
Abstract
Rapid development of high-throughput sequencing methods and metagenomics revealed a diverse world of microbiota associated with multicellular organisms. Although recent discoveries indicate that freshwater invertebrates are hosts for specific bacteria, it is still unknown if this specificity is driven by host-derived factors or by the environment, especially in animals with diapause in ephemeral habitats, where parents and offspring are separated in time and space. In this work, using both low-throughput molecular approach and Next-generation sequencing of 16S ribosomal RNA gene, we present a taxonomic analysis of bacteria associated with two species of non-marine ostracods Sclerocypris tuberculata and Potamocypris mastigophora raised from diapausing eggs and co-cultured in laboratory conditions. Our analysis showed that despite sharing the same environment, each ostracod host developed distinct bacterial communities. The major difference was caused by the dominance of the family Comamonadaceae (Betaproteobacteria) in P. mastigophora and the Aeromonadaceae (Gammaproteobacteria) in S. tuberculata. Furthermore, prediction of metabolic pathways in metagenomes, revealed that microbiota of P. mastigophora exhibit higher number of sequences associated with the membrane transport and xenobiotics biodegradation and metabolism. Our study not only provides an insight into microbiota of non-marine ostracods but also shows that different ostracod species host functionally distinct bacterial communities.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Microorganisms are the most abundant group of living organisms and are found in any ecosystem on Earth. It has been known for decades, that apart from living in the environment, microorganisms inhabit surface and body cavities of multicellular organisms (Wernegreen, 2012). Studies of these associations, however, using classical microbiology methods were cumbersome and provided limited information about microbial communities. The true advances in the field of microbe-host associations were made with the advent of novel methods of DNA sequencing and metagenomics, which revealed the complex microbial communities inhabiting animals’ bodies (Zinger et al., 2012; Garza & Dutilh, 2015). Nevertheless, despite the availability of powerful tools to describe microbial communities within a host, the biological significance of these associations remains unknown for understudied groups (Waldor et al., 2015).
The community of microorganisms inhabiting a defined environment, such as body cavities of a multicellular organism, is defined as microbiota (Marchesi & Ravel, 2015). The beneficial role of microbiota for human and animal health is well established (Sommer & Bäckhed, 2013). Research devoted to elucidate the importance of microbiota-host associations in several experimental models (human, mouse, Drosophila) revealed that commensal bacteria influence the host’s immunity, development, metabolism, nutritional habits, mating and behavior (Turnbaugh et al., 2006; Rhee et al., 2009; Sharon et al., 2010; Shin et al., 2011; Foster & McVey Neufeld, 2013; David et al., 2014). However, despite providing valuable examples of microbiota-host interactions, human and other vertebrate models due to the complexity of their microbial communities are cumbersome in elucidating discrete interactions (Waldor et al., 2015). In contrast, invertebrates provide a more convenient models to study microbiota-host interactions, due to their large diversity, often less complex morphology, ease of work and possibility to culture large number of individuals under laboratory conditions (Chaston & Goodrich-Blair, 2010; Kostic et al., 2013).
Freshwater invertebrates have been shown to play a major role in decomposition of organic material, nutrient cycling and energy flow, bioerosion and bioturbation of bottom sediment (Adámek & Maršálek, 2013; Prather et al., 2013). Several benthic species by feeding on microorganisms constitute a trophic link between microbial primary production and higher trophic levels (Schmid-Araya & Schmid, 2000) and are an important factor enhancing decomposition of organic matter in sediments (Nascimento et al., 2012; Chambord et al., 2017). On the other hand, growth rate of benthic fauna is affected by bottom-up effects through bacterial abundance (Gaudes et al., 2013). Although many aspects and biological role of zoobenthos-bacteria associations still remain obscure, recent studies have revealed that meiofauna have an important effect on marine benthic ecosystem processes, e.g. increases bacterial denitrification (Bonaglia et al., 2014), reduces bacterial mineralization of organic pollutants (Näslund et al., 2010) or contribute to macroalgae decomposition and nutrient recycling (Herrera et al., 2017). Yet, little is known about such interactions in freshwaters compared to marine environments. Halpern & Senderovich (2015) for instance found that microbiota of chironomid egg masses and larvae degrade various toxicants, enabling their host to live in polluted environments. There are also examples of important biological role of relationships between microbiota and planktonic freshwater invertebrates, like those on Daphnia studies (e.g. Freese & Schink, 2011; Sison-Mangus et al., 2015; Macke et al., 2017).
Considering the above, microbiota and their freshwater benthic hosts may exhibit complex relationships and meiofauna may play a critical role in maintenance and spreading of microbes. However, information about the specificity of bacterial communities associated with zoobenthos is lacking and further studies are needed to answer questions about host-specificity or environment-specificity of meiofauna-associated microbiota.
One of the most ubiquitous groups of freshwater benthic arthropods is Ostracoda (Martens et al., 2008). To date, ostracods served mainly as a study model for environmental and climate reconstruction, stratigraphy, toxicology or evolution of sexual reproduction (Butlin et al., 1998; Park & Smith, 2003; Horne et al., 2012; Smith et al., 2015). In inland waters, ostracods can be found in every kind of water body, including ephemeral habitats, where they are often a dominant invertebrate group (Smith et al., 2015). As other taxa associated with temporary waters, ostracods have an ability to rapidly populate available water bodies during aquatic phase by hatching from resting eggs resistant to adverse environmental conditions (Rossi et al., 2012; Vandekerkhove et al., 2013). This ability coupled with other adaptations (e.g. short life cycle, parthenogenetic reproduction) makes ostracods one of the pioneering organisms in temporary freshwaters (Martins et al., 2009; Olmo et al., 2016).
Despite their ubiquity, the ecological role of ostracods in a food chain is not fully understood. It is likely that they play a key role in decomposition of organic matter in the benthic area, either through direct feeding on detritus or by feeding on detritus-associated bacteria (Benincà et al. 2008; Mesquita-Joanes et al., 2012). Decomposition of complex organic matter often requires aid from intestinal bacteria, as it is observed in termites and herbivores (Engel & Moran, 2013). However, besides five papers that record intracellular bacteria in several non-marine ostracod species using either transmission electron microscopy (Vandekerckhove, 1998) or DNA sequencing (Baltanás et al., 2007; Mioduchowska et al., 2018; Çelen et al., 2019; Schön et al., 2019) there are only two studies describing whole microbial communities in one marine (Jarett et al., 2013) and four non-marine (Schön et al., 2019) ostracod species. The latter two studies (Jarett et al., 2013; Schön et al., 2019) suggest considerable inter- and intraspecific variation in the taxonomic composition of ostracod microbiota.
Considering their abundance in aquatic environments, the lack of information on ostracods-associated microbiota is surprising. Moreover, taking into account their exceptional ability to withstand adverse environmental conditions, as well as the ability to restore population when water is available, characterization of ostracod-microbiota relationships can shed light into how animals form associations with microbiota after dormancy. An interesting question is if diapausing ostracods transmit microbes into the resting stage, or otherwise either selectively develop a specific microbial community or randomly associate with bacteria present in the ambient environment.
In this work, we present the first insight into the microbiota associated with two taxonomically different species of ostracods, Sclerocypris tuberculata and Potamocypris mastigophora. Using 16S rRNA gene sequencing, we analyzed bacterial community profiles associated with individuals hatched from resting eggs and co-cultured in the same artificial environment mimicking natural conditions. We hypothesized that (1) the two ostracod species will develop distinct bacterial community compositions and that (2) metabolic pathways associated with bacteria present in the two ostracods will be also different.
Methods
Sediment collection, ostracod culturing and sampling for microbial community profiling
A sample of dry sediment was collected from the surface of a temporary shallow salt lake within the vast Makgadikgadi depression in Botswana (lat. 20° 08′ 02″ S, long. 25° 33′ 41″ E, alt. 934 m ASL) at the end of the dry season (17 Sept. 2012). Upon arrival to the laboratory, the sediment was stored dry in the dark at ca. 16–18 °C for 14 months until further processing. On 22 Nov. 2013 a part of the sediment sample was placed in 2 l glass aquarium (2–3 cm deep substratum), inundated with bi-carbonate rich mineral water to a depth of ca. 10 cm and incubated in a temperature- and light-controlled room at 23 ± 1 °C and under 12:12 h (light:dark) photoperiod, allowing ostracods as well as algae and bacteria (the food for ostracods) to rear and grow from original draught resistant eggs and spores. The culture of raised ostracods was maintained permanently in self-sustaining (not isolated from the air) microcosm for ca. 1 year before sampling for the microbiome study started, keeping more or less fixed water level and occasionally providing spinach and potato starch as additional food. Out of seven ostracod species which were successfully raised, two most abundant and with stable population size were selected for the microbiome study: Sclerocypris tuberculata (Sars) (= Sclerocypris sarsi Martens) known to occur only in south Africa, and Potamocypris mastigophora (Methuen) widely distributed in Africa (Martens, 1984). Although both species belong to the family Cyprididae, the most diverse and species-rich group of non-marine ostracods (Meisch et al., 2019), they differ considerably in morphology and size; S. tuberculata is 3.6–3.8 mm long and belongs to the subfamily Megalocypridinae, while P. mastigophora is only ca. 0.5 mm long and belongs to the subfamily Cypridopsinae. Sampling of ostracods for microbial community profiling was started on 4 Dec. 2014, and five samples were collected over a 7-week-period to estimate the stability of bacterial communities during this period.
DNA extraction
Due to the size differences, for total DNA isolation from each of the five samples two females and two males of Sclerocypris tuberculata or ten individuals of Potamocypris mastigophora were used. Prior to homogenization, ostracods were washed four times in sterile TBS (150 mM NaCl, 20 mM Tris pH 7.4) to remove water-derived bacteria. After the last wash the TBS was removed and 200 μl of lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris pH 7.8) were added. Individuals were ground using disposable tissue homogenizers, followed by addition of proteinase K (to final concentration of 50 μg/ml) and incubation at 55 °C for 1 h. Next homogenate was treated once with phenol: chloroform and once with chloroform. DNA was precipitated with one volume of isopropanol in presence of 2.5 M ammonium acetate and 10 μg/ml linear acrylamide. DNA was re-suspended in 30 μl of TE buffer and stored at − 20 °C for further analysis. The quantification of DNA was performed with NanoDrop 2000 spectrophotometer and Qubit 2.0 fluorometer.
16S rRNA gene based clone library preparation
For amplification of 16S rRNA fragment we utilized DNA primers which provide the broadest spectrum of bacterial genera and which amplify 465 bp fragment, spanning variable regions V3-V4 in prokaryotic 16S rRNA gene (Klindworth et al., 2013). Primers sequences were: S-D-Bact-0341-b-S-17 5′-CCTACGGGNGGCWGCAG-3′ and S-D-Bact-0785-a-A-21 5′-GACTACHVGGGTATCTAATCC-3′. Routine PCR was performed with Q5 Hot Start High-Fidelity DNA Polymerase (New England Biolabs) using 10 ng of total DNA from ostracods as the template. The reaction conditions were as suggested by producer except that annealing temperature was set at 55 °C and elongation time was 20 s. For amplification, a negative control was included and PCR was considered successful when no band was detected in the control reaction. Primers were removed by adding an equal volume of PEG solution (20% PEG 8000, 2.5 M NaCl) to precipitate PCR amplicons. Purified PCR amplicons were ligated into pJET1.2 (Thermo Scientific) and the ligation mix was transformed into Escherichia coli DH5a cells. Colonies were screened for the presence of inserts with colony PCR (using pJet2.1 sequencing primers: forward -CGACTCACTATAGGGAGAGCGGC and reverse -AAGAACATCGATTTTCCATGGCAG) and positive clones were used for plasmid isolation. DNA of a total of 192 plasmids was isolated using one-tube protocol by Lezin et al. (2011). Plasmids were subjected to Sanger sequencing using the pJet1.2 forward sequencing primer (Thermo Scientific) at Macrogen Europe. Sequences were identified using SILVA online tool and microbial BLAST (Johnson et al., 2008; Quast et al., 2013). Sequences are available in GenBank, under accession numbers KX359408–KX359580.
16S rRNA gene high-resolution melt analysis (HRM)
General differences in bacterial communities between two samples can be assessed by analysis of 16S rRNA gene, which is used as a barcode for bacterial species identification. Here we used a rapid and cost-efficient method, the high-resolution melting (HRM) analysis (Hjelmsø et al., 2014). The HRM is based on the monitoring of melting point (Tm) of a double-stranded DNA (dsDNA) fragment in small temperature increments and in the presence of dsDNA-specific fluorescent dye. Upon melting of dsDNA, the fluorescence signal is lost and measurement of fluorescence during temperature increase allows for detection of differences in Tm caused by nucleotide substitutions. In case of mixture of DNA fragments, the result is an average profile of melting curve for all fragments and it can be considered as melting fingerprint of the sample. Thus if two samples differ in the composition of 16S rRNA sequences, they will show distinct Tm profiles which are specific for either sample. For HRM analysis, we used the same primers as for cloned library preparation (S-D-Bact-0341-b-S-17 and S-D-Bact-0785-a-A-21). Optimization experiments were performed to determine of DNA template. Amplification was performed using Fast Start SYBR Green Master Mix (Roche) and 1 ng of total DNA template, in total volume of 10 μl. Each reaction was performed in four technical replicates. All HRM experiments were carried out on Light Cycler 96 (Roche). Data were initially analyzed using Light Cycler 96 software and exported to text file for further analysis.
454 pyrosequencing
For the Next-Generation Sequencing (NGS) experiment, pooled samples from the first 3 weeks (week 0, 1 and 3) and pooled samples from the remaining weeks (week 5 and 7) were used for further analysis. In total, two samples for Potamocypris mastigophora (P1 and P2, respectively) and four samples for Sclerocypris tuberculata (S1 and S2 for females and S3 and S4 for males) were prepared. This corresponds to 20 individuals of Sclerocypris tuberculata (10 for males and 10 for females) and 50 individuals of Potamocypris mastigophora. Genomic DNA (80–200 ng) was prepared and sent to Macrogen Inc. Korea for sequencing. Processing and data analysis was performed with QIIME pipeline (Caporaso et al., 2010), chimeric sequences were detected and removed using Chimera Slayer (Haas et al., 2011), taxonomic assignment of operational taxonomic units (OTUs) were acquired at 97% identity level from GreenGenes v13_05 (DeSantis et al., 2006). Raw data are deposited in Sequence Read Archive (SRA) under accession number SRP076066 and are also available upon request.
Data analysis
Alpha diversity metrics (including total number of reads, observed number of OTUs, number of identified bacterial taxa, Chao 1 of predicted number of OTUs, Good’s Coverage Estimator, Shannon and Simpson’s diversity indices and Pielou’s evenness index) and rarefaction curves were generated from OTU table in the BIOM format using R and the Vegan package (Oksanen et al., 2015). Principal coordinate analysis (PCO), Multi-dimensional scaling (MDS) and UPGMA hierarchical clustering were performed to visualize differences in bacterial communities between ostracod species, while SIMPER (Similarity Percentage) analysis was used to identify bacterial OTU contributions to (dis-)similarities within and between ostracods. Bray–Curtis similarity coefficient was based on standardized (percentage abundance) bacterial data. These analyses were conducted with the R package Vegan and PRIMER 7 and PERMANOVA+software (Anderson et al., 2008). Kyoto Encyclopedia of Genes and Genomes KEGG (Kanehisa & Goto, 2000) was used as reference in analysis of microbe-ostracod metabolic pathways (hierarchical levels 2 and 3). KEGG pathway prediction was performed with PICRUST (Langille et al., 2013). To test statistical interspecific differences in bacterial community structure and KEGG metabolic pathways between the two studied ostracod species, Permutational analysis of variance (PERMANOVA) on the Bray–Curtis similarity matrix was used with the Monte Carlo P values [p(MC)]. This is preferred over the permutation P value method when the number of unique permutations is too small and insufficient to make statistical inferences at a significant level of 0.05 (see Anderson and Robinson (2003) for details). The procedure was performed using PRIMER 7 and PERMANOVA+software (Anderson et al., 2008). For descriptive and univariate statistics (mean values, standard deviation, normality and equal variance tests, Student’s t test, Mann–Whitney test) SigmaPlot ver. 11.0 (Systat Software, San Jose, CA) was used.
Results
Profiling bacterial communities associated with ostracods
In the first step, we assessed if HRM is suitable for analysis of ostracod-associated bacterial communities using annealing temperature gradient on representative samples. Since HRM is based on PCR amplification of the 16S rRNA gene fragment using universal primers, the annealing temperature could potentially introduce a bias towards amplification of specific sequences. However, our results indicate that the studied samples showed different but sample-specific profiles in all tested temperatures (Fig. 1A). Furthermore, these profiles were constant also for other samples collected over the period of the experiment (Fig. 1B). Importantly, both ostracod species showed HRM profiles clearly distinct from the profile obtained for samples from aquarium water (Fig. 1A, B), indicating the presence of different bacteria species.

Profiling bacterial communities associated with ostracods. A Primer annealing temperature gradient for HRM analysis: PCR reaction was performed at annealing temperatures distributed over 12-step gradient between 50 and 62 °C; fluorescence difference was normalized to the water samples (gray lines). B HRM analysis of microbial community stability: color intensity increases with the week of the experiment; water samples were used as the baselines. C Distribution of bacterial classes and families (percentage of sequences) identified through cloned library analysis for two studied ostracod species
Small-scale bacteria identification through 16S rRNA gene clone library sequencing
To identify differences in bacterial community composition between the hosts, we prepared a cloned library of the amplified PCR f16S rRNA fragment. For each library, 96 clones were sequenced. After removal of sequences with low quality (based on sequence quality score > 20 and visual inspection), 82 and 85 sequences were obtained for Sclerocypris tuberculata and Potamocypris mastigophora, respectively (Fig. 1C). Sequences were compared with the NCBI database using BLAST. The most obvious difference between the hosts was observed in the distribution of classes of the phylum Proteobacteria. In Sclerocypris tuberculata the majority of sequences were classified as Alphaproteobacteria (63 out of the total 82 sequences), while in Potamocypris mastigophora number of sequences classified as Alphaproteobacteria and Betaproteobacteria was similar (42/85 and 38/85, respectively). Class Gammaproteobacteria was represented by a smaller number of sequences, 8/82 and 5/85 in Sclerocypris tuberculata and Potamocypris mastigophora, respectively. Sequences classified to the class Cytophagia of the phylum Bacteroidetes were present only in the Sclerocypris tuberculata samples (Fig. 1C).
454 sequencing of 16S rRNA gene amplicons
Cloned library analysis allowed for the initial identification of bacteria associated with either Sclerocypris tuberculata or Potamocypris mastigophora and for assessment of their distribution in the studied ostracods. Differences observed with this low-throughput approach were the inspiration for high-throughput analysis of microbiota associated with ostracods. Sequencing resulted in 170788 reads which after quality filtering yielded in total 126311 sequences with an average length of 413 bp. With the exception of the sample S2 from females of S. tuberculata, which yielded only 1797 sequences, the other obtained libraries were of similar size and on average with 24903 ± 5203.3 standard deviation (SD) sequences per sample (Table 1). The reason for poor quality of the sample S2 is unknown, thus results of further testing differences between the two species were presented as both including and excluding this sample. Consistently with the number of sequences obtained for each library, the total number of OTUs identified at 97% identity level was the lowest for the sample S2 (only 1281 OTUs). For the remaining samples, the number of identified OTUs exceeded 7900 (mean ± SD = 10112 ± 1728.9; Table 1). However, both the mean number of sequences and OTUs did not differ statistically between the two ostracod species, irrespective of whether the sample S2 was included or excluded (Table 1). Good's coverage estimated that > 98% of bacterial OTUs was detected, indicating adequate sampling effort (Table 1). The mean Chao1 index values for Sclerocypris tuberculata (112.7 ± 8.91) and for Potamocypris mastigophora (114.0 ± 5.59) were similar and statistically insignificant (Table 1). However, microbiota communities of Sclerocypris tuberculata appeared significantly more diverse than those of Potamocypris mastigophora as indicated by both the Shannon and Simpson’s indexes (Table 1). Nevertheless, rarefaction curves did not reach saturation, indicating that depth of sequencing was not exhaustive and a higher number of sequencing reads per sample would be more appropriate to fully cover the microbiota diversity (Fig. 2).

Rarefaction curves based on the number of OTUs identified for each sequenced ostracod sample (P1 and P2—Potamocypris mastigophora, S1 and S2—Sclerocypris tuberculata females, S3 and S4—S. tuberculata males)
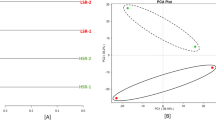
In terms of similarity between samples from different hosts, the PCO and MDS indicated that bacterial communities were different in a species-dependent manner (Fig. 3). We used SIMPER analysis to identify OTUs which contributed the most to the overall difference between the ostracod hosts (Fig. 4). The highest difference was caused by OTU 530377 classified as the genus Hydrogenophaga of the family Comamonadaceae, which constituted on average 47% of sequences in the samples from Potamocypris mastigophora, while in those from S. tuberculata (regardless of including or excluding the sample S2 in the analysis)—less than 0.2% (Fig. 4). The opposite distribution was observed for OTU 778059 classified as belonging to the family Aeromonadaceae (31–33% of sequences in the samples of S. tuberculata versus < 0.2% in P. mastigophora). Another OTU showing higher abundance in Potamocypris mastigophora was OTU 136160 (12% vs. < 0.06% in S. tuberculata) classified as belonging to the family Comamonadaceae. These three OTUs contributed 57% to the observed differences (Fig. 4). The average Bray–Curtis dissimilarity between P. mastigophora and S. tuberculata samples was high (81%), compared to the within-species variability (average similarity of P. mastigophora samples was 93%, while that for S. tuberculata samples 60% when including the S2 sample). The PERMANOVA analysis confirmed the significance of the described above divergence pattern and showed that two ostracod hosts differed significantly in bacterial community structure (Pseudo-F > 11.8, 0.01 < p(MC) ≤ 0.05; Table 2).

Ordination of the ostracod samples by Principal coordinates analysis PCO (main scatter plot) and Multi-dimensional scaling MDS (inset) based on the identified microbiota dataset. Each point in the PCO plot represents 16S rRNA data from a single ostracod sample (sample codes as in Fig. 2) and is shown as a three-sector circle displaying relative percentages of three bacterial taxa (OTU) which contribute most to the PCO pattern and were identified by the SIMPER analysis as OTUs which were the best discriminators between the two ostracod species. MDS plot (inset) displays bootstrap averages (blue squares for S. tuberculata and orange circles for P. mastigophora), overall bootstrap averages (black symbols) and bootstrap region with 95% nominal coverage for S. tuberculata

Shade plot (or heat map) and hierarchical UPGMA clustering of relative abundances of 10 bacterial taxa identified by the SIMPER analysis as main OTUs accounting for dissimilarity among the two ostracod species. Codes for ostracod samples as in Fig. 2
Metabolic pathways in ostracod-associated bacterial communities
Our results indicated that bacterial communities in the tested ostracods had host-specific taxonomic structure and composition. However, based solely on the taxonomy of microbiota it is difficult to conclude about possible biological or ecological significance of these associations. To get insight into these issues, we analyzed the data using PICRUST software, allowing for prediction of metagenomes and bacterial functional profiles from 16S rRNA sequencing data. Interestingly, processes showing the highest difference between the ostracod hosts belonged to two KEEG classes of metabolic pathways of the second hierarchical level: (1) Membrane transport and (2) Xenobiotics Biodegradation and Metabolism (Fig. 5). Overall profiles of metabolic pathways were specific for either host, since P. mastigophora and S. tuberculata formed separate UPGMA clusters (Fig. 5), similarly as it was observed for OTU distribution (Fig. 4). Indeed, these differences appeared statistically significant in the PERMANOVA analysis (Pseudo-F = 9.998, p(MC) = 0.018, Table 2). Detailed analysis of the metabolic processes showed that genes encoding membrane transport (ABC transporters) and xenobiotics biodegradation and metabolism (benzoate degradation) were enriched in samples from Potamocypris mastigophora as compared with those from Sclerocypris tuberculata (Fig. 5).

Shade plot and hierarchical UPGMA clustering of relative abundances of sequences associated with the corresponding metabolic pathways of the KEGG hierarchical level 2, which were identified by the SIMPER analysis as main categories accounting for dissimilarity among the two studied ostracod species. Codes for ostracod samples as in Fig. 2
Discussion
In recent years, we could observe a constantly growing number of studies describing bacterial communities associated with different organisms. While mammals are the main object of interest, there is also a growing number of studies on microbiota associated with invertebrates, including also those from aquatic environments. The vast majority of studies, however, are biased in favor of macroinvertebrates, e.g. Porifera (Lee et al., 2011), Anthozoa (Pogoreutz et al., 2017), Hydrozoa, Polychaeta, Hirudinea (Chaston & Goodrich-Blair, 2010), Gastropoda (Takacs-Vesbach et al., 2016), Malacostraca (Skelton et al., 2017) or Insecta (Ayayee et al., 2018). Available data indicate that also freshwater planktonic invertebrates are associated with specific microorganisms (Qi et al., 2009; Tang et al., 2011). Yet there is a paucity of studies on microbial communities of meiobenthic animals inhabiting inland waters, including ostracods.
In our experiment, we aimed at determining if co-cultured ostracods develop distinct or similar microbial communities. In in the first step we utilized the high-resolution melt (HRM) analysis which has been shown to be useful in generating general profiles of soil bacteria communities (Hjelmsø et al., 2014). Despite some shortcomings, like all genotyping methods (e.g. physical limitations in melting curve distinction for alternative variants and some technical issues which can affect the performance of the analysis, see Słomka et al., 2017 for details), HRM can be successfully used for monitoring the stability of bacterial community in long-term experiments and to select the most valuable samples for more expensive high-throughput sequencing. This is a rapid, relatively simple and specific approach. Using HRM we were able to estimate differences in bacterial community profiles between ostracods and the ambient water. We also showed that Sclerocypris tuberculata and Potamocypris mastigophora associate with bacterial communities displaying different HRM profiles (Fig. 1A, B). However, HRM does not provide information about which bacterial taxa are different in the studied samples. Therefore, to identify potential differences in bacterial taxa composition and confirm differences between the hosts, in the next step we prepared 16S rRNA gene clone library for the used PCR fragment. Although the fragment of 16S rRNA gene used for the analysis was relatively short and the throughput of the study was low, our results showed that the two studied ostracod species were associated with clearly different bacterial communities (Fig. 1C).
Further analysis with high throughput sequencing allowed identifying taxonomic composition of bacterial communities for both co-habiting ostracod species and demonstrated that the ostracod-associated microbiota remained taxonomically highly consistent in the same ostracod species but they significantly differed among the species (Table 2, Figs. 3 and 4). In general, bacteria present in the studied ostracods were mostly of the phyla Proteobacteria and Bacteroidetes. The SIMPER analysis identified the families Comamonadaceae (Betaproteobacteria) and Aeromonadaceae (Gammaproteobacteria) as contributing most to the difference between ostracod-associated microbiota (Fig. 4). These families were the most abundant taxa in Potamocypris mastigophora and Sclerocypris tuberculata, respectively. Only 17 Actinobacteria sequences were found in P. mastigophora samples. Actinobacteria are generally considered as environmental bacteria and are abundant in soil and freshwater samples (Newton et al., 2011).
Although both approaches, the cloned library and 454 pyrosequencing successfully showed clear differences between the studied ostracod species in their bacterial communities, the two approaches did not give exactly similar results. As expected, the difference in relative abundance at the phylum level was almost negligible (Fig. S1A) but it increased at lower taxonomic levels (see Figs. S1B, C). In case of Potamocypris mastigophora the dissimilarity pattern is consistent at the class and family level and results mostly from differences in the relative abundances of the families Caulobacteraceae (Alphaproteobacteria) and Comamonadaceae (Betaproteobacteria), the latter having higher percentage in the 454 sequencing approach (Fig. S1C). In case of Sclerocypris tuberculata the most significant difference between the results from the two approaches appeared to be due to very low detectability of the family Aeromonadaceae (Gammaproteobacteria) when using the cloned library approach (in Fig. S1C showed as other Gammaproteobacteria). Nevertheless, even if the analysis of bacterial composition on lower taxonomic levels was uncertain using the cloned library, the discrepancies in bacteria associated with the studied ostracods revealed by this low-throughput approach provided a stimulus to the high-throughput analysis.
Our findings of the phyla with the highest relative percentages in the whole bacterial communities associated with the studied ostracods are in agreement with the study of Schön et al., (2019) who show that bacteria belonging to Proteobacteria and Bacteroidetes dominate also in four other non-marine ostracod species: combined percentage mean ± SD = 82.4% ± 7.16%, N = 9 allopatric samples (based on Schön et al., 2019) versus 99.3% ± 0.52%, N = 6 in our studies (Fig. S2). These highest-abundance shared phyla may represent a core microbiota for non-marine ostracods, while other phyla (of low abundance and/or unshared, as e.g. Acidobacteria present in one specimen of Heterocypris incongruens (Ramdohr) or Fusobacteria in one specimen of Darwinula stevensoni (Brady & Robertson) studied by Schön et al., 2019) may be facultative and more variable representatives of ostracod microbiome. Substantial differences between hosts (also within one species) were nevertheless observed in mutual percentage abundance of the two dominant phyla in the data of Schön et al. (2019). Proteobacteria abundances ranged between as high as 96.2% in one specimen of Eucypris virens (Jurine) (which is comparable with our data on both S. tuberculata and P. mastigophora, see Fig S2) and as low as 9.4% in one specimen of H. incongruens. There is also significant inter- and intraspecific variability in the distribution of classes of the two phyla and at lower taxonomic levels (Schön et al., 2019). Jarett et al. (2013) studying epibiotic microbial community of two specimens of marine benthic myodocopid ostracods tentatively assigned to the genus Rutiderma also recorded very large variability between the two ostracod samples (Bray–Curtis within-species similarity = only 5%). One ostracod sample was dominated by bacteria of the phylum Proteobacteria (81.7%), similarly to samples of S. tuberculata and P. mastigophora of our study, but the relative abundance of the phylum Bacteroidetes was only 0.5%. On the other hand, the most abundant bacterial phyla of the second specimen studied by Jarett et al. (2013) were Planctomycetes (53.8%) known to dominate marine biofilms (Jarett et al., 2013) and Cyanobacteria (39.1%). Such significant variability in bacteria community composition within and between ostracod species studied so far may well be due to the fact that all ostracod samples were processed in total and DNA was extracted from whole individuals. Therefore, diverse facultative epibiotic bacteria residing on ostracod carapace or different gut microbiota known to vary depending on current type of ingested food may represent significant portion of and contribute towards the whole biodiversity of ostracod-associated microbiota. Definitely, additional replicate samples of other species, including different sexes and developmental stages with separation of the carapace from the body as well as dissection of different body parts (digestive tract, reproductive organs) will be necessary in future studies.
The results on composition and variation of ostracod-associated bacteria communities (Jarett et al., 2013; Schön et al., 2019 and the present study) are in accordance with studies on microbiota of planktonic freshwater invertebrates (Qi et al., 2009; Tang et al., 2011). In three species of Daphnia, the vast majority of bacterial taxa (88–98%), similar to our results (79–99%), were also assigned to the phylum Proteobacteria, of which most sequences belonged to the family Comamonadaceae of the class Betaproteobacteria (Qi et al., 2009). The analysis of Daphnia metagenomes revealed that laboratory strains of Daphnia had highly similar but not identical composition of bacterial species. The largest differences were observed at the lowest taxonomic levels, namely at the species and strain levels (Qi et al., 2009). However, specificity of bacteria association with invertebrates, including planktonic crustaceans, can also be observed at higher taxonomic levels (e.g. Tang et al., 2011).
Although distinct species-specific microbial communities have been revealed in aquatic hosts from different locations in a number of previous studies (e.g. Hentschel et al., 2002), there have been only few projects assessing whether different co-habiting (i.e. sharing the same place either in the wild or in laboratory conditions) aquatic animal species harbor unique microbial communities. Papers meeting the formulated above criterion have supported species-specific host-microbiota associations using molecular methods in e.g. marine sponges (Lee et al., 2011; Gloeckner et al., 2013), octocorals (Holm & Heidelberg, 2016) and freshwater amphibians (McKenzie et al., 2012). Gloeckner et al. (2013) detected consistent species-specific microbial communities in five Mediterranean sponge species of the same genus, Holm & Heidelberg (2016)—in two Eastern Pacific gorgonian octocoral species of the same genus, whereas Lee et al. (2011)—in three Red Sea sponge species belonging to three different families. These studies confirmed that the microbiota appeared mostly unaffected by environmental factors. In the study on skin bacteria of three amphibian species from pond habitats in Colorado, USA, McKenzie et al., (2012) proved that host species was highly significant predictor of microbiota similarity, while coexistence within the same pond was insignificant. Further field and experimental approaches on a wider group of aquatic animals, including ostracods, are needed to determine if host specificity of microbiota inhabiting defined host body parts is a general phenomenon.
The other question which remains to be explored is the mechanism by which the two studied ostracod species established distinctive bacterial communities after diapause. In our experimental system microbes could derive either from the same pool of available bacterial inocula present in the sediment (horizontal transmission) or from inocula of maternal origin persisting in association with the resting eggs (vertical transmission). Although the occurrence of high host specificity of microbes would indirectly support the latter source, in our opinion, the existence of mechanisms selectively favoring development of unique bacteria communities from the ambient environment cannot be excluded, even if such mechanism should not be expected when the host is dependent on acquisition of beneficial microbiota from the environment after the diapause. Recently, Mushegian et al., (2018) have found that bacteria which are required for the successful development of Daphnia after dormancy are of environmental origin, although vertically transmitted microbiota can occur in ephippia housing the resting eggs. In any case, forming the microbiota-ostracod associations after diapause is a stimulating study area that needs further experiments and field observations to test if ostracods coevolved with specific symbiotic bacteria acquired vertically or evolved dependence on horizontally acquired more opportunistic and taxonomically variable microbiota. The latter hypothesis may be expected to be valid for diapausing ostracods, because considering longevity and capacity for long-distance dispersal of their eggs, environmental conditions experienced by mother and offspring could be different, so that maternal bacteria may not be optimal for the offspring.
Our results showed that bacterial communities in the cultured ostracods were different in their OTUs dominance structure but sharing several taxa. Therefore, the question arises what factors drive differences in ostracod-associated microbiota. It is generally known that microbiota form complex communities with unique metabolic properties, which benefit the host (Sommer & Bäckhed, 2013). In our work, we took advantage of available tools for predicting metabolic pathways from 16S rRNA sequencing data. Surprisingly, in concert with significant differences in bacterial taxonomic structure and composition, we found that microbiota associated with the studied ostracods had also statistically distinct metabolic profiles (Fig. 5, Table 2). In the light of these results, it is possible that different species of ostracods by hosting specific groups of bacteria allow for local concentration of certain metabolic pathways present in their microbiota. Such hypothesis was already proposed and partially verified by other researchers, suggesting that relationships between bacteria and meiofauna or zooplankton are crucial for nutrient flow in aquatic ecosystems (Tang et al., 2010, 2011; Bonaglia et al., 2014).
The role of meiofauna in the bacterial decomposition of complex compounds of natural or industrial origin is twofold. It has been shown that abundance of meiofauna negatively correlates with mineralization of naphthalene by bacteria (Näslund et al., 2010). On the other hand, meiofauna increases denitrification driven by bacteria in marine sediments (Bonaglia et al., 2014). In our experiments, we found that ostracods are potential hosts for bacteria with metabolic pathways involved in benzoate and aminobenzoate degradation. Although these data are inferred from 16S rRNA gene sequencing and the presence of particular bacteria or capability for (amino-)benzoate degradation has to be confirmed, our findings underscore a potential role of ostracods in bioremediation of toxic pollutants or pharmaceuticals. Furthermore, besides degradation of potential pollutants, other metabolic pathways such as isoflavonoid biosynthesis or starch and sucrose metabolism were also present in our ostracod-associated microbiota. These findings correlate with data showing that presence of microcrustaceans increases the rate of plant material decomposition in freshwater (Chambord et al., 2017). Further studies will allow determining if ostracod-associated microbiota indeed has the metabolic potential suggested in this and other studies. Interestingly, since some ostracod species are relatively easy to culture in the laboratory, it creates interesting opportunity to study the metabolic potential of otherwise unculturable bacteria (Vartoukian et al., 2010; Stewart, 2012).
In this report, we provide the first insight into host-specificity of microbiota associated with non-marine ostracods. However, the most surprising finding of our study was that together with divergence in bacterial communities’ structure and composition, bacterial metabolic pathways present in the two ostracod species were also different. It is important to note that the aquarium where the culture was maintained, although mimicking natural conditions, was an artificial environment, thus bacteria species detected in this work may differ from bacteria associated with Sclerocypris tuberculata and Potamocypris mastigophora in their natural habitat. Nevertheless, the main conclusion of our work is that both species associate with taxonomically and metabolically different bacteria. One of the implications of our findings is the need to preserve the biodiversity of aquatic ecosystems as it can be the main factor allowing for the development of fully functional trophic networks, supported by the metabolic capacity of ostracod-associated microbiota. Further studies focused on functional profiling of bacterial communities associated with ostracods, especially in the natural environment, will elucidate the importance of these interactions. Assessing whether field studies bring results comparable with those got in laboratory experiments with respect to particular mechanisms or taxa would be especially enlightening.
References
Adámek, Z. & B. Maršálek, 2013. Bioturbation of sediments by benthic macroinvertebrates and fish and its implication for pond ecosystems: a review. Aquaculture International 21: 1–17.
Anderson, M. J. & J. Robinson, 2003. Generalized discriminant analysis based on distances. Australian & New Zealand Journal of Statistics 45: 301–318.
Anderson, M. J., R. N. Gorley & K. R. Clarke, 2008. PERMANOVA+ for PRIMER: guide to software and statistical methods. PRIMER-E, Plymouth.
Ayayee, P. A., C. R. Cosgrove, A. Beckwith, A. A. Roberto & L. G. Leff, 2018. Gut bacterial assemblages of freshwater macroinvertebrate functional feeding groups. Hydrobiologia 822: 157–172.
Baltanás, A., M. Zabal-Aguirre, M. Pita & C. Lopez-Fernandez, 2007. Wolbachia identified in a new crustacean host: an explanation of the prevalence of asexual reproduction in non-marine ostracods? Fundamental and Applied Limnology 169: 217–221.
Benincà, E., J. Huisman, R. Heerkloss, K. D. Jöhnk, P. Branco, E. H. Van Nes, M. Scheffer & S. P. Ellner, 2008. Chaos in a long-term experiment with a plankton community. Nature 451: 822–825.
Bonaglia, S., F. J. A. Nascimento, M. Bartoli, I. Klawonn, V. Brüchert, 2014. Meiofauna increases bacterial denitrification in marine sediments. Nature Communications 5: 5133.
Butlin, R. K., I. Schön & K. Martens, 1998. Asexual reproduction in nonmarine ostracods. Heredity 81: 473–480.
Caporaso, J. G., J. Kuczynski, J. Stombaugh, K. Bittinger, F. D. Bushman, E. K. Costello, N. Fierer, A. G. Peña, J. K. Goodrich, J. I. Gordon, G. A. Huttley, S. T. Kelley, D. Knights, J. E. Koenig, R. E. Ley, C. A Lozupone, D. McDonald, B. D. Muegge, M. Pirrung, J. Reeder, J. R. Sevinsky, P. J. Turnbaugh, W. A. Walters, J. Widmann, T. Yatsunenko, J. Zaneveld & R. Knight, 2010. QIIME allows analysis of high- throughput community sequencing data. Nature Methods 7: 335–336.
Çelen, E., O. Külköylüoğlu, M. Yavuzatmaca, D. Akdemir & O. Yılmaz, 2019. First evidence of Cardinium (Sphingobacteria) in non-marine ostracods from Turkey. Journal of Crustacean Biology 39: 547–551.
Chambord, S., M. Tackx, E. Chauvet, G. Escolar & F. Colas, 2017. Two microcrustaceans affect microbial and macroinvertebrate‐driven litter breakdown. Freshwater Biology 62: 530–543.
Chaston, J. & H. Goodrich-Blair, 2010. Common trends in mutualism revealed by model associations between invertebrates and bacteria. FEMS Microbiology Reviews 34: 41–58.
David, L. A., C. F. Maurice, R. N. Carmody, D. B. Gootenberg, J. E. Button, B. E. Wolfe, A. V. Ling, A. S. Devlin, Y. Varma, M. A. Fischbach, S. B. Biddinger, R. J. Dutton & P. J. Turnbaugh, 2014. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505: 559–563.
DeSantis, T. Z., P. Hugenholtz, N. Larsen, M. Rojas, E. L. Brodie, K. Keller, T. Huber, D. dalevi, P. Hu & G. L. Andersen, 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology 72: 5069–5072.
Engel, P. & N. A. Moran, 2013. The gut microbiota of insects - diversity in structure and function. FEMS Microbiology Reviews 37: 699–735.
Foster, J. A. & K. A. McVey Neufeld, 2013. Gut-brain axis: how the microbiome influences anxiety and depression. Trends in Neurosciences 36: 305–312.
Freese, H. M. & B. Schink, 2011. Composition and stability of the microbial community inside the digestive tract of the aquatic crustacean Daphnia magna. Microbial Ecology 62: 882–894.
Garza, D. R. & B. E. Dutilh, 2015. From cultured to uncultured genome sequences: Metagenomics and modelling microbial ecosystems. Cellular and Molecular Life Sciences 72: 4287–4308.
Gaudes, A., I. Muñoz & T. Moens, 2013. Bottom-up effects on freshwater bacterivorous nematode populations: A microcosm approach. Hydrobiologia 707: 159–172.
Gloeckner, V., U. Hentschel, A. V. Ereskovsky & S. Schmitt, 2013. Unique and species-specific microbial communities in Oscarella lobularis and other Mediterranean Oscarella species (Porifera: Homoscleromorpha). Marine Biology 160: 781–791.
Haas, B. J., D. Gevers, A. M. Earl, M. Feldgarden, D. V. Ward, G. Giannoukos, D. Tabbaa, S. K. Highlander, E. Sodergren, B. Methé, T. Z. DeSantis, Human Microbiome Consortium, J. F. Petrosino, R. Knight & B. W. Birren, 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Research 21: 494–504.
Halpern, M. & Y. Senderovich, 2015. Chironomid microbiome. Microbial Ecology 70: 1–8.
Hentschel, U., J. Hopke, M. Horn, A. B. Friedrich, M. Wagner, J. Hacker J. & B. S. Moore, 2002. Molecular evidence for a uniform microbial community in sponges from different oceans. Applied and Environmental Microbiology 68: 4431–4440
Herrera, L. M., C. X. García-Laviña, J. J. Marizcurrena, O. Volonterio, R. Ponce de León & S. Castro-Sowinski, 2017. Hydrolytic enzyme-producing microbes in the Antarctic oligochaete Grania sp. (Annelida). Polar Biology 40: 947–953.
Hjelmsø, M. H., L. H. Hansen, J. Bælum, L. Feld, W. E. Holben & C. S. Jacobsen, 2014. High-resolution melt analysis for rapid comparison of bacterial community compositions. Applied and Environmental Microbiology 80: 3568–3575.
Holm, J. B. & K. B. Heidelberg, 2016. Microbiomes of Muricea californica and M. fruticosa: comparative analyses of two co-occurring Eastern Pacific octocorals. Frontiers in Microbiology 7: 917.
Horne, D.J., J. Holmes, J. Rodriguez-Lazaro & F. Viehberg (eds), 2012. Ostracoda as proxies for Quaternary climate change. Elsevier, Amsterdam.
Jarett, J., C. Fiore, C. Mazel & M. Lesser, 2013. Fluorescent epibiotic microbial community on the carapace of a Bahamian ostracod. Archives of Microbiology 195: 595–604.
Johnson, M., I. Zaretskaya, Y. Raytselis, Y. Merezhuk, S. McGinnis & T. L. Madden, 2008. NCBI BLAST: a better web interface. Nucleic Acids Research 36: 5–9.
Kanehisa, M. & S. Goto, 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acid Research 28: 27–30.
Klindworth, A., E. Pruesse, T. Schweer, J. Peplies, C. Quast, M. Horn & F. O. Glöckner, 2013. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Research 41: 1–11.
Kostic, A.D., M. R. Howitt & W. S. Garrett, 2013. Exploring host-microbiota interactions in animal models and humans. Genes & Development 27: 701–718.
Langille, M. G., J. Zaneveld, J. G. Caporaso, D. McDonald, D. Knights, J. A. Reyes, J. C. Clemente, D. E. Burkepile, R. L. Vega Thurber, R. Knight, R. G. Beiko & C. Huttenhower, 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology 31: 814–821.
Lee, O. O., Y. Wang, J. Yang, F. F. Lafi, A. Al-Suwailem & P.-Y. Qian, 2011. Pyrosequencing reveals highly diverse and species-specific microbial communities in sponges from the Red Sea. ISME Journal 5: 650–664.
Lezin, G., Y. Kosaka, H. J. Yost, M. R. Kuehn & L. Brunelli, 2011. A one-step miniprep for the isolation of plasmid DNA and lambda phage particles. PLoS ONE 6(8):e23457.
Macke, E., M. Callens, L. De Meester & E. Decaestecker, 2017. Host-genotype dependent gut microbiota drives zooplankton tolerance to toxic cyanobacteria. Nature Communications 8: 1608.
Marchesi, J.R. & J. Ravel, 2015. The vocabulary of microbiome research: a proposal. Microbiome 3: 31.
Martens, K. 1984. Annotated checklist of non-marine ostracods (Crustacea, Ostracoda) from African inland waters. Documentation Zoologique au Musée Royal de l’Afrique Centrale 20: 1–51.
Martens, K., I. Schön, C. Meisch & D. J. Horne, 2008. Global diversity of ostracods (Ostracoda, Crustacea) in freshwater. Hydrobiologia 595: 185–193.
Martins, M. J. F., J. Vandekerkhove, F. Mezquita, O. Schmit, J. Rueda, G. Rossetti & T. Namiotko, 2009. Dynamics of sexual and parthenogenetic populations of Eucypris virens (Crustacea: Ostracoda) in three temporary ponds. Hydrobiologia 636: 219–232.
McKenzie, V. J., R. M. Bowers, N. Fierer, R. Knight & C. L. Lauber, 2012. Co-habiting amphibian species harbor unique skin bacterial communities in wild populations. The ISME Journal 6: 588–596.
Meisch C., R. J. Smith & K. Martens, 2019. A subjective global checklist of the extant non-marine Ostracoda (Crustacea). European Journal of Taxonomy 492: 1–135.
Mesquita-Joanes, F., A. J. Smith & F. A. Viehberg, 2012. The ecology of Ostracoda across levels of biological organisation from individual to ecosystem: A review of recent developments and future potential. In Horne, D. J., J. Holmes, J. Rodriguez-Lazaro & F. A. Viehberg (eds), Ostracoda as proxies for Quaternary climate change. Elsevier, Amsterdam: 15–35.
Mioduchowska, M., M. J. Czyż, B. Gołdyn, A. Kilikowska, T. Namiotko, T. Pinceel, M. Łaciak & J. Sell, 2018. Detection of bacterial endosymbionts in freshwater crustaceans: the applicability of non-degenerate primers to amplify the bacterial 16S rRNA gene. PeerJ 6: e6039.
Mushegian, A. A., J.-C. Walser, K. E. Sullam & D. Ebert, 2018. The microbiota of diapause: How host-microbe associations are formed after dormancy in an aquatic crustacean. Journal of Animal Ecology 87: 400–413.
Nascimento, F. J., J. Näslund & R. Elmgren, 2012. Meiofauna enhances organic matter mineralization in soft sediment ecosystems. Limnology and Oceanography 57: 338–346.
Näslund, J., F. J. Nascimento & J. S. Gunnarsson, 2010. Meiofauna reduces bacterial mineralization of naphthalene in marine sediment. The ISME Journal 4: 1421–1430.
Newton, R. J., S. E. Jones, A. Eiler, K. D. McMahon & S. Bertilsson, 2011. A guide to the natural history of freshwater lake bacteria. Microbiology and Molecular Biology Reviews 75: 14–49.
Oksanen, J., F. G. Blanchet, R. Kindt, P. Legendre, P. R. Minchin, R. B. O’Hara, G. L. Simpson, P. Solymos, M. H. H. Stevens & H. H. Wagner, 2015. Vegan: community ecology package. R package vegan vers. 2.2–1. https://vegan.r-forge.r-project.org
Olmo, C., X. Armengol, M. Antón-Pardo & R. Ortells, 2016. The environmental and zooplankton community changes in restored ponds over 4 years. Journal of Plankton Research 38: 490–501.
Park, L. E. & A. J. Smith (eds), 2003. Bridging the gap: trends in the ostracode biological and geological sciences. The Paleontological Society Special Publications 9: 1–290.
Pogoreutz, C., N. Rädecker, A. Cárdenas, A. Gärdes, C. Wild & C. R. Voolstra, 2017. Dominance of Endozoicomonas bacteria throughout coral bleaching and mortality suggests structural inflexibility of the Pocillopora verrucosa microbiome. Ecology and Evolution 8: 2240–2252.
Prather, C. M., S. L. Pelini, A. Laws, E. Rivest, M. Woltz, C. P. Bloch, I. Del Toro, C.-K. Ho, J. Kominoski, T. A. S. Newbold, S. Parsons & A. Joern, 2013. Invertebrates, ecosystem services and climate change. Biological Reviews 88: 327–348.
Qi, W., G. Nong, J. F. Preston, F. Ben-Ami & D. Ebert, 2009. Comparative metagenomics of Daphnia symbionts. BMC Genomics 10: 172.
Quast, C., E. E. Pruesse, P. Yilmaz, J. Gerken, T. Schweer, P. Yarza, J. Peplies & F. O. Glöckner, 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Research 41: D590-D596.
Rhee, S. H., C. Pothoulakis & E. A. Mayer, 2009. Principles and clinical implications of the brain–gut–enteric microbiota axis. Nature Reviews Gastroenterology & Hepatology 6: 306–314.
Rossi, V., D. Albini, G. Benassi & P. Menozzi, 2012. To rest in hydration: hatching phenology of resting eggs of Heterocypris incongruens (Crustacea: Ostracoda). Fundamental and Applied Limnology 181: 49–58.
Schmid-Araya, J. M. & P. E. Schmid, 2000. Trophic relationships: Integrating meiofauna into a realistic benthic food web. Freshwater Biology 44: 149–163.
Schön, I., T. Kamiya, T. Van den Berghe, L. Van den Broecke & K. Martens, 2019. Novel Cardinium strains in non-marine ostracod (Crustacea) hosts from natural populations. Molecular Phylogenetics and Evolution 130: 406–415.
Sharon, G., D. Segal, J. M. Ringo, A. Hefetz, I. Zilber-Rosenberg & E. Rosenberg, 2010. Commensal bacteria play a role in mating preference of Drosophila melanogaster. Proceedings of the National Academy of Sciences of the United States of America 107: 20051–20056.
Shin, S. C., S. H. Kim, H. You, B. Kim, A. C. Kim, K. A. Lee, J. H. Yoon, J. H. Ryu & W. J. Lee, 2011. Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 334: 670–674.
Sison-Mangus, M. P., A. A. Mushegian & D. Ebert, 2015. Water fleas require microbiota for survival, growth and reproduction. ISME Journal 9: 59–67.
Skelton, J., K. M. Geyer, J. T. Lennon, R. P. Creed & B. L. Brown, 2017. Multi-scale ecological filters shape the crayfish microbiome. Symbiosis 72: 159–170.
Słomka, M., M. Sobalska-Kwapis, M. Wachulec, G. Bartosz & D. Strapagiel, 2017. High Resolution Melting (HRM) for high-throughput genotyping – limitations and caveats in practical case studies. International Journal of Molecular Sciences 18: 2316.
Smith, A. J., D. J. Horne, K. Martens & I. Schön, 2015. Class Ostracoda. In Thorp, J. & D. C. Rogers (eds), Ecology and general biology: Thorp and Covich’s freshwater invertebrates. Academic Press, London: 757–780.
Sommer, F. & F. Bäckhed, 2013. The gut microbiota – masters of host development and physiology. Nature Reviews Microbiology 11: 227–238.
Stewart, E. J., 2012. Growing unculturable bacteria. Journal of Bacteriology 194: 4151–4160.
Takacs-Vesbach, C., K. King, D. Van Horn, K. Larkin & M. Neiman, 2016. Distinct bacterial microbiomes in sexual and asexual Potamopyrgus antipodarum, a New Zealand freshwater snail. PLoS ONE 11: e0161050.
Tang, K. W., C. Dziallas, H. P. Grossart, 2011. Zooplankton and aggregates as refuge for aquatic bacteria: Protection from UV, heat and ozone stresses used for water treatment. Environmental Microbiology 13: 378–390.
Tang, K. W., V. Turk & H. P. Grossart, 2010. Linkage between crustacean zooplankton and aquatic bacteria. Aquatic Microbial Ecology 61: 261–277.
Turnbaugh, P. J., R. E. Ley, M. A. Mahowald, V. Magrini, E. R. Mardis & J. I. Gordon, 2006. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444: 1027–1031.
Vandekerckhove, T. T. M., 1998. Cytoplasmic bacteria in Heterocypris incongruens (Crustacea, Ostracoda). In Martens, K. (ed), Sex and parthenogenesis: Evolutionary ecology of reproductive modes in non-marine ostracods. Backhuys Publishers, Leiden: 119–125.
Vandekerkhove, J., K. Martens, G. Rossetti, F. Mesquita-Joanes & T. Namiotko, 2013. Extreme tolerance to environmental stress of sexual and asexual resting eggs of Eucypris virens (Crustacea, Ostracoda). Freshwater Biology 58: 237–247.
Vartoukian, S. R., R. M. Palmer & W. G. Wade, 2010. Strategies for culture of ‘unculturable’ bacteria. FEMS Microbiology Letters 309: 1–7.
Waldor, M. K., G. Tyson, E. Borenstein, H. Ochman, A. Moeller, B. B. Finlay, H. H. Kong, J. I. Gordon, K. E. Nelson, K. Debbagh & H. Smith, 2015. Where next for microbiome research? PLoS Biology 13: 1–9.
Wernegreen, J. J., 2012. Endosymbiosis. Current Biology 22: R555–R561.
Zinger, L., A. Gobet & T. Pommier, 2012. Two decades of describing the unseen majority of aquatic microbial diversity. Molecular Ecology 21: 1878–1896.
Acknowledgements
We would like to thank Ms. Paulina Nejfert for assistance with bioinformatics analysis. Paweł Olszewski was supported by START scholarship from the Foundation for Polish Science (FNP). The HRM analysis was carried out on Light Cycler 96 provided by Roche Poland. This study was partly funded by the University of Gdansk under internal Grant No. 530-L155-D249-19 attributed to Tadeusz Namiotko. We also thank two anonymous reviewers for their many insightful comments which have greatly improved this paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The experiments were performed in accordance with the Polish Act on Experiments on Animals and the EU Directive 2010/63/EU on the protection of animals used for scientific purpose.
Additional information
Handling editor: Koen Martens.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
BIOM table with bacterial OTUs distribution in ostracod samples accordingly to the similarity with the Greengenes database (Supplementary Table S1) as well as data on functional analysis of ostracod-associated metagenomes generated by PICRUST from the BIOM table analyzed subsequently using STAMP (Supplementary Table S2) are available as Electronic Supplementary Material together with two figures (Figs. S1 and S2). Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Olszewski, P., Bruhn-Olszewska, B., Namiotko, L. et al. Co-cultured non-marine ostracods from a temporary wetland harbor host-specific microbiota of different metabolic profiles. Hydrobiologia 847, 2503–2519 (2020). https://doi.org/10.1007/s10750-020-04269-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-020-04269-z