
Abstract
The distributional patterns of Arctic species are commonly affected by the recurring Pleistocene glaciations, which contributed to transient or permanent genetic isolation. Here we explore the phylogeography of the climate-sensitive Arctic fairy shrimp Branchinecta paludosa, which has a circumpolar range of distribution, with certain southern alpine outreaches. We sequenced the mitochondrial cytochrome oxidase I subunit from samples collected at ten Nearctic and nine Palaearctic sites, including southern alpine populations. A handful of ambiguous bases in certain sequences strongly suggested heteroplasmy, possibly being reported for the first time in anostracans. Evolutionary analysis of the sequence variations showed a temporal divergence coinciding with the flooding of the Beringia land bridge. Sequence alignment with outgroup taxa for phylogenetic analysis showed three distinct major clades, reflecting geographical isolation. The most divergent clade, from isolated alpine ponds in the Rocky Mountains, probably represents a different and undescribed species. Two other major clades corresponded to the geographical areas of Nearctic and Palaearctic. Finally, the southern Palaearctic outstretch showed genetic separation, most likely representing a geographical and climatic isolated relict population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Arctic offers an important field of research for understanding dispersal, vicariance and recent responses to climate change. The historical fusion of Palaearctic and Nearctic regions via the Beringia land bridge, which persisted through the Miocene and during the Pleistocene, allowed an intercontinental flux of species (Cook et al., 2005). Transgressions and flooding of Beringia in late Pliocene and in the Holocene disturbed the connection, as recurrent glaciations caused closing and re-opening of landmasses and a complex dance of expansion and retraction around the Arctic Sea for the affected biota (Hultén, 1937; Waltari et al., 2007). Beringia remained a large refugium for cold-adapted species during glacial periods, followed by rapid release into previously glaciated and pristine areas in Holocene (Weider & Hobæk, 2000; Abbott & Brochmann, 2003). In addition, several local refugia have been identified by regional haplotypes of Arctic plants (Tremblay & Schoen, 1999), especially from the North-Eastern archipelago of the Nearctic and in the North-Western coast of North Europe.
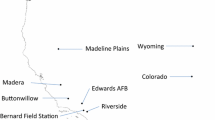
The Beringia land bridge was dominated by cold steppe climate, favouring ephemeral ponds and temporal water bodies, suitable for short-lived freshwater crustaceans. These invertebrates disperse over land by drought- and frost-resistant resting eggs by means of wind or zoochory, and may persist in dormancy for decades (Hairston, 1996). Fairy shrimps as branchinectids (Anostraca, Branchiopoda) are particularly well adapted to such habitats (Eriksen & Belk, 1999; Lindholm, 2014). The genus is present in both the New World and in most of Eurasia, but the majority of species are narrowly limited to southwestern Nearctic and the Cordillera (Eriksen & Belk, 1999; Rogers, 2006), and many are endemic, sometimes solely known from a few sites (Fugate, 1992, 1996; Rogers, 2006). The Arctic fairy shrimp (Branchinecta paludosa O.F. Müller 1788) offers the most distinct biogeographic exception. It is widespread in small cold water lakes and ponds around the Arctic Sea, with southern outreaches along the Rocky Mountains and in the alpine regions of South Norway and Sweden, and a last isolated outpost in the Tatra Mountains (Slovak republic; Fig. 1). Fugate (1992) compared morphology and allozymes for 15 Nearctic and two Eurasian branchinectids and considered B. paludosa as the bridging species between the Nearctic and Palaearctic lineages, implying that Old world Branchinecta species should form a phylogenetic cluster of relatively closely related species.

Geographical distribution of the circumpolar Arctic fairy shrimp (Branchinecta paludosa). Red–black bullet points mark the 10 Nearctic sampled geographic regions, blue–black bullet points mark the 9 Palaearctic sampled geographic regions and green shading shows the main known distribution range of B. paludosa
The aim of this study is to reconstruct the phylogeographic history of B. paludosa from its hypothesized South Nearctic origin into the Arctic and to its present biogeographical region of northern Europe, and to connect its molecular differentiation to regional glacial history. We quantify genetic polymorphism from various sites around the Arctic Sea and its southern outreaches, in order to test the hypothesis that the species originated from the Rocky Mountains and expanded into the Palaearctic across the Beringian land bridge. Phylogeographic knowledge is of particular relevance in this case, as the distribution of B. paludosa has been shown to be particularly vulnerable to warming, hence possessing features which makes it a suitable indicator for recent climate change (Lindholm et al., 2012, 2015, 2016).
Methods
We analysed samples of B. paludosa deriving from different sources accumulated over more than 20 years. Material was available from collections made in Russia (1994), Northern Norway (1995; 2012) and Canada (1999). The samples from alpine South Norway were collected in 2011–2012. In addition, we obtained samples from Sweden collected in 2001, from the Tatra Mountains (Slovakia) in 2002 and from Wyoming (U.S.A.) in 2012. Collections were made by hand-held sweep nets in tundra and mountain ponds, and conserved directly in ethanol. Long-term storage was done using 96% ethanol and storing at 4°C.
DNA extraction was performed using Mole Tissue kit on the GeneMole instrument (Mole Genetics, discontinued). Briefly, 1 individual, using either the head and thorax or the entire animal, was incubated in 100 µl Mole Lysis with 2 µl Proteinase K at 65°C for 1 h. The lysate was processed according to the manufacturer’s instructions. DNA was quantified on a NanoDrop™ 1000 Spectrophotometer (Thermo Scientific, Bonn, Germany), and diluted in Tris EDTA buffer (Fluka) to a standard concentration of 8 ng/µl. PCR amplifications were performed using a CFX96 BioRad thermocycler (Bio-Rad, Hercules, CA, USA) in 15 μl reaction volume containing 7.5 μl SsoFast or iProof Master Mix (Bio-Rad), 0.1 μM of each primers and 2.5 μl sample (8 ng/μl DNA). Reaction volume was completed with sterile deionised water.
We amplified a fragment of the mitochondrial COI gene with primers LCO1490 and HCO2198 (Folmer et al., 1994). PCR amplifications were carried out under the following conditions: a denaturing step for 2 min at 98°C, followed by 40 cycles of 98°C for 10 s, 43 or 41°C for 30 s and 72°C for 20 s followed by a final extension at 72°C for 2 min. PCR products were electrophoresed in 1.4% agarose (Top Vision LE GQ, Thermo Scientific) gel in 2X TAE buffer (VWR) and visualized with GelRed (Biotium) staining. Extracts that failed to amplify at an annealing temperature of 43°C were re-run with annealing temperature set at 41°C.
Cycle sequencing was performed in both directions using amplification primers and BigDye Terminator v3.1 kit (Life technologies, Applied Biosystems). 1 µl PCR template was used with 0.5 μl Terminator mix, 0.32 μl 10 μΜ forward or reverse primer and 1.75 μl Terminator ×5 buffer in a final volume of 10 μl. Cycle sequencing was performed using an ABI 7500 qPCR machine (Life technologies, Applied Biosystems) as follows: 96°C for 1 min followed by 28 cycles of 96°C for 10 s, 41°C for 5 s and 60°C for 4 min. Products from cycle sequencing were purified using BigDye XTerminator Purification kit (Life technologies, Applied Biosystems) adding to each PCR sample well 10 μl XTermination solution and 45 μl Sam solution, final volume of 65 μl. The PCR plate was then sealed and vortexed for 30 min prior to being processed by an ABI3730XL DNA analyser (Life technologies, Applied Biosystems). Sequence alignments and graphical presentation of the sequencing trace files were performed using CodonCode Aligner v4.0.4 (CodonCode corporation) and BioEdit v7.1.9 software (Hall, 1999). The resulting sequences plus outgroup sequences were aligned in MEGA6 using the ClustalW and Muscle algorithms (Tamura et al., 2013).
Forty nine individuals of B. paludosa from 35 different populations and 19 different geographical regions (10 Nearctic and 9 Palaearctic) were included in the phylogeographic analysis and sequenced for the mitochondrial COI gene (Fig. 1; Table 1). One additional B. paludosa sequence from the North West Territories, Canada (Genbank AF209064), was also included. For the outgroup, we sequenced one B. tolli individual collected in the Indigirka delta (Eastern Siberia), and included three American Branchinecta species from GenBank (accession numbers in parentheses): B. lynchi (FJ439749; FJ439751), B. sandiegonensis (FJ439689; FJ439697) and B. lindahli (FJ439744; FJ439748), all from the Western USA. Phylogenetic trees were rooted by an Artemiopsis stefanssoni sequence (Genbank AF209062). This taxon was chosen to represent the Chirocephalidae, which (including the Polyartemiinae) forms a sister clade to the Branchinectidae (deWaard et al., 2006; Sun et al., 2006).
Evolutionary divergence between and within phylogenetic clades was estimated by the Kimura 2-parameter model in MEGA6, applying a gamma shape parameter of 0.77 as estimated in MEGA6 for the K2P model. All codon positions were included, while ambiguous sites were excluded from the analyses. The aligned sequences (excluding Artemiopsis) were pasted into ABGD (Automatic Barcode Gap Discovery) (Puillandre et al., 2012) at http://wwwabi.snv.jussieu.fr/public/abgd/abgdweb.html using default priors, to check for gaps in nucleotide differentiation based on the Kimura 2-parameter distance measure.
We tested nucleotide substitution models in jModeltest v2.1.3 (Guindon & Gascuel, 2003; Darriba et al., 2012), based on all 58 aligned sequences (including A. stefanssoni and seven outgroup sequences). While a transition model (the TIM3 model with invariant sites) scored slightly better by Akaike and Bayesian Information criteria, we chose the General Time Reversible model with invariant sites and gamma-distributed rate categories (GTR+I+G) for phylogenetic analyses. This model had the best log-likelihood score, and scored only slightly lower on the Akaike and Bayesian Information criteria. The rationale for preferring the GTR+I+G model was that it could be easily implemented in both MrBayes and BEAST (see below). jModelTest indicated a proportion of invariant sites of 0.580 and a gamma shape parameter of 1.356 for the GTR+I+G model. The rate matrix was A → C = 0.012, A → G = 0.645, A → T = 0.022, C → G = 0.037, C → T = 0.298, G → T = 0.010. Phylogenetic analyses included neighbour-joining (NJ) in MEGA6 and maximum likelihood (PhyML v. 3.0 implemented in jModelTest v2.1.3) analyses, followed by a Bayes Inference (BI) analysis using MrBayes v.3.2 (Ronquist et al., 2012). The PhyML analysis was based on parameters from jModelTest v2.1.3 given above. The BI analysis was run with one cold and three heated chains for 2 million generations, with invariant sites and parameters of the GTR substitution model estimated by the program. Trees were sampled every 1,000 generation, and the first 25% of trees discarded as burn-in. This analysis converged well to an average standard deviation of split frequencies of 0.0074, with a proportion of invariant sites at 0.482, a gamma shape parameter of 0.968 and the rate matrix A → C = 0.014, A → G = 0.650, A → T = 0.027, C → G = 0.035, C → T = 0.258, G → T = 0.014 (mean values for all parameters).
Divergence times between clades were estimated by applying strict molecular clocks in BEAST v1.7.5 (Drummond et al., 2012). Initially, we tested for a strict clock model with a stepping stone analysis in MrBayes v. 3.2, which compares unrestricted and restricted MCMC model runs by marginal likelihoods. The models were run for 2 × 106 generations, sampling every 50,000. This analysis supported the strict clock model with marginal log-likelihoods of −3723 and −3,813 for restricted and unrestricted runs, respectively. In the BEAST runs, we only included the 50 B. paludosa sequences, since our primary focus was on divergence times within B. paludosa. BEAST analyses were run with the GTR+G+I substitution model. Due to the reduction of the data set, the following parameter settings were used for the BEAST analyses: gamma shape parameter and proportion invariant were 1.14 and 0.61, respectively; base frequencies were A 0.2415, T 0.3355, C 0.2265, G 0.1965 and rate matrix were A → C = 0.014, A → G = 0.271, A → T = 0.003, C → G = 0.019, G → (set to 0.001 due to restriction in BEAST). In accordance with Reniers et al. (2013), model assumptions were set to constant population size with exponential prior distribution and strict molecular clock sequence divergence rates of 1.4 and 2.6% as boundary values, and inclusion of 2.0% for visualization. These boundary divergence rates were selected to represent a range previously shown to be appropriate (Schwentner et al., 2012; Reniers et al., 2013) based on previous estimates for decapod crustaceans (Knowlton et al., 1993; Knowlton & Weigt, 1998; Schubart et al., 1998). Each BEAST profile was run three independent times for 10 million generations, logging every 1,000, on the computer cluster (www.lifeportal.uio.no) at the University of Oslo. After evaluation of individual and combined runs with Tracer v1.6 (Rambaut & Drummond, 2004), and discarding 10% burn-in, tree files were combined using LogCombiner v1.8.0 (Drummond & Rambaut, 2007). A maximum clade credibility tree was constructed with TreeAnnotator v1.8.0 (Drummond & Rambaut, 2007) and visualized in FigTree v1.4.0 (Rambaut, 2008).
We constructed a haplotype network using SplitsTree v. 4.13.1 (Huson & Bryant, 2006). In this analysis, all ambiguous sites (see Results about heteroplasmies) were automatically disregarded by the program, which resulted in 604 nt long alignments. For easy visualization, we chose a parsimony splits network, with branch supports evaluated by 500 bootstraps in SplitsTree.
Results
Alignment of the 58 COI sequences, including the eight sequences taken from GenBank (See Table 1), was unambiguous, and required no indels. ClustalW and Muscle yielded identical alignments of 658 bp. The full alignment contained 251 variable sites (38.1%). Considering only B. paludosa, the number of variable sites was 134 (20.4%). Base composition (excluding outgroup species and the 13 individuals with heteroplasmic positions) was generally rich in T (33.7%) and poor in G (19.6%). This pattern was most pronounced in codon third positions (35.0% T; 13.1% G), while the first position was richer in G (29.2%). Second positions were dominated by T (42.0%) and C (27.9%). Three identical sequences from Wyoming were distinguished by 42.3% A and only 6.8% G in codon third positions. In the full alignment, 196 positions were phylogenetically informative, while this number shrunk to 105 when only the 49 B. paludosa sequences were considered.
We were unable to resolve a number of ambiguous bases in certain sequences (particularly three isolates from one population in Sweden and two isolates from one population in the Tatra Mountains, Slovakia). In spite of re-amplification and sequencing in both directions, the electropherograms consistently showed dual peaks at these positions (see Appendix S1 in Supporting Information), strongly suggesting, to our surprise, heteroplasmy. Heteroplasmic nucleotide positions were mainly Y or R (45.2 and 38.1%, respectively, see Table S2), which corresponds to transitions, the most common mutation type. These ambiguities were retained in the alignment, but these positions were neglected in pairwise distance estimates (pairwise deletion). In total, 54 heteroplasmic positions occurred 84 times in 13 individuals (see S3), of which 52 occurred individually on a codon, usually on the third position, while the last 2 occurred each combined with another one on the same codon (2 heteroplasmic sites in a codon) (See Table S2). Most of these positions, 51 out of 54, were synonymous coding for the same amino acid. Finally, no stop codons were found in any of the 50 sequences. The total sequence length without the heteroplasmic positions was 604 nucleotides.
Results of phylogenetic analyses (NJ, ML and BI) were congruent in their topologies of major clades and most minor clusters. BI and ML support values are shown in Fig. 2. Relationships among Branchinecta species in the outgroup were not well resolved, whilst B. paludosa formed a distinct cluster in all analyses. Within B. paludosa, two strongly supported clusters separated the Wyoming isolates from all others, which further grouped into one Nearctic and one Palaearctic clade. Several subclades were also well supported within the Nearctic and Palaearctic clades. In the Nearctic clade, one subclade included animals from the Western Canadian Arctic (i.e. two sites on the mainland, and others on Banks, Melville and Victoria Islands) plus isolates from Churchill. This group also included a sequence from Genbank lacking a precise geographic reference (beyond North West Territories; Table 1). A second Nearctic subclade included animals from Ellesmere and Devon Islands, one animal from Thule (Greenland) and one from Melville Island. Thus, Melville Island was the only site where haplotypes from both North Nearctic subclades were detected. Among Palaearctic haplotypes, all tundra lineages formed a poorly resolved cluster at the base of the clade. This diffuse group also included two individuals from the Tatra Mountains, and one individual from Sweden (HG797704; Fig. 2). All individuals from South Norway plus two individuals from Sweden clustered in a well-supported crown clade within the Palaearctic clade. Apart from this South Fennoscandian subclade, no clear geographic patterns could be discerned among Northern Palaearctic lineages.

Bayesian phylogeny of Branchinecta paludosa differentiation based on the mitochondrial COI gene. The alignment contains 58 sequences of length 658 nt with 126 polymorphic nt positions, 105 of which were phylogenetically informative (excluding the outgroup). Support values shown on the nodes of the tree are Bayesian posterior probabilities/bootstrap support from a maximum likelihood analysis (see text). Four additional Branchinecta species form the outgroup, while the tree was rooted using a COI sequence from Artemiopsis stefanssoni. Circles indicate sequences produced for this study, whereas squares are sequences obtained from Genbank. Colours are used to identify clusters corresponding to geographical regions. Blue colour is used for Palaearctic with three different shades to identify South Norway + Sweden (dark blue), Finnmark + Russian regions (light blue) and Tatra Mountains (violet). Individual HG797704 did not cluster with other individuals from South Norway and Sweden (see discussion), and is singled out by using a blue empty circle. Red colour is used for Nearctic with two shades to identify West (dark red) from East (light red–orange). One geographic region, Melville Island, has its 2 individuals associated to the West and the East clusters (see discussion) and they are singled out by using empty orange circles
The parsimony splits analysis (Fig. 3) resulted in a haplotype network which reflected the same general clustering of sequences into the Palaearctic, North Nearctic and Wyoming clades, and also supported the South Fennoscandian clade (albeit with lower support than the BI and ML analyses (Fig. 3). In this approach (in which ambiguous sites were eliminated), the HG797704 individual from Sweden fell into the central North Palaearctic node (Fig. 3), together with the Tatra isolates and one Russian isolate from Shirokostan.

A parsimony splits network of B. paludosa haplotypes. The network was generated in SplitsTree v. 4, based on COI sequences with ambiguities removed (length 604 bp). Bootstrap support values (500 bootstraps) are given for branches connecting the three clades. Colour coding corresponds to Fig. 2 (dark blue South Fennoscandian clade; light blue North Palaearctic clade; red Western Nearctic clade; orange East Nearctic clade). Node sizes are proportional to the number of sequences represented by the node (1–9). The central node within the North Palaearctic clade includes one sequence from Russia (Shirokostan peninsula), two sequences from the Tatra Mountains and HG797704 from Sweden. The central node within the South Fennoscandian clade includes nine sequences from South Norway and Sweden, while all haplotypes that radiate from this node are Norwegian. Inset shows the same network including sequences from Wyoming
A barcode gap analysis indicated a distinct gap in sequence divergence between 0.06 and 0.09 (Fig. 4), which separated Wyoming from the main B. paludosa group. The latter group (K2P distances below 0.07) showed a bimodal frequency distribution of genetic distances, which reflects the Palaearctic and Nearctic clades. Divergence estimates between the Wyoming population and other B. paludosa ranged from 0.12 to 0.13 (K2P distances, Table 2), which suggests a specific or at least subspecific differentiation between this lineage and B. paludosa. P distance estimates between the same clades ranged from 0.099 to 0.106 (Table 2). The K2P differentiation between North Nearctic and North Palaearctic B. paludosa averaged 0.0485 (Tables 2, 3).

Barcode gap analysis based on sequences from B. paludosa, B. tolli, B. lindahli, B. lynchi and B. sandiegonensis, depicting frequency distribution of Kimura 2-parameter distances
For three geographic groups (North Nearctic, North Palaearctic and South Fennoscandia), we estimated mean within-group sequence evolutionary divergence (Table 4). The Nearctic lineage group had slightly higher within-group divergence than the North Palaearctic group, while divergence was much lower within the South Fennoscandia clade.
The results obtained with BEAST also provided a Bayesian Inference tree (Fig. 5). The major features of the resulting topology were the same as in the Mr Bayes analysis, except for the clustering of North Palaearctic lineages (Figs. 2, 5). For instance, the HG797704 lineage here clustered within the North Palaearctic clade, rather than at the base of the clade. The main interest of this analysis were the estimated times of divergence between clades. A late Miocene split was indicated between the Wyoming lineage and all other B. paludosa (15–8 million years). Further, genetic distances indicate 4.6–2.5 million years of isolation between the Nearctic and Palaearctic clades, corresponding to late Pliocene/onset of the Pleistocene. Within the Nearctic clade, an early-middle Pleistocene divergence was indicated between the two main subclades. The South Fennoscandian clade appears to have diverged from the North Palaearctic group 1.1–0.6 million years (mid Pleistocene; Fig. 5).

BEAST phylogenetic differentiation of Branchinecta paludosa from its circumpolar distribution range, including southern alpine regions. Analyses were run with strict molecular clocks. Ages in square brackets represent the time range defined from 1.4 and 2.6% divergence rates, shown on a tree based on 2.0% divergence rate. Bold numbers indicate posterior probability of nodes and horizontal blue bars indicate 95% probability intervals. Only nodes of significance for geographic and previous climatic significances are highlighted. Collapsed sequences are coloured in accordance with Fig. 2
Discussion
Data quality
We amplified the COI region of Branchinecta paludosa, which has proved useful in resolving geographic structuring within a number of different species, including branchiopod crustaceans (Cox & Hebert, 2001; Reniers et al., 2013). A possible pitfall associated with our procedure could be the erroneous use of Nuclear Mitochondrial Pseudogenes (NUMTS) sequences instead of the functional mitochondrial gene sequences (Song et al., 2008). As the COI mitochondrial gene encodes for a protein, most NUMTS can be detected by finding indels or in-frame stop codons. This last characteristic makes COI a better choice than ribosomal genes, as these also may be duplicated into NUMTS, which are harder to detect, as they are not translated. In this study, no indels were present in any of the 50 new sequences. Further, no stop codons were found either, even when taking into account the 84 identified heteroplasmic positions. This last point is consistent with the presence of functional genes from mitochondria.
Our dataset is based on a limited number of samples from locations scattered over a wide geographical range. Additional sampling would certainly have contributed to a better phylogenetic resolution, as would the inclusion of additional molecular markers. Notwithstanding these reservations, the data revealed some clear-cut phylogeographic patterns, supported by two independent analyses.
Origin of Branchinecta paludosa
The genus Branchinecta is apparently of pre-Gondwanan origin (Belk & Schram, 2001; Sun et al., 2006; Rogers & Coronel, 2011), yet most species are limited to warm-arid Nearctic (Eriksen & Belk, 1999; Brendonck et al., 2008), and a large number have a limited distribution or are local endemics, known from solely a few ponds. High sensitivity against predation and interspecific competition (Eriksen & Belk, 1999; Lindholm, 2014) and limited dispersal abilities (Bohonak, 1998; Bohonak & Jenkins, 2003) are the suggested causes for their habitat conservatism.
The wide geographical range of B. paludosa is more of an exception, but morphological, biochemical and spatial data point to an origin from North-Western USA also in this case (Fugate, 1992).
Its northward exodus is corroborated by our data. In fact, our findings indicate that the Wyoming clade represents a taxon close to, but separated from, B. paludosa. This surprising conclusion remains tentative, as our sample derives from a single locality, and only one haplotype was detected. Nonetheless, the genetic distance (13% K2P) that distinguishes this clade equals those observed among other branchinectid species (10–14%, (Vandergast et al., 2009), and among species of other anostracan genera (Reniers et al., 2013). A recently described species, B. serrata, known only from a single alpine pond in Wyoming, is morphologically very similar to B. paludosa (Rogers, 2006). Moreover, another morphologically similar species, B. kaibabensis, has a narrow range in Arizona. Both taxa have previously been misidentified as B. paludosa (Saunders et al., 1993; Belk & Fugate, 2000; Rogers, 2006). How B. paludosa and the Wyoming lineage relate to these taxa is unknown, but it seems possible that the Holarctic B. paludosa originated from an ancestor that diversified into several species, and among them only B. paludosa expanded into a wide range. Further studies including all these taxa would be necessary to resolve their relationships. The split between Wyoming and the circumpolar clade dates back to the second half of Miocene, between 15 and 8 million years ago. This estimate is within the range suggested by Cox (2003), who considered the time of divergence for the same lineage split to 9.7 (16S rDNA) and 6.7 (COI) million years. The middle Miocene climatic transition was characterized by gradual cooling and drought, combined with a shift from forest to grassland in vast areas of the Northern Nearctic (Flower & Kennett, 1994). Brtek & Thiery (1995) showed that forested regions are effective barriers of dispersal for branchinectids. Cooling combined with a shift to open grassland and the proliferation of ephemeral water bodies could have facilitated a northward dispersal and corroborated the lineage splitting during the following period.
The temporal origin of the circumpolar clade remains uncertain, as dating divergences by the strict clock method without fossil references may be problematic. Relative time estimates are reasonably stable for shallow phylogenies (Brown & Yang, 2011; Ho & Duchene, 2014), and the obtained divergence rates of 1.4 and 2.6% million years−1 largely fall within the confidence intervals of our 2% estimates. We therefore consider the BEAST calculations as reasonable. The suggested times of main splits fit well with key events of climate, transgressions and onset of the Pleistocene.
The BEAST analysis points to Pliocene as time for the last common ancestor of the Palaearctic and Nearctic clades. B. paludosa must accordingly have been present in Beringia prior to the oceanic transgressions which gradually submerged Beringia and separated the two continents during late Pliocene (Marincovich & Gladenkov, 1999; Gladenkov et al., 2002). Such a key position of Beringia for pre-Pleistocene splits is consistent with what Bernatchez & Dodson (1994) reported for whitefish (Coregonus sp.), Abbott & Comes (2004) for Saxifraga oppositifolia and Eidesen et al. (2007) reported for Cassiope tetragona.
The Nearctic clade
The North Nearctic clade of B. paludosa reveals a relatively deep Pleistocene split between Eastern high Arctic Canada (Devon and Ellesmere Il., Churchill) plus Greenland and Central/Western Arctic Canada (Victoria and Banks Il., Albert Bay, Cape Bathurst), which is likely to reflect isolation in two different glacial refugia, perhaps through several glacial cycles. The Western clade (Victoria and Banks Il., Albert Bay, Cape Bathurst) may represent the Beringian refuge, while the eastern group (Devon and Ellesmere Il., Churchill and Thule) may derive from several potential refugia. For instance, pollen analyses have indicated possible minor refugia in the Northern central Canadian archipelago (Ellesmere Island), in coastal areas of Greenland and coastal fringes of eastern North America (Heaton et al., 1996; Tremblay & Schoen, 1999). An eastern high-arctic glacial refuge was also suggested for members of the Daphnia pulex complex (Weider & Hobæk, 2003). On Melville Island in the North-Western Arctic, we detected one Western and one Eastern haplotype. The eastern haplotype may derive from a rare post-glacial dispersal event, but could also indicate that post-glacial admixture of western and eastern lineages is more widespread than we have detected. The presumed Beringian clade is also present at the Hudson Bay (Churchill), which possibly reflects a post-glacial expansion via continental water corridors across the continent NW to SE following the rapid Wisconsinan melt down. A similar pattern has also been reported in Daphnia tenebrosa (Weider & Hobæk, 2003), arctic charr Salvelinus alpinus (Wilson et al., 1996) and whitefish (Bernatchez & Dodson, 1994). The B. paludosa sequences we obtained from Churchill match perfectly with sequences from this region available in the BOLD database (http://www.boldsystems.org/index.php).
The Palaearctic clade
The phylogenetic analyses did not reveal consistent structuring within the Palaearctic clade, except for the South Fennoscandian clade. This clade contained many isolates from South Norway. To check whether this relative over-representation influenced on topology and/or support values of, a BI analysis was also performed using only four isolates from South Norway (B12, B33, B20 and B173, see Table 1) instead of 18, with the same settings as previously. The resulting topology was identical to the full BI tree, and the support values for all subclades were the same or in a few cases slightly higher. Thus, over-representation of South Norwegian isolates did not influence on the phylogenetic results.
The lack of resolution within the Palaearctic clade may be due to a higher incidence of heteroplasmies within this clade (see below). When ambiguous sites were removed in the haplotype network analysis, the Palaearctic clade appeared with two central nodes corresponding to the North Palaearctic and the South Fennoscandian clades, from each of which a series of haplotypes radiate. An alternative explanation may be that Pleistocene glaciations were more fragmented and never covered a major fraction of the Northern Palaearctic, and large areas from Eastern Siberia to Central Europe remained more or less ice free. Large areas were probably habitable for B. paludosa even during the glacials, and isolation was presumably less severe than in the heavily glaciated Nearctic, hence retarding vicariance differentiation in the Palaearctic. It is noteworthy that the distinct South Fennoscandian cluster is associated with a region that was repeatedly and heavily glaciated. Today, these populations are geographically isolated from the Arctic main range by 600–700 km (Lindholm et al., 2015). A long-standing dispute has concerned the possibility for nunatak refugia during the Weichsel glaciation in this area (Segerström & von Stedingk, 2003; Westergaard et al., 2011; Parducci et al., 2012). Our BEAST analysis indicates isolation of the South Fennoscandia clade since well before the Weichsel. However, this does not necessarily imply a continuous occupancy of the area, which may have been colonized by a lineage that survived the Weichsel somewhere else, like the periglacial European tundra. The pattern shares similarities with that found for lemming by Lagerholm et al. (2014). In addition to the deep splits, shallow regional clades seemingly later emerged on both continents, reflecting the Pleistocene dispersal patterns of B. paludosa around the Arctic Sea.
Two non-mutually exclusive trends have been suggested in Holocene dispersal of freshwater crustacean (Reniers et al., 2013): Invading species from southern, ice-free latitudes will comprise low genetic diversity and limited geographical structure (De Gelas & De Meester, 2005), while species dispersing from different local refugia possess larger genetic differentiation, as shown for rotifers by Gómez et al. (2007). The star-like pattern of the South Fennoscandia clade in the haplotype network, as well as a low nucleotide divergence within this clade, is rather consistent with a small founding population and a lack of connectivity with the main range. This pattern is further reinforced by a much lower allelic richness at several microsatellite loci among the South Norway and Sweden populations compared to populations in the Arctic (Hobæk, Anglès d’Auriac and Lindholm, unpublished data).
Saunders et al. (1993) suggested frequent input of resting eggs from the north, transported by migrating waterfowl, as a possible mechanism in maintaining isolated populations south of the northern circumpolar range, such as in Wyoming. Geographically, the Wyoming populations as well as others further north in Alberta, Canada, are situated along a major north/south flyway. The South Norway and Sweden populations are also geographically isolated from the north, and the Tatra Mountains even more so. Nonetheless, our results do not indicate that long-distance bird-mediated dispersal from the Arctic plays a major role in maintaining these populations. In South Norway and Sweden, all but one individual belong to a cluster that most probably became isolated from the Arctic range before the last glaciation. The single outlier individual from Sweden seems to derive from a northern lineage, suggesting that rare long-range dispersal events may occur, although additional corroborating data would be required to ascertain this conclusion. The Tatra population appears to be closely linked with the main range, and is possibly of more recent origin than the South Fennoscandian group.
It is noteworthy that the Siberian Arctic endemic B. tolli appears to be only distantly related to B. paludosa. It seems clear that this taxon represents an independent and probably much earlier colonization of the Palaearctic. Fugate (1992) suggested that at least two additional Eurasian species (B. ferox and B. orientalis) could have diverged from the B. paludosa lineage during its expansion in the Old World. Molecular data are well suited to test this hypothesis, but none are so far available.
Heteroplasmy has been associated to the aetiology of many mitochondrial diseases (Wallace & Chalkia, 2013) and used in forensics (Coble et al., 2009). Although at first mainly found in mammals, it has recently been described in other animal groups as well, such as fish (Artamonova et al., 2015; Dudu et al., 2012) and crustaceans (Doublet et al., 2008). Heteroplasmy may be underestimated and erroneously interpreted as an experimental artefact, and possibly ignored as the nucleotide showing the highest intensity may be selected by default. If mitochondrial heteroplasmies really are more common than previously assumed, the associated need for properly integrating this additional variation information in phylogenetic analyses will increase.
References
Abbott, R. J. & C. Brochmann, 2003. History and evolution of the arctic flora: in the footsteps of Eric Hulten. Molecular Ecology 12: 299–313.
Abbott, R. J. & H. P. Comes, 2004. Evolution in the Arctic: a phylogeographic analysis of the circumarctic plant, Saxifraga oppositifolia (Purple saxifrage). New Phytologist 161: 211–224.
Artamonova, V., A. Kucheryavyy & A. Makhrov, 2015. Nucleotide sequence diversity of the mitochondrial cytochrome oxidase subunit I (COI) gene of the Arctic lamprey (Lethenteron camtschaticum) in the Eurasian part of the range. Hydrobiologia 757: 1–12.
Belk, D. & M. Fugate, 2000. Two new Branchinecta (Crustacea: anostraca) from the Southwestern United States. The Southwestern Naturalist 45: 111–117.
Belk, D. & F. R. Schram, 2001. A new species of Anostracan from the Miocene of California. Journal of Crustacean Biology 21: 49–55.
Bernatchez, L. & J. J. Dodson, 1994. Phylogenetic relationships among Palearctic and Nearctic Whitefish (Coregonus sp.) populations as revealed by mitochondrial DNA variation. Canadian Journal of Fisheries and Aquatic Sciences 51: 240–251.
Bohonak, A. J., 1998. Genetic population structure of the fairy shrimp Branchinecta coloradensis (Anostraca) in the Rocky Mountains of Colorado. Canadian Journal of Zoology 76: 2049–2057.
Bohonak, A. J. & D. G. Jenkins, 2003. Ecological and evolutionary significance of dispersal by freshwater invertebrates. Ecology Letters 6: 783–796.
Brendonck, L., D. C. Rogers, J. Olesen, S. Weeks & W. Hoeh, 2008. Global diversity of large branchiopods (Crustacea: Branchiopoda) in freshwater. In Balian, E. V., C. Lévêque, H. Segers & K. Martens (eds), Freshwater Animal Diversity Assessment. Developments in Hydrobiology, Vol. 198. Springer, New York: 167–176.
Brown, R. & Z. Yang, 2011. Rate variation and estimation of divergence times using strict and relaxed clocks. Bmc Evolutionary Biology 11: 271.
Brtek, J. & A. Thiery, 1995. The geographic-distribution of the European Branchiopods (Anostraca, Notostraca, Spinicaudata, Laevicaudata). Hydrobiologia 298: 263–280.
Coble, M. D., O. M. Loreille, M. J. Wadhams, S. M. Edson, K. Maynard, C. E. Meyer, H. Niederstatter, C. Berger, B. Berger, A. B. Falsetti, P. Gill, W. Parson & L. N. Finelli, 2009. Mystery solved: the identification of the two missing Romanov children using DNA analysis. PLoS One 4: e4838.
Cook, J. A., E. P. Hoberg, A. Koehler, H. Henttonen, L. Wickström, V. Haukisalmi, K. Galbreath, F. Chernyavski, N. Dokuchaev, A. Lahzuhtkin, S. O. MacDonald, A. Hope, E. Waltari, A. Runck, A. Veitch, R. Popko, E. Jenkins, S. Kutz & R. Eckerlin, 2005. Beringia: intercontinental exchange and diversification of high latitude mammals and their parasites during the Pliocene and quaternary. Mammal Study 30: S33–S44.
Cox, A. J., 2003. Freshwater Phylogeography: The impact of Life History Traits on the Post-glacial Dispersal of Zooplankton in North America. University of Guelph, Ottawa.
Cox, A. J. & P. D. N. Hebert, 2001. Colonization, extinction, and phylogeographic patterning in a freshwater crustacean. Molecular Ecology 10: 371–386.
Darriba, D., G. L. Taboada, R. Doallo & D. Posada, 2012. jModelTest 2: more models, new heuristics and parallel computing. Nature Methods 9: 772.
De Gelas, K. & L. De Meester, 2005. Phylogeography of Daphnia magna in Europe. Molecular Ecology 14: 753–764.
deWaard, J. R., V. Sacherova, M. E. A. Cristescu, E. A. Remigio, T. J. Crease & P. D. N. Hebert, 2006. Probing the relationships of the branchiopod crustaceans. Molecular Phylogenetics and Evolution 39: 491–502.
Doublet, V., C. Souty-Grosset, D. Bouchon, R. Cordaux & I. Marcadé, 2008. A thirty million year-old inherited heteroplasmy. PLoS One 3: e2938.
Drummond, A. & A. Rambaut, 2007. BEAST: bayesian evolutionary analysis by sampling trees. Bmc Evolutionary Biology 7: 214.
Drummond, A. J., M. A. Suchard, D. Xie & A. Rambaut, 2012. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution 29: 1969–1973.
Dudu, A., S. E. Georgescu, P. Berrebi & M. Costache, 2012. Site heteroplasmy in the mitochondrial cytochrome b gene of the sterlet sturgeon Acipenser ruthenus. Genetics and Molecular Biology 35: 886–891.
Eidesen, P. B., T. Carlsen, U. Molau & C. Brochmann, 2007. Repeatedly out of Beringia: Cassiope tetragona embraces the Arctic. Journal of Biogeography 34: 1559–1574.
Eriksen, C. H. & D. Belk, 1999. Fairy Shrimps of California’s Puddles, Pools and Playas. Mad River Press, Eureka, CA.
Flower, B. P. & J. P. Kennett, 1994. The middle Miocene climatic transition: East Antarctic ice sheet development, deep ocean circulation and global carbon cycling. Palaeogeography, Palaeoclimatology, Palaeoecology 108: 537–555.
Folmer, O., M. Black, W. Hoeh, R. Lutz & R. Vrijenhoek, 1994. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology 3: 294–299.
Fugate, M., 1992. Speciation in the Fairy Shrimp Genus Branchinecta (Crustacea: Anostraca) from North America. University of California, Riverside.
Fugate, M., 1996. Branchinecta of North America: population structure and its implications for conservation practice. In Witham, C. W., E. Bauder, D. Belk, W. Ferren & R. Ornduff (eds), Ecology, Conservation, and Management of Vernal Pool Ecosystems—Proceedings from a 1996 Conference 1996. California Native Plant Society, Sacramento, CA: 140–146.
Gladenkov, A. Y., A. E. Oleinik, L. Marincovich & K. B. Barinov, 2002. A refined age for the earliest opening of Bering Strait. Palaeogeography Palaeoclimatology Palaeoecology 183: 321–328.
Gómez, A., J. Montero-Pau, D. H. Lunt, M. Serra & S. Campillo, 2007. Persistent genetic signatures of colonization in Brachionus manjavacas rotifers in the Iberian Peninsula. Molecular Ecology 16: 3228–3240.
Guindon, S. & O. Gascuel, 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology 52: 696–704.
Hairston, N. G., 1996. Zooplankton egg banks as biotic reservoirs in changing environments. Limnology and Oceanography 41: 1087–1092.
Hall, T. A., 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41: 95–98.
Heaton, T. H., S. L. Talbot & G. F. Shields, 1996. An ice age refugium for large mammals in the Alexander Archipelago, southeastern Alaska. Quaternary Research 46: 186–192.
Ho, S. Y. & S. Duchene, 2014. Molecular-clock methods for estimating evolutionary rates and timescales. Molecular Ecology 23: 5947–5965.
Hultén, E., 1937. Outline of the History of Arctic and Boreal Biota During the Quaternary Period; Their Evolution During and After the Glacial Period as Indicated by the Equiformal Progressive Areas of Present Plant Species Bokförlags aktiebolaget Thule edn. Lehre J Cramer, New York.
Huson, D. H. & D. Bryant, 2006. Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution 23: 254–267.
Knowlton, N. & L. A. Weigt, 1998. New dates and new rates for divergence across the Isthmus of Panama. Proceedings of the Royal Society B: Biological Sciences 265: 7.
Knowlton, N., L. Weigt, L. Solorzano, D. Mills & E. Bermingham, 1993. Divergence in proteins, mitochondrial DNA, and reproductive compatibility across the isthmus of Panama. Science 260: 1629–1632.
Lagerholm, V. K., E. Sandoval-Castellanos, D. Ehrich, N. I. Abramson, A. Nadachowski, D. C. Kalthoff, M. Germonpre, A. Angerbjorn, J. R. Stewart & L. Dalen, 2014. On the origin of the Norwegian lemming. Molecular Ecology 23: 2060–2071.
Lindholm, M., 2014. Morphologically conservative but physiologically diverse: the mode of stasis in Anostraca (Crustacea: Branchiopoda). Evolutionary Biology 41: 1–5.
Lindholm, M., F. Stordal, S. J. Moe, D. O. Hessen & P. Aass, 2012. Climate-driven range retraction of an Arctic freshwater crustacean. Freshwater Biology 57: 2591–2601.
Lindholm, M., D. O. Hessen, P. J. Færøvig, B. Rognerud, T. Andersen & F. Stordal, 2015. Is distribution of cold stenotherms constrained by temperature? The case of the Arctic fairy shrimp (Branchinecta paludosa O.F. Müller 1788). Journal of Thermal Biology 53: 46–52.
Lindholm, M., R. Wolf, A. Finstad & D. O. Hessen, 2016. Water browning mediates decimation of tha Arctic fairy shrimp Branchinecta paludosa. Freshwater Biology 61: 340–347.
Marincovich, L. & A. Y. Gladenkov, 1999. Evidence for an early opening of the Bering Strait. Nature 397: 149–151.
Parducci, L., T. Jørgensen, M. M. Tollefsrud, E. Elverland, T. Alm, S. L. Fontana, K. D. Bennett, J. Haile, I. Matetovici, Y. Suyama, M. E. Edwards, K. Andersen, M. Rasmussen, S. Boessenkool, E. Coissac, C. Brochmann, P. Taberlet, M. Houmark-Nielsen, N. K. Larsen, L. Orlando, M. T. P. Gilbert, K. H. Kjær, I. G. Alsos & E. Willerslev, 2012. Glacial survival of boreal trees in Northern Scandinavia. Science 335: 1083–1086.
Puillandre, N., A. Lambert, S. Brouillet & G. Achaz, 2012. ABGD, automatic barcode gap discovery for primary species delimitation. Molecular Ecology 21: 1864–1877.
Rambaut, A., 2008. Tree Figure Drawing Tool Version 1.2. Institute of Evolutionary Biology, University of Edinburgh, Edinburgh.
Rambaut, A. & A. J. Drummond, 2004. Tracer. University of Oxford, Oxford.
Reniers, J., B. Vanschoenwinkel, N. Rabet & L. Brendonck, 2013. Mitochondrial gene trees support persistence of cold tolerant fairy shrimp throughout the Pleistocene glaciations in both southern and more northerly refugia. Hydrobiologia 714: 155–167.
Rogers, D. C., 2006. Three new species of Branchinecta (Crustacea: Branchiopoda: Anostraca) from the Nearctic. Zootaxa 1126: 35–51.
Rogers, D. C. & J. S. Coronel, 2011. A redescription of Branchinecta pollicifera Harding, 1940, and its placement in a new genus (Branchiopoda: Anostraca: Branchinectidae). Journal of Crustacean Biology 31: 717–724.
Ronquist, F., M. Teslenko, P. van der Mark, D. L. Ayres, A. Darling, S. Hohna, B. Larget, L. Liu, M. A. Suchard & J. P. Huelsenbeck, 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61: 539–542.
Saunders, J. F., D. Belk & R. Dufford, 1993. Persistence of Branchinecta paludosa (Anostraca) in Southern Wyoming, with notes on zoogeography. Journal of Crustacean Biology 13: 184–189.
Schubart, C. D., R. Diesel & S. B. Hedges, 1998. Rapid evolution to terrestrial life in Jamaican crabs. Nature 393: 363–365.
Schwentner, M., B. V. Timms & S. Richter, 2012. Flying with the birds? Recent large-area dispersal of four Australian Limnadopsis species (Crustacea: Branchiopoda: Spinicaudata). Ecology and Evolution 2: 1605–1626.
Segerström, U. & H. von Stedingk, 2003. Early-Holocene spruce, Picea abies (L.) Karst., in west central Sweden as revealed by pollen analysis. The Holocene 13: 897–906.
Song, H., J. E. Buhay, M. F. Whiting & K. A. Crandall, 2008. Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proceedings of the National Academy of Sciences 105: 13486–13491.
Sun, X., Q. Yang & Y. Shen, 2006. Jurassic radiation of large Branchiopoda (Arthropoda: Crustacea) using secondary structure-based phylogeny and relaxed molecular clocks. Progress in Natural Science 16: 292–302.
Tamura, K., G. Stecher, D. Peterson, A. Filipski & S. Kumar, 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution 30: 2725–2729.
Tremblay, N. O. & D. J. Schoen, 1999. Molecular phylogeography of Dryas integrifolia: glacial refugia and postglacial recolonization. Molecular Ecology 8: 1187–1198.
Vandergast, A. G., D. A. Wood, M. Simovich & A. J. Bohonak, 2009. Identification of co-occurring Branchinecta fairy shrimp species from encysted embryos using multiplex polymerase chain reaction. Molecular Ecology Resources 9: 767–770.
Wallace, D. C. & D. Chalkia, 2013. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harbor Perspectives in Biology 5: a021220.
Waltari, E., R. J. Hijmans, A. T. Peterson, Á. S. Nyári, S. L. Perkins & R. P. Guralnick, 2007. Locating Pleistocene refugia: comparing phylogeographic and ecological Niche model predictions. PLoS One 2: e563.
Weider, L. J. & A. Hobæk, 2000. Phylogeography and arctic biodiversity: a review. Annales Zoologici Fennici 37: 217–231.
Weider, L. J. & A. Hobæk, 2003. Glacial refugia, haplotype distributions, and clonal richness of the Daphnia pulex complex in arctic Canada. Molecular Ecology 12: 463–473.
Westergaard, K. B., I. G. Alsos, M. Popp, T. Engelskjon, K. I. Flatberg & C. Brochmann, 2011. Glacial survival may matter after all: nunatak signatures in the rare European populations of two west-arctic species. Molecular Ecology 20: 376–393.
Wilson, C. C., P. D. N. Hebert, J. D. Reist & J. B. Dempson, 1996. Phylogeography and postglacial dispersal of arctic charr Salvelinus alpinus in North America. Molecular Ecology 5: 187–197.
Acknowledgments
This work was financed by the Biodiversity Strategic Institute Initiative at the Norwegian Institute for Water Research. We sincerely thank the following colleagues for providing us with Branchinecta samples: Veronika Sacherova, Bob Musselman, Lawrence J. Weider, Marcin Wojwodzic, Ingemar Näslund and Tor Erik Eriksen.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Diego Fontaneto
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lindholm, M., d’Auriac, M.A., Thaulow, J. et al. Dancing around the pole: holarctic phylogeography of the Arctic fairy shrimp Branchinecta paludosa (Anostraca, Branchiopoda). Hydrobiologia 772, 189–205 (2016). https://doi.org/10.1007/s10750-016-2660-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-016-2660-7