκB-α expression was decreased (p < 0.05). Interestingly, these expression patterns were significantly reversed in the OE + CGA and H/R + CGA groups compared to that of the OE and H/R groups (p < 0.05). These results further indicate that CGA could reduce HIRI-induced sterile inflammation by inhibiting the TLR-4/NF-κB signaling pathway.
CGA Reduced ROS Expression, Mitochondrial Damage, and Apoptosis In Vitro
To further prove our hypothesis, we established a hepatocyte hypoxia model for 6 h and reoxygenation model for 12 h to simulate HIRI, and examined ROS levels, mitochondrial damage, and apoptosis related indicators. First, L02 cells were divided into NC, H/R, and H/R+CGA low, medium, and high dose groups, the latter were pretreated with 12.5, 50 and 200 µM CGA for 48 h. The H/R and H/R+CGA groups were established as H/R models, and cleaved-caspase3 protein was detected in each group by western blotting. The results showed that the levels of cleaved-caspase3 protein in the H/R and H/R+CGA groups were significantly higher than those in the NC group (p < 0.05), and the levels of cleaved-caspase3 protein in the H/R+CGA 50 and 200 µM groups were significantly lower than those in the H/R group (p < 0.05), most significantly in the 200 µM group (p < 0.01, Fig. 6A). The results suggest that both medium and high doses of CGA may have protective effects on H/R-induced apoptosis, with the best effect observed at a CGA concentration of 200 µM. Therefore, 200 µM CGA was chosen for subsequent experiments to explore the specific underlying mechanism.

CGA reduced ROS expression, mitochondrial damage, and apoptosis in vitro.
Next, L02 cells were stained with a DCFH-DA fluorescent probe and detected by flow cytometry (Fig. 6B). The ROS fluorescence intensity was higher in the H/R + CGA and H/R groups than in the NC group (p < 0.01). Furthermore, the ROS fluorescence intensity was decreased in the H/R + CGA group compared to that of the H/R group (p < 0.01). The results suggest that CGA inhibits the production of ROS in hepatocytes during H/R.
Next, MitoTracker Red CMXRos and Annexin V-FITC fluorescent probes were used to detect the mitochondrial damage and apoptosis of L02 cells (Fig. 6C). Compared to that of the NC group, MitoTracker fluorescence intensity in the H/R + CGA and H/R groups was significantly lower, whereas the apoptotic cells were significantly higher (p < 0.01). Conversely, MitoTracker fluorescence intensity was increased, and apoptosis was lower in the H/R + CGA group than in the H/R group (p < 0.01). We also detected the expression of mitochondrial apoptosis pathway-related proteins (Fig. 6D), and found that BCL-2 expression was significantly lower in the H/R group than in the NC group (p < 0.05). However, BCL-2 was significantly upregulated in the H/R + CGA group (p < 0.05) compared to that of the H/R group. Compared with that of the NC group, Bax, Cyt-c, cleaved-caspase9 and cleaved- caspase3, ENDOG, and AIF expression was significantly increased in the H/R group (p < 0.05); however, their expression was significantly decreased in the H/R + CGA group compared to that in the H/R group (p < 0.05). The results show that CGA could alleviate hepatocyte mitochondrial damage by reducing ROS production in hepatocytes during HIRI, thereby inhibiting the mitochondria-mediated apoptotic pathway and reducing hepatocyte injury.
DISCUSSION
HIRI is an important cause of liver failure after liver surgery or transplantation; however, its underlying mechanism is highly complex. Kupffer cells are liver macrophages that play an important role in the development of HIRI. During the early stages of reperfusion after ischemia, Kupffer cells alter their metabolic behavior and produce a large amount of ROS [34]. In addition, endogenous antioxidants such as SOD and GSH are inactivated or removed during I/R, leading to reduced ROS scavenging [1]. Increased ROS levels can damage macromolecules and lipids and produce MDA. In addition, large amounts of ROS cause cellular chain reactions leading to inflammatory responses, cell death, and even organ failure [35]. Du Y et al. [36] showed that aloin and γ-oryzanol, which have antioxidant effects, had protective effects against HIRI, and the mechanism was related to ROS scavenging and increasing SOD and GSH levels. CGA also alleviated cerebral I/R injury in a rat model, and its mechanism is linked to an increase in SOD and GSH levels and a decrease in ROS and MDA levels [37]. In this study, we demonstrated that the damage following HIRI is caused by ROS. Notably, serum ALT and AST levels increased, SOD and GSH levels decreased, and ROS and MDA levels increased following HIRI. In addition, liver tissues displayed cellular edema, vacuolation, lamellar necrosis, massive inflammatory cell infiltration, and extensive hemorrhage associated with inflammation and apoptosis. Furthermore, CGA pretreatment decreased the levels of ALT, AST, MDA, and ROS while increasing SOD and GSH levels, as well as alleviating the pathological damage of liver tissue, suggesting that CGA has good antioxidant effects.
In our study, following HIRI, inflammatory markers such as HMGB1, TNF-α, and IL-1β were elevated, and hepatocytes were injured, showing that inflammatory processes were responsible for cellular injury. HMGB1 has been linked to the onset of sterile inflammation in numerous studies [38]. In a sepsis model, CGA also protects mice from sepsis by blocking the release of HMGB1 [39]. In this study, we found that HMGB1 expression was significantly increased in liver tissue after HIRI, and that TNF-α, IL-1β expression was also increased. These effects could be reversed by CGA, suggesting that CGA may have an anti-inflammatory effect in HIRI. As previously mentioned, HMGB1 normally binds to nuclear DNA in its deacetylated form; however, during I/R, it is acetylated (Ac-HMGB1) and secreted extracellularly, and ROS and interferon regulatory factor 1 (IRF-1) are involved in the secretion and translocation of HMGB1. Aucubin was reported to inhibit ROS and IRF-1 expression during HIRI in rats, resulting in reduced levels of HMGB1 and Ac-HMGB1 [23]. In this study, we found that ROS, IRF-1, and Ac-HMGB1 levels were significantly increased in the rat after HIRI, and their levels were significantly decreased following CGA pretreatment. This suggests that CGA can protect against HIRI by inhibiting the ROS/IRF-1 pathway and thus reducing the early active secretion of HMGB1.
Most studies have shown that HMGB1 promotes inflammation by binding to TLR-4 receptors and activating downstream pathways [40, 41]. Moreover, the TLR-4/NF-κB signaling pathway-mediated inflammation is vital in the development of HIRI [42]. In this process, activated TLR-4 receptor binds to the myeloid differentiation factor 88 (MyD88) adaptor protein, which in turn activates the nuclear factor kappa-B P65 (NF-κB P65, P65) signaling pathway and induces the expression of inflammatory factors such as IL-1β and TNF-α [43]. Furthermore, the blockade of TLR-4 expression may help alleviate HIRI. In this study, we showed that the protein levels of the TLR-4/NF-κB pathway (TLR-4, MyD88, P65, P-P65, P-IκB-α, TNF-α, and IL-1β) increased following HIRI in the rat model, whereas the IκB-α level decreased. However, CGA pretreatment was able to reverse these effects, indicating that CGA may act via the TLR-4/NF-κB pathway. To verify this result, we examined the effect of L02 cells overexpressing TLR-4 and L02 cells subjected to H/R to simulate HIRI. Both TLR-4 overexpression and H/R injury increased TLR-4, MyD88, P65, P-P65, and P-IκB-α levels and decreased IκB-α levels, whereas CGA pretreatment decreased TLR-4, MyD88, P65, P-P65, and P-IκB-α levels, and increased IκB-α levels, suggesting that CGA can protect against HIRI by alleviating the inflammatory response via the HMGB1/TLR-4/NF-κB axis.
Recent evidence has indicated that apoptosis is the main mode of I/R-induced hepatocyte death [14, 44]. ROS-induced oxidative stress plays a key role in cell apoptosis and promotes mitochondrial damage. In this research, we discovered a large rise in ROS following HIRI, as well as a corresponding increase in apoptotic cells in both in vivo and in vitro experiments, and a significant decrease after pretreatment with CGA, suggesting that CGA alleviated hepatocyte apoptosis. Recent studies have revealed that mitochondrial function plays a significant role in HIRI development [15, 45]. In addition, animal studies have shown that infusing active mitochondria before reperfusion can dramatically reduce HIRI and that blocking apoptosis can minimize the degree of I/R injury in the liver and heart [16, 17, 46]. In our research, we found that after H/R, mitochondria were greatly reduced and apoptotic cells increased, which was reversed by CGA pretreatment, implying that CGA may protect against HIRI by reducing mitochondrial damage. Furthermore, the BCL-2 protein family regulates the intrinsic apoptosis pathway, with certain members being pro-apoptotic (Bad and Bax) and others being anti-apoptotic (BCL-2 and BCL-XL). The susceptibility of cells to apoptotic stimuli is determined by the balance of these proteins [20]. Chitosan pretreatment was found to protect rats from HIRI by increasing BCL-2 expression while decreasing Bax expression [47]. In addition, Bax knockout mice livers have better tolerance to I/R injury [48]. In this study, we found that after HIRI, BCL-2 expression decreased and ROS and Bax increased in vivo and in vitro, whereas CGA pretreatment reversed this effect. This suggests that CGA protects mitochondria by reducing ROS production as well as affecting BCL-2 and Bax expression.
Mitochondrial damage occurs when mitochondria are continuously stimulated by ROS and pro-inflammatory factors, thereby disrupting the balance between proapoptotic and antiapoptotic factors. Pro-apoptotic proteins initiate apoptosis by forming mitochondrial outer membrane permeability complexes that release Cytochrome C (Cyt-c) into the cytoplasm [49]. This then activates the caspase cascade to generate apoptosis bodies and initiate endogenous apoptosis. Several studies have recently shown that inhibiting Cyt-c spillover and caspase activation could help alleviate HIRI [50, 51]. In our study, we found that HIRI boosted Cyt-c overflow and activated downstream caspases (caspase9, cleaved-caspase9, caspase3, cleaved-caspase3). CGA pretreatment decreased Cyt-c spillover and reduced caspase activation, thereby alleviating hepatocyte apoptosis caused by the mitochondria-mediated caspase-dependent pathway in vivo and in vitro. Mitochondria also mediate the caspase-independent apoptosis pathway and play an important role in the mitochondrial apoptosis pathway, including ENDOG and AIF, which can cause chromatin DNA cleavage, chromatin condensation, and DNA degradation [52, 53]. Recently, in a model of cerebral I/R injury, the Gualou Guizhi decoction, an herbal medicine, could protect against brain injury by inhibiting the expression of ENDOG and AIF. In our study, we found that ENDOG and AIF were upregulated after HIRI but downregulated after CGA pretreatment in vivo and in vitro, thereby alleviating hepatocyte apoptosis.
Together, we have proved that oxidative stress, aseptic inflammation and mitochondria mediated apoptosis are the causes of HIRI and the protective effect of CGA on HIRI in vivo and in vitro. However, there are still shortcomings in our experiments, such as not detecting the levels of IRF-1 and AC-HMGB1 at the cellular level, and not upregulating the mitochondrial apoptosis pathway by the over-expression of agonists or key proteins. These observations provide a comprehensive overview of the protective mechanism of CGA against HIRI and suggest that CGA could be used to treat HIRI; however, further studies are warranted to elucidate the detailed underlying mechanism.
CONCLUSION
In conclusion, the findings of this study demonstrate that CGA can decrease HIRI (Fig. 7) by (1) reducing liver transaminase activity and histopathological changes in the liver; (2) inhibiting oxidative stress; (3) reducing the active secretion of HMGB1 and regulating TLR-4/NF-κB signaling pathway to reduce the expression of inflammatory factors; and (4) inhibiting the mitochondria-mediated apoptosis pathway.

A potential mechanism model for CGA regulation of oxidative stress, inflammatory response and mitochondria-mediated apoptosis in HIRI.
Availability of Data and Materials
The experimental data used to support this study can be provided upon request.
Abbreviations
- AIF:
-
Apoptosis inducing factor
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- Bax:
-
BCL2-Associated X
- BCL-2:
-
B-cell lymphoma-2
- CGA:
-
Chlorogenic acid
- Cyt-c:
-
Cytochrome C
- ENDOG:
-
Endonuclease G
- GSH:
-
Glutathione
- HIRI:
-
Hepatic ischemia–reperfusion injury
- HMGB1:
-
High mobility group protein B1
- IL-1β:
-
Interleukin-1β
- IRF-1:
-
Interferon regulatory factors 1
- IκB-α:
-
I kappa B alpha
- MDA:
-
Malondialdehyde
- MyD88:
-
Myeloid differentiation factor 88
- NF-κB P65:
-
Nuclear factor kappa-B P65
- P-IκB-α:
-
Phospho-I kappa B alpha
- P-P65:
-
Phospho nuclear factor kappa-B P65
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
- TLR-4:
-
Toll-like receptors 4
- TNF-α:
-
Tumor necrosis factor alpha
References
Li, J., R.J. Li, G.Y. Lv, et al. 2015. The mechanisms and strategies to protect from hepatic ischemia-reperfusion injury. European Review for Medical and Pharmacological Sciences 19: 2036–2047.
Huang, Z., T. Mou, Y. Luo, et al. 2020. Inhibition of miR-450b-5p ameliorates hepatic ischemia/reperfusion injury via targeting CRYAB. Cell Death & Disease 11: 455.
Ricca, L., A. Lemoine, F. Cauchy, et al. 2015. Ischemic postconditioning of the liver graft in adult liver transplantation. Transplantation 99: 1633–1643.
Elias-Miró, M., M. Jiménez-Castro, J. Rodés, et al. 2013. Current knowledge on oxidative stress in hepatic ischemia/reperfusion. Free radical research 47: 555–568.
Zheng, J., L. Chen, T. Lu, et al. 2020. MSCs ameliorate hepatocellular apoptosis mediated by PINK1-dependent mitophagy in liver ischemia/reperfusion injury through AMPKα activation. Cell death & disease 11: 256.
Choudhury, S., S. Ghosh, S. Mukherjee, et al. 2016. Pomegranate protects against arsenic-induced p53-dependent ROS-mediated inflammation and apoptosis in liver cells. The Journal of nutritional biochemistry 38: 25–40.
Zhang, G.Y., D. Lu, S.F. Duan, et al. 2018. Hydrogen sulfide alleviates lipopolysaccharide-induced diaphragm dysfunction in rats by reducing apoptosis and inflammation through ROS/MAPK and TLR4/NF-B signaling pathways. Oxidative Medicine and Cellular Longevity 2018: 9647809.
Liu, Y., L. Yang, K. Tao, et al. 2014. Protective effects of hydrogen enriched saline on liver ischemia reperfusion injury by reducing oxidative stress and HMGB1 release. BMC Gastroenterology 14: 12.
Dhupar, R., J. Klune, J. Evankovich, et al. 2011. Interferon regulatory factor 1 mediates acetylation and release of high mobility group box 1 from hepatocytes during murine liver ischemia-reperfusion injury. Shock 35: 293–301.
Doyle, K., and F.A. Fitzpatrick. 2010. Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function. The Journal of biological chemistry 285: 17417–17424.
Tsung, A., J. Klune, X. Zhang, et al. 2007. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. The Journal of experimental medicin 204: 2913–2923.
Ai, M., S. Lin, M. Zhang, et al. 2021. Cirsilineol attenuates LPS-induced inflammation in both in vivo and in vitro models via inhibiting TLR-4/NFkB/IKK signaling pathway. Journal of Biochemical and Molecular Toxicology 35: e22799.
Ben-Ari, Z., O. Avlas, R. Fallach, et al. 2012. Ischemia and reperfusion liver injury is reduced in the absence of Toll-like receptor 4. Cellular physiology and biochemistry 30: 489–498.
Wu, B., W. Qiu, P. Wang, et al. 2007. p53 independent induction of PUMA mediates intestinal apoptosis in response to ischaemia-reperfusion. Gut 56: 645–654.
Yamada, Y., K. Nakamura, J. Abe, et al. 2015. Mitochondrial delivery of Coenzyme Q10 via systemic administration using a MITO-Porter prevents ischemia/reperfusion injury in the mouse liver. Journal of controlled release 213: 86–95.
Lin, H., S. Liu, H. Lai, et al. 2013. Isolated mitochondria infusion mitigates ischemia-reperfusion injury of the liver in rats. Shock 39: 304–310.
Zhang, S., S. Jiang, H. Wang, et al. 2018. SIRT6 protects against hepatic ischemia/reperfusion injury by inhibiting apoptosis and autophagy related cell death. Free Radical Biology & Medicine 115: 18–30.
Casillas-Ramírez, A., I. Mosbah, F. Ramalho, et al. 2006. Past and future approaches to ischemia-reperfusion lesion associated with liver transplantation. Life sciences 79: 1881–1894.
Zhang, T., J. Gu, J. Guo, et al. 2019. Renalase attenuates mouse fatty liver ischemia/reperfusion injury through mitigating oxidative stress and mitochondrial damage via activating SIRT1. Oxidative Medicine and Cellular Longevity 2019: 7534285.
Siddiqui, W., A. Ahad, and H. Ahsan. 2015. The mystery of BCL2 family: Bcl-2 proteins and apoptosis: An update. Archives of toxicology 89: 289–317.
Wang, H., L. Guo, Y. Wang, et al. 2021. Isoflurane upregulates microRNA-9–3p to protect rats from hepatic ischemia-reperfusion injury through inhibiting fibronectin type III domain containing 3B. Cell cycle 1–13.
Zhou, Y., Z. Tan, H. Huang, et al. 2021. Baicalein pre-treatment alleviates hepatic ischemia/reperfusion injury in mice by regulating the Nrf2/ARE pathway. Experimental and therapeutic medicine 22: 1380.
Zhang, S., Z. Feng, W. Gao, et al. 2020. Aucubin attenuates liver ischemia-reperfusion injury by inhibiting the HMGB1/TLR-4/NF-κB signaling pathway, oxidative stress, and apoptosis. Frontiers in pharmacology 11: 544124.
Lu, H., Z. Tian, Y. Cui, et al. 2020. Chlorogenic acid: A comprehensive review of the dietary sources, processing effects, bioavailability, beneficial properties, mechanisms of action, and future directions. Comprehensive reviews in food science and food safety 19: 3130–3158.
Naveed, M., V. Hejazi, M. Abbas, et al. 2018. Chlorogenic acid (CGA): a pharmacological review and call for further research. Biomedicine & pharmacotherapy 97: 67–74.
Kim, H., J. Pan, S. Kim, et al. 2018. Chlorogenic acid ameliorates alcohol-induced liver injuries through scavenging reactive oxygen species. Biochimie 150: 131–138.
Shi, H., L. Dong, J. Jiang, et al. 2013. Chlorogenic acid reduces liver inflammation and fibrosis through inhibition of toll-like receptor 4 signaling pathway. Toxicology 303: 107–114.
Ali, N., S. Rashid, S. Nafees, et al. 2017. Protective effect of chlorogenic acid against methotrexate induced oxidative stress, inflammation and apoptosis in rat liver: An experimental approach. Chemico-biological interactions 272: 80–91.
Shi, H., A. Shi, L. Dong, et al. 2016. Chlorogenic acid protects against liver fibrosis in vivo and in vitro through inhibition of oxidative stress. Clinical nutrition 35: 1366–1373.
Wu, D., C. Bao, L. Li, et al. 2015. Chlorogenic acid protects against cholestatic liver injury in rats. Journal of pharmacological sciences 129: 177–182.
Yang, J., R. Wu, X. Qiang, et al. 2009. Human adrenomedullin and its binding protein attenuate organ injury and reduce mortality after hepatic ischemia-reperfusion. Annals of surgery 249: 310–317.
Gao, W., Z. Feng, S. Zhang, et al. 2020. Eucommia ulmoides anti-inflammatory and antioxidant effect of polysaccharide in hepatic ischemia-reperfusion injury by regulating ROS and the TLR-4-NF-B PATHWAY. BioMed research international 2020: 1860637.
Suzuki S, L. Toledo-Pereyra, F. Rodriguez, et al. 1993. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury. Modulating effects of FK506 and cyclosporine. Transplantation 55: 1265–1272.
Jaeschke, H., A. Bautista, Z. Spolarics, et al. 1992. Superoxide generation by neutrophils and Kupffer cells during in vivo reperfusion after hepatic ischemia in rats. Journal of leukocyte biology 52: 377–382.
Chen, L., K. Ma, H. Fan, et al. 2021. Exogenous hydrogen sulfide protects against hepatic ischemia/reperfusion injury by inhibiting endoplasmic reticulum stress and cell apoptosis. Experimental and therapeutic medicine 22: 799.
Du, Y., F. Zhong, H. Cheng, et al. 2021. γ The dietary supplement -oryzanol attenuates hepatic ischemia reperfusion injury via inhibiting endoplasmic reticulum stress and HMGB1/NLRP3 inflammasome. Oxidative medicine and cellular longevity 2021: 4628050.
Liu, D., H. Wang, Y. Zhang, et al. 2020. Protective effects of chlorogenic acid on cerebral ischemia/reperfusion injury rats by regulating oxidative stress-related Nrf2 pathway. Drug Des Devel Ther 14: 51–60.
Yang, H., H. Wang, and U. Andersson. 2020. Targeting inflammation driven by HMGB1. Frontiers in immunology 11: 484.
Lee, C.H., S.J. Yoon, and S.M. Lee. 2013. Chlorogenic acid attenuates high mobility group box 1 (HMGB1) and enhances host defense mechanisms in murine sepsis. Molecular medicine (Cambridge, Mass) 18: 1437–1448.
Ni, Y., H. Chen, H. Nie, et al. 2021. HMGB1: An overview of its roles in the pathogenesis of liver disease. Journal of leukocyte biology 110 (5): 987–998.
Deng, C., L. Zhao, Z. Yang, et al. 2022. Targeting HMGB1 for the treatment of sepsis and sepsis-induced organ injury. Acta Pharmacologica Sinica 43 (3): 520–528.
Sherif, I.O., and N.H. Al-Shaalan. 2018. Vildagliptin attenuates hepatic ischemia/reperfusion injury via the TLR4/NF-B signaling pathway. Oxidative Medicine and Cellular Longevity 2018: 3509091.
Liu, T., M. Zhang, H. Niu, et al. 2019. Astragalus polysaccharide from Astragalus Melittin ameliorates inflammation via suppressing the activation of TLR-4/NF-κB p65 signal pathway and protects mice from CVB3-induced virus myocarditis. International Journal of Biological Macromolecules 126: 179–186.
Lin, Y., M. Sheng, Y. Weng, et al. 2017. Berberine protects against ischemia/reperfusion injury after orthotopic liver transplantation via activating Sirt1/FoxO3α induced autophagy. Biochemical and Biophysical Research Communications 483: 885–891.
Zhuonan, Z., G. Sen, J. Zhipeng, et al. 2015. Hypoxia preconditioning induced HIF-1α promotes glucose metabolism and protects mitochondria in liver I/R injury. Clinics and Research in Hepatology and Gastroenterology 39: 610–619.
Liu, H., X. Jing, A. Dong, et al. 2017. Overexpression of TIMP3 protects against cardiac ischemia/reperfusion injury by inhibiting myocardial apoptosis through ROS/Mapks pathway. Cellular Physiology and Biochemistry 44: 1011–1023.
Saleh, H., and H.M. El-Shorbagy. 2020. Chitosan protects liver against ischemia-reperfusion injury via regulating Bcl-2/Bax, TNF-α and TGF-β expression. International journal of biological macromolecules 164: 1565–1574.
Ben-Ari, Z., O. Pappo, Y. Cheporko, et al. 2007. Bax ablation protects against hepatic ischemia/reperfusion injury in transgenic mice. Liver transplantation 13: 1181–1188.
Green, D., and G.J.S. Kroemer. 2004. The pathophysiology of mitochondrial cell death. Science 305: 626–629.
Bi, J., J. Zhang, Y. Ren, et al. 2019. Irisin alleviates liver ischemia-reperfusion injury by inhibiting excessive mitochondrial fission, promoting mitochondrial biogenesis and decreasing oxidative stress. Redox Biology 20: 296–306.
Chen, H.H., Y.T. Chen, C.C. Yang, et al. 2016. Melatonin pretreatment enhances the therapeutic effects of exogenous mitochondria against hepatic ischemia-reperfusion injury in rats through suppression of mitochondrial permeability transition. Journal of Pineal Research 61: 52–68.
Hangen, E., K. Blomgren, P. Bénit, et al. 2010. Life with or without AIF. Trends in biochemical sciences 35: 278–287.
Li, L., X. Luo, and X. Wang. 2001. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412: 95–99.
Acknowledgements
Thanks to the Biochemistry and Molecular Biology Department of Zunyi Medical University for providing us with the laboratory.
Funding
This research was supported by the National Natural Science Foundation of China (NO. 81660688).
Author information
Authors and Affiliations
Contributions
Kai Li and Zanjie Feng contributed in the study design, practical work, manuscript writing and revision. Cijun Peng participated in research design and discussion. Liusong Wang, Xuan Ma, Lei Wang, Kangwei Liu and Xin Geng participated in the implementation of some experiments.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
All animal experiment was approved by the Laboratory Animal Welfare & Ethics Committee of Zunyi Medical University (approval no: KLLY(A)-2020-029).
Consent for Publication
All the authors have read the manuscript and agreed to submit the paper to the journal.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, K., Feng, Z., Wang, L. et al. Chlorogenic Acid Alleviates Hepatic Ischemia–Reperfusion Injury by Inhibiting Oxidative Stress, Inflammation, and Mitochondria-Mediated Apoptosis In Vivo and In Vitro. Inflammation 46, 1061–1076 (2023). https://doi.org/10.1007/s10753-023-01792-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-023-01792-8