
Abstract
The renin-angiotensin-aldosterone-system (RAAS) plays a central role in the pathophysiology of heart failure and cardiorenal interaction. Drugs interfering in the RAAS form the pillars in treatment of heart failure and cardiorenal syndrome. Although RAAS inhibitors improve prognosis, heart failure–associated morbidity and mortality remain high, especially in the presence of kidney disease. The effect of RAAS blockade may be limited due to the loss of an inhibitory feedback of angiotensin II on renin production. The subsequent increase in prorenin and renin may activate several alternative pathways. These include the recently discovered (pro-) renin receptor, angiotensin II escape via chymase and cathepsin, and the formation of various angiotensin subforms upstream from the blockade, including angiotensin 1–7, angiotensin III, and angiotensin IV. Recently, the direct renin inhibitor aliskiren has been proven effective in reducing plasma renin activity (PRA) and appears to provide additional (tissue) RAAS blockade on top of angiotensin-converting enzyme and angiotensin receptor blockers, underscoring the important role of renin, even (or more so) under adequate RAAS blockade. Reducing PRA however occurs at the expense of an increase plasma renin concentration (PRC). PRC may exert direct effects independent of PRA through the recently discovered (pro-) renin receptor. Additional novel possibilities to interfere in the RAAS, for instance using vitamin D receptor activation, as well as the increased knowledge on alternative pathways, have revived the question on how ideal RAAS-guided therapy should be implemented. Renin and prorenin are pivotal since these are at the base of all of these pathways.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The heart and kidney are in close interaction with each other. In patients with heart disease, concomitant renal disease is an important prognostic factor, and vice versa, patients with renal disease often suffer and die from cardiac diseases. Morbidity and mortality associated with cardiorenal failure (cardiorenal syndrome) remains very high, and several “cardiorenal connectors” have been described to explain why decreased function of one organ leads to dysfunction of the other [1, 2]. In this cardiorenal connector concept, the renin-angiotensin-aldosterone-system (RAAS) plays a pivotal role.
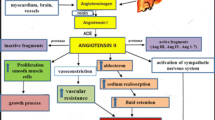
The RAAS is a key regulatory system of cardiovascular (CV), renal, and adrenal function, which maintains body fluid and electrolyte balance, as well as arterial pressure. The classical RAAS consists of a circulating endocrine system in which the principal effector hormone is angiotensin (ANG) II. The conversion of angiotensinogen to ANG I by renin is the first and rate-limiting step in the RAAS. Although activation of this system may be appropriate as an initial response to hypoperfusion (as in early stages of cardiac and renal disease), chronic activation of the RAAS is a major contributing factor to the pathogenesis and progression of CV and renal disease. Blockade of the RAAS with angiotensin-converting enzyme inhibitors (ACEi), angiotensin receptor blockers (ARBs), and aldosterone receptor antagonists aldosterone antagonists (ARAs) has become the cornerstone of treatment in patients at various stages of heart and kidney disease, from early disease (hypertension and diabetes mellitus) to advanced severe end-organ cardiorenal failure (including nephropathy, heart failure, and combined cardiorenal failure).
However, in clinical and experimental studies, the suppression of ANG II and/or aldosterone generation by ACEi and/or ARBs does not result in persistently decreased ANG II and aldosterone plasma levels. Through various “escape” mechanisms, ANG II and aldosterone levels may return to pretreatment levels or even exceed them [3]. One of the factors that may contribute to the reduced effectiveness of RAAS blockers is the compensatory increase in renin and ANG I levels in response to interruption of the negative feedback activity of ANG II signaling. Although deemed unharmful for a long time, recent analyses of clinical trials with RAAS inhibitors suggested that sustained elevations of renin levels are clearly associated with worse outcome [4, 5]. Moreover, recent evidence suggests that these increases in renin may have additional, non-RAAS-dependent effects, since renin has been shown to interact with the recently discovered (pro-) renin receptor [6].
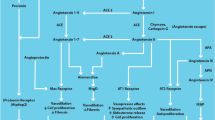
In this review, we will focus on the role of renin and its inactive precursor prorenin in the progression of cardiorenal disease, with an emphasis on direct renin inhibition and effects beyond the classical RAAS such activation of the (pro-) renin receptor ((P)RR) Fig. 1.

Renin receptor pathway
Prorenin and renin chemistry, physiology, and pathophysiology
Structure and synthesis
Renin is an enzyme that belongs to a family of aspartic proteases that also includes pepsin, cathepsin, and chymosin [7]. It consists of 350 amino acids that form 2 homologous lobes with an active side located in the cleft between them. The active site has 2 aspartic acid residues and, in contrast to the other aspartic proteases, is highly specific for its substrate angiotensinogen due to a distinct subpocket (S3sp). Prorenin is the inactive proenzyme form of renin characterized by an additional 43-amino acid prosegment covering the enzymatic cleft [8, 9].
The first step in renin synthesis is the production of preprorenin in the juxtaglomerular cells. In the cisterns of the endoplasmic reticulum, the signal peptide is cleaved off and prorenin is formed and directed to the cis-Golgi cisterns [10]. Prorenin can be either excreted immediately or converted to renin, by cleaving off the prosegment, and stored in dense core vesicles [11, 12]. The amount of prorenin that is cleaved into renin is relatively low, being around 25% [13]. In addition several extrarenal sites have been identified that produce prorenin, whereas renin production is mostly limited to the kidney. Therefore, circulating prorenin levels are 5- to 10-fold higher than those of renin [14]. The prorenin/renin ratio is, however, not a constant. In diabetes mellitus for instance, the ratio is markedly increased.
Regulation of (pro-) renin release
Given the important homeostatic actions of the RAAS on the cardiovascular system, it is not surprising that the synthesis and secretion of (pro-) renin are tightly controlled. Prorenin release is continuous and depends on the level of gene activation, transcription efficiency in the individual cell and the total number of (pro-) renin-producing cells. This is unaltered by acute stimuli, but may be influenced by chronic stimuli. Interestingly, chronic stimuli also cause more prorenin to be converted to renin, decreasing the prorenin/renin ratio [15]. The most striking exception to this rule is diabetes with end-organ damage, in which prorenin levels are increased out of proportion to renin [16, 17]. This increase is not apparent in patients with uncomplicated diabetes. It appears that this may be due to decreased clearance of prorenin from the circulation and/or increased production from extrarenal sources [18]; however, the exact mechanisms remain to be elucidated. The increase in prorenin levels in diabetes with end-organ damage suggests that not only renin but also prorenin levels are important markers of (tissue) RAAS activation.
In contrast to prorenin, renin release is not continuous. In a rat afferent arteriole, the spontaneous discharge rate of a single renin containing granule was observed on average once every 5 min [19]. Unlike prorenin, renin release can be increased rapidly in response to acute stimuli, by secretion of stored renin from the dense core vesicles [15]. The rate of renin excretion can be increased through several stimuli. The four main stimuli for renin release are: (1) decreased stretch in the baroreceptors of the afferent arteriole, (2) decreased sodium chloride delivery to the macula densa, (3) activation of renal sympathetic nerves and stimulation of β-adrenergic receptors, and (4) decreased negative feedback signaling through ANG II [20].
The transcription control of renin gene expression has been studied extensively by many groups. Numerous studies have elucidated various transcription factors, among which CREB is the best characterized. CREB regulates renin gene transcription by binding to specific and non-specific (cis and trans) regulatory elements in the 5′ UTR and 3′ UTR of the renin gene [21–23]. However, as the main second messenger for renin secretion is increase in cAMP [24], all mechanisms increasing cAMP may stimulate renin release, including prostaglandins E2 and I2 [25, 26], adrenomedullin [27], dopamine [28], and the neurohormones CGRP [29] and PACAP [30]. Interestingly, cytosolic calcium attenuates renin release. Since calcium usually facilitates exocytosis, its inhibitory effect on renin secretion has been coined as the “calcium paradox of renin release” [31]. Recent data suggest that this effect is mediated through the reduction of cytosolic cAMP by calcium [32, 33]. Calcium-mobilizing hormones such as ANG II, endothelins, or vasopressin may thus inhibit the secretion of renin [34–36].
Other mechanisms regulating renin release are under investigation. We recently reviewed the important role of nuclear hormone receptors in renin regulation [37]. Epidemiological studies show an inverse relationship between vitamin D and plasma renin activity, suggesting an inhibitory effect of vitamin D on renin secretion [38]. One of the mechanisms put forward is that the effect of vitamin on renin secretion is mediated through an increase in intracellular calcium [39]. Other experimental studies show that the vitamin D receptor (VDR) binds retinoid X receptor forming a heterodimer that competes with other regulators for elements in the renin promoter. By this, it suppresses renin transcription [37, 40–43]. In mice with total disruption of the gene encoding for the VDR, renal renin mRNA levels are threefold higher than in wild-type mice, and plasma ANG II is increased 2.5-fold. As the angiotensinogen levels show no difference between both groups, the increase in ANG II is attributed to increased renin activation [40].
In contrast to the extensive literature on transcriptional regulation of renin, there is almost no knowledge on genetic regulation of renin. Newton-Cheh et al. [44] showed that aldosterone-to-renin ratio is heritable and that modest linkage to chromosome 11p exists, but was not associated with 17 common variants in the renin gene. We are currently conducting genome-wide association studies (GWAS) to link genetic variants to renin levels.
Finally, it is important to notice that most drugs used to treat CV and renal disease can influence renin levels (Table 1). ACEi and ARB, for example, are used to block the RAAS; however, by doing so, they also block the ANG II-negative feedback and therefore cause an increase in renin levels. A rise in renin is also observed after administration of diuretics, mainly due to reduced circulatory volume and neurohormonal feedback. In addition loop diuretics can stimulate renin release by the inhibition of macula densa sodium transport in the kidney, which mimics a situation of low sodium delivery to the macula densa and thus elicits renin secretion [45]. In contrast, beta-blockers reduce renin levels through suppression of beta-adrenergic stimulation of the kidney [46]. It is important to dissect these effects from the physiological response, when measuring RAAS activation.
The prorenin/renin receptor or (P)RR
The general assumption that prorenin is merely an inactive precursor of renin has been challenged by the recent discovery of the (pro-) renin receptor ((P)RR). This receptor has been localized in various tissues, like brain, kidney, and heart, specifically in vascular smooth muscle cells in human heart and kidney, in glomerular mesangial cells and in distal and collecting tubular cells in the kidney. (P)RR binds both renin and prorenin [6]. Upon binding of prorenin to (P)RR, the prosegment covering the active site of prorenin becomes unfolded, and the enzymatic cleft exposed, activating prorenin in a non-proteolytic way [14] (Fig. 1). In addition upon binding to the (P)RR, the enzymatic activity of renin is increased [47] This renders the receptor an important regulator of tissue RAAS activity [17].
Interestingly, there is also evidence that the (P)RR may exert (angiotensin independent) effects by the activation of an intracellular postreceptor cascade. The cascade includes the activation of mitogen-activated protein kinase (MAPK), ERK1, ERK2, and phosphorylation of heat shock protein 27 (HSP27), leading to enhanced synthesis of DNA, plasminogen activator inhibitor-1 (PAI-1), collagen-1, fibronectin, and transforming growth factor-β1 (TGFβ1) [48–50]. This suggests an important role for (P)RR in the tissue remodeling process and provides a mechanism through which enzymatically inactive prorenin may exert an effect. It is of note however that in the absence of a specific inhibitor, we currently lack data whether (P)RR is pathophysiologically relevant and a potential target for treatment.
Plasma renin concentration versus activity
Although measurement of renin has long been used to assess RAAS activation, prorenin levels may be of interest as well. In addition to absolute renin and prorenin levels, the prorenin/renin ratio may provide useful information to dissect various pathways stimulating the RAAS and potentially guide therapy.
Traditionally, renin levels have been estimated by measuring its enzymatic activity. Plasma renin activity (PRA) is expressed as the amount of angiotensinogen that is converted to ANG I per time unit. This method is, however, also dependent on the amount of angiotensinogen. Other methods are measurement of active renin concentration (ARC or APRC) with an antibody directed against the active site of renin. These techniques show a high correlation and measure both renin and activated prorenin, but not inactive prorenin. Some authors also refer to ARC using the term plasma renin concentration. It is, however, important to make the distinction with total plasma renin concentration (TPRC), which includes inactive prorenin [51] The amount of prorenin is usually determined as the difference between TPRC and ARC or PRA, but can also be measured directly [52]. The distinction between PRA/ARC and TPRC has become even more important with the discovery of the (P)RR and the development of direct renin inhibitors that can block the active site of renin. The distinction between PRA and ARC is less clear, but may be of importance when angiotensinogen levels are the rate-limiting factor and in evaluating the effects of the (P)RR on renin activity. In an observational study in heart failure patients, ARC appeared to be a better predictor than PRA [53], underscoring the importance to select the appropriate measuring technique for the population of interest.
Evidence for a pivotal role of renin in heart and/or renal failure
Although the RAAS first has been described decades ago, multiple new pathways and mechanisms are continuously being discovered (Fig. 2). Renin plays a central role in all of these pathways. By blocking the action or reducing the plasma levels of ANG II, renin and prorenin production is increased leading to accumulation of the upstream and parallel RAAS components, including ANG I, ANG 1–7, and ANG IV.

Renin angiotensin system extended, (P)RR (pro-) renin receptor, MAPK mitogen-activated protein kinase, ERK extracellular-signal-regulated kinase, HSP heat shock protein, ANG angiotensin, AT1R/AT2R/AT4R angiotensin receptor type 1/2/4, BKR bradykinin receptor, ACE angiotesin converting enzyme, MME neprilysin
Several publications have shown a link between renin levels and cardiovascular and/or renal disease [54, 55]. It was assumed that renin acts through the activation of the classical circulating and local RAAS systems and by default could not exert direct effects or effects outside the main cascade. Discovery of the (P)RR and various angiotensin subforms has challenged this view, and there is increasing evidence that renin plays an important role in the development of CV and renal disease independent of the classical RAAS. Both observational and interventional studies have sought to dissect the effect of renin from the classical RAAS.
Prognostic value of plasma renin activity/concentration
Several studies have been published examining the association of renin with cardiovascular disease. In interpreting these results, it is important to consider the background medication, since these have a strong influence on renin levels and can obscure the results, but also the technique used to measure renin differs in the various studies. The first major prospective study on the association between renin and the incidence of cardiovascular disease has been conducted by Alderman et al. in 1991. They measured PRA and sodium excretion in 1,717 patients with an untreated systolic blood pressure ≥160 mmHg or diastolic blood pressure ≥95 mmHg or on antihypertensive medication. Patients had not taken their antihypertensive drugs 4 weeks prior to the PRA and sodium measurements. Comparison of the high- versus low-renin group showed that high PRA was associated with increased risk for incident myocardial infarction even after adjustment for age, sex, race, cholesterol, smoking, glucose, blood pressure, and use of beta-blockers [54]. Several years later, Meade et al. conducted a study in 803 untreated normotensive patients. In this cohort, however, the relationship between renin (here:ARC) and CV events could not be confirmed [56]. A recent report from the Framingham Heart Study has shown an association of renin (here: PRA) with short-term all-cause mortality <3 years, but not long-term mortality or CV disease (myocardial infarction, unstable angina pectoris, stroke, or congestive heart failure). [55] The relationship may have been obscured, however, by the use of various antihypertensive drugs, including ACEi, diuretics, and beta-blockers. Another study described patients with coronary artery disease and reported that high renin (PRA > 2,30 ng/ml/h) was associated with cardiac morbidity and mortality [57]. In interpreting these epidemiological data, ANG II was generally thought to be the main culprit [58]; however, in heart failure patients, high PRA was also associated with mortality in both patients on ACEi or ARB [4, 53]. Part of the observed associations may be explained by ANG II and aldosterone breakthrough [59, 60], but it is important notice that other mechanisms may play a role, such as direct effects of renin through the (P)RR.
This distinction is crucial in the development of new treatment for CV disease. Although the classical RAAS may (temporarily) be blocked by ACEi, ARB, and ARAs and has been shown to improve prognosis, the subsequent rise of renin levels is worrisome, considering it is associated with adverse prognosis. Unfortunately, the aforementioned studies by their observational design cannot answer the question whether renin is a risk factor or indicator reflecting neurohormonal activation due to compromised circulation. Hopefully, interventional studies with direct renin inhibitors and renin receptor blockers will provide the answer.
Renin in cardiorenal interaction
Heart failure (HF) is often complicated by decreased renal blood flow and a subsequent decrease in glomerular filtration rate (GFR) [61, 62]. Decreased renal function is one of the strongest predictors of mortality in patients with advanced HF [61] In these patients, RAAS is not only activated to maintain systemic circulatory volume, but mainly to maintain GFR [63]. Initiation of RAAS blockade is therefore often associated with an initial decrease in GFR. Long-term RAAS activation, however, negatively influences renal function, among others through fluid and salt retention and subsequent increase in congestion [62] This is supported by the bidirectional relationship that is observed between renin levels and renal function. In addition, ANG II exerts various potentially harmful effects including proliferative and profibrotic effects. Activation of RAAS may therefore lead to a downward spiral, which can potentially be stopped by RAAS blockade. The potential benefit of interfering in the RAAS is supported by the observation in the Val-Heft trial that double RAAS blockade in patients with heart failure is especially beneficial in patients with kidney disease [64]. PRA and APRC often are increased in patients with HF and renal dysfunction [61, 65]. Caution has to be taken, however, to extrapolate these results to all patients with kidney disease, since the ONTARGET and VALIANT trial showed harmful effects of double RAAS blockade in patients with resp. atherosclerotic vascular disease and left ventricular dysfunction directly after myocardial infarction [66, 67]. Here, some RAAS activation may well be a necessary compensatory mechanism. Interesting to notice is that in contrast to patients with an estimated GFR (eGFR) < 60 ml/min/1,73 m2, patients with proteinuria had decreased PRA levels.
The aforementioned trials however interfered downstream of ANG I and did not target renin activity or concentration, since heart failure and kidney disease are associated with increased renin levels, especially in the presence of ACEi or ARB. Targeting PRA may prove especially effective in these patients. The role of RAAS in cardiorenal interaction however is complex. According to the primary injury, as well as the stage of disease, activation can be either compensatory or harmful. Therapies targeting PRA or PRC may provide important additional evidence.
The (pro-) renin receptor in cardiorenal disease
Evidence regarding the role of the (P)RR in CV and kidney disease is slowly becoming available. Studies with transgenic animals have shown evidence that (P)RR might be related to CV and renal diseases. Rats with a ubiquitous, yet moderate overexpression of (P)RR develop proteinuria and progressive nephropathy, despite normal blood pressure, suggesting a direct pathological role of (P)RR in renal damage [68]. Rat models with a strong overexpression of human (P)RR in vascular smooth muscle cells showed a progressive increase in systolic blood pressure and heart rate at 4 months of age, although kidney function remained normal [69].
Mice that lack the gene encoding the (P)RR are not viable. Recently, mice were generated with cardiorestricted deletion of (P)RR; these mice died because of cardiomyocyte cell death due to acidification. This does not come as a complete surprise as the (P)RR not only binds renin and prorenin, but also exerts other functions, notably acting as a vacuolar ATPase and regulator of cellular pH [70]. Furthermore, (P)RR also plays an important role in neural development through renin-independent mediation of wnt signaling [71] Pharmacological inhibition of the (P)RR with a (P)RR-blocker could therefore have provided conclusive evidence on the role of the receptor in cardiac and renal disease. Development of a drug blocking the activation of (P)RR by renin proved to be very difficult, since structure–function studies and crystallographic data are lacking. Recently, a ‘handle region peptide’ (HRP) has been discovered, which allegedly blocks the (P)RR [72]. Several studies have suggested that HRP may prevent and even reverse diabetic nephropathy in diabetic rodents and diminish cardiac fibrosis in stroke-prone spontaneously hypertensive rats [73, 74]. However, other studies have shown less promising results: HRP did not show any beneficial effect in high-renin low-prorenin Goldblatt rats [75] or mice overexpressing prorenin [76]. The effectiveness of the HRP in blocking the prorenin receptor is questioned, since in vitro studies showed that prorenin binding to (P)RR was not prevented by HRP [77]. In summary, although in the future, the (P)RR may provide a novel opportunity to treat CV and renal disease, and at this point, the exact function of the (P)RR in CV and kidney disease remains to be elucidated. In addition, caution has to be taken in blocking (P)RR, since it appears to exerts renin-independent effects beyond the cardiorenal system.
Renin blockade
As mentioned above, both ACEi and ARB increase renin levels due to the loss of negative feedback of ANG II on renin release. Despite the aforementioned clues that high PRA might play a role in the progression of cardiac disease in patients on RAAS blockade, conclusive evidence is missing. Directly blocking the active site of renin may provide important information. It was already in 1980 that the first studies were performed with a renin inhibitor [78]; however, the effectiveness of this renin inhibitor was poor, mainly due to lack of specificity [79]. Recently, an orally active renin inhibitor, aliskiren, has become commercially available, and several other direct renin inhibitors are in development. Numerous studies are now trying to establish the potentials for this treatment in CV and renal disease. Despite low bioavailability, aliskiren blocks the active site of renin and effectively lowers PRA [80], thus providing very useful information on the role of PRA outside the classical RAAS pathway.
Since aliskiren blocks PRA, it acts upstream of ACEi, ARB of ARA and is believed to block the RAAS more completely. Its exact effects on ANG 1–7, ANG 1–9, ANG 1–5, ANG III, and ANG IV formation, however, have not been studied in detail. Aliskiren blocks the active site of renin and can thus block both renin and non-proteolitically activated prorenin. Therefore, it has the potential to block both circulating and tissue RAAS. This has been supported by the observation that aliskiren blocks tissue RAAS more effectively that ACEi and ARB [81].
These assumptions have been supported by the observation that 3 months of aliskiren 150 mg once daily provided additional blood pressure lowering on top op of an ACEi, ARB, or diuretic [82], and it reduced PRA, urinary aldosterone, and BNP on top of ‘optimal’ therapy in stable HF patients in the ALOFT trial [83]. Furthermore, aliskiren reduced LV mass as much as Losartan, and the combination reduced LV mass slightly more, however not statistically significant in patients with hypertension and left ventricular hypertrophy [84]. Unfortunately, the ASPIRE study did not show any improvement in echocardiographic measurements in patients with left ventricular dysfunction after myocardial infarction when treated with aliskiren on top of beta-blockers and ACEi or ARB [85], neither did the ALOFT trial in stable HF patients. This may however be due to the short follow-up time.
Blocking PRA results in an increase in PRC. In diabetic TG(mRen-2)27 rats, aliskiren did not prevent renin binding of (pro-) renin to the (P)RR nor did it block the intracellular cascades, and therefore, the intracellular cascades may even increase due to higher PRC. Although an aliskiren-induced suppression of gene expression of (P)RR was observed in vivo, this was not observed in human mesangial cells in vitro. The most likely explanation is that in vivo high-(pro) renin levels inhibit (P)RR expression via negative feedback [86]. The exact mechanism, however, remains to be elucidated, and whether the increased PRC results in harmful effects is still subject of debate [87].
Several studies are now on the way evaluating the effects of aliskiren in patients with both systolic and diastolic heart failure. The Atmosphere [88] is currently investigating the effects of aliskiren compared to and on top of Enalapril on morbidity and mortality in patients with systolic heart failure, and the ASTRONAUT study is investigating the effect of aliskiren in the acute HF setting [89]. Studies on patients with diastolic heart failure are on their way as well. The evidence for a potential effect of renin blockade in diastolic heart failure is, however, scarce. Patients with diastolic heart failure tend to have a higher PRA than healthy controls, although not as high as patients with systolic HF [90]; however, this may be due to concomitant medication. Moreover, ACEi and ARB have proven little benefit in these patients so far [91–93]. The effects of direct renin inhibition remain to be investigated.
There is also some evidence that direct renin inhibition may improve renal function in patients with heart failure by improving effective renal plasma flow (ERPF). In a normotensive population, direct renin inhibition has been shown to have stronger beneficial effects on renal hemodynamics in comparison with ACEi [94]. There was also an increase in ERPF observed in patients with diabetes type I treated with direct renin inhibitors [95]. Whether these results can be achieved in patients with heart failure and decreased renal function is currently under investigation in the ARIANA-CHF-RD trial (clinicaltrials.gov id NCT00881439). As mentioned above, the reactive rise of PRC in patients on direct renin inhibition raises some concern. Its effects, however, are unknown, since medication specifically lowering TPRC is missing.
There are a few agents that in addition to other effects lower renin. First, beta-blockers have been proven beneficial in treatment of the entire spectrum of CV disease. Part of their beneficial effect is attributed to the decrease in TPRC and PRA as a consequence of inhibition of the beta1-adrenergic receptors in the JG cells [96]. Indeed, several post hoc analyses of beta-blocker trials in patients with heart failure showed that the beta-blockers lower PRA [97].
Another therapy aimed to lower PRA and/or TPRC is activation of the VDR. Several experimental studies show that the selective vitamin D receptor activator paricalcitol effectively reduces renin transcript levels and PRA in mice [40, 98]. Furthermore, in Dahlt-salt sensitive rats [99] and in Spontaneously Hypertensive Rats (SHR) [100], paricalcitol treatment attenuated the development of hypertensive cardiomyopathy, which was ascribed at least in part due to lower renin levels. In rat model of nephropathy, paricalcitol lowered proteinuria associated with the inhibition of the RAAS [101]. These promising experimental results have been backed-up by small-scale clinical observations. In small-scale clinical studies, administration of 1,25(OH)2D3 showed reductions in PRA, ANG II levels, BP, and myocardial hypertrophy [41, 102]. Kong et al. also showed in a human pilot study in chronic hemodialysis patients that treatment with VDR activators lowered PRA in human subjects. The recently published VITAL study [103] confirmed the antiproteinuric effects of paricalcitol in patients with CKD stage 3 and 4. The authors did not observe changes in aldosterone and PRA, but stated that the trial was not designed to measure effects on the RAAS. Currently, the “Study to Investigate the Effects of Vitamin D Administration on Plasma Renin Activity in Patients With Stable Chronic Heart Failure (Vit D-CHF)” trial investigates the effect of high-dose vitamin D on plasma renin activity in chronic heart failure patients (clinicaltrials.gov id: NCT01092130).
Finally, more experimental approaches like gene therapy with antisense oligos directed against renin are tested for their value to reduce renin levels [104].
Future perspectives
Development of ACEi, and later ARBs, has substantially improved the prognosis of patients with heart failure and/or kidney disease. Morbidity and mortality, however, remain high. Renin levels are generally elevated in these patients because of chronic activation of the RAAS and compensatory increases in response to chronic use of RAAS inhibitors. There is ample evidence that this might be harmful. The development of renin inhibitors increases our arsenal to modulate the RAAS and might provide evidence as to whether increased renin activity is a cause for autonomic progression of CV and kidney disease or merely a risk indicator.
The development of new RAAS blockers also renews the question whether RAAS-guided therapy may prove additional benefit. There has been a vigorous debate whether drug choice for the treatment of hypertension should be based on PRA. Although currently PRA-based treatment has not made it to the guidelines, the development of new drugs however also provides us with new tools to block the RAAS at different sites and promote better understanding of the RAAS and alternative pathways.
The discovery of the (P)RR has also raised the question as to effects of increased renin levels under renin blockade and the potential of blocking the receptor itself. There have been reports that a (P)RR-blocker has been developed, and it may be a effective treatment in CV and renal disease [73, 74]. However, these results have not be confirmed by other study groups, and more research is needed to fully understand the role of (P)RR in CV disease.
Questions also arise about the role of prorenin in cardiac and renal disease. The ALTITUDE-trial is currently investigating the effect of a direct renin inhibitor in patients with diabetes that usually have excessive levels of prorenin [105] It is believed that prorenin may influence prognosis by activating tissue RAAS, which can potentially be blocked by direct renin inhibitors, but it may also exert a direct effect through the (pro-) renin receptor. Results of this trial may provide useful information for future investigations.
In conclusion, the RAAS is an important regulatory mechanism in heart failure and kidney disease. The system, however, is not static and blockade of one of the components results in upregulation of other pathways. Important questions to be answered are which pathways are deleterious and which may provide beneficial effect in different patient populations. Renin plays a pivotal role in the activation of the RAAS and many of the alternative pathways and will most likely become an important new target for the treatment of CV and renal disease. Results of ongoing trials with direct renin inhibitors as well as new studies examining the different effects of renin concentration and activity will hopefully provide conclusive evidence on the role of renin in cardiorenal disease.
References
Bongartz LG, Cramer MJ, Doevendans PA, Joles JA, Braam B (2005) The severe cardiorenal syndrome: ‘Guyton revisited’. Eur Heart J 26(1):11–17
Ronco C, Haapio M, House AA, Anavekar N, Bellomo R (2008) Cardiorenal syndrome. J Am Coll Cardiol 52(19):1527–1539
van de Wal RM, Plokker HW, Lok DJ, Boomsma F, van der Horst FA, van Veldhuisen DJ et al (2006) Determinants of increased angiotensin II levels in severe chronic heart failure patients despite ACE inhibition. Int J Cardiol 106(3):367–372
Latini R, Masson S, Anand I, Salio M, Hester A, Judd D et al (2004) The comparative prognostic value of plasma neurohormones at baseline in patients with heart failure enrolled in Val-HeFT. Eur Heart J 25(4):292–299
Rouleau JL, Packer M, Moye L, de Champlain J, Bichet D, Klein M et al (1994) Prognostic value of neurohumoral activation in patients with an acute myocardial infarction: effect of captopril. J Am Coll Cardiol 24(3):583–591
Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD (2002) Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest 109(11):1417–1427
Guruprasad K, Dhanaraj V, Groves M, Blundell T (1995) Aspartic proteinases: The structures and functions of a versatile superfamily of enzymes. Perspect Drug Discov Des 2(3):329–341
Sielecki AR, Hayakawa K, Fujinaga M, Murphy ME, Fraser M, Muir AK et al (1989) Structure of recombinant human renin, a target for cardiovascular-active drugs, at 2.5 A resolution. Science 243(4896):1346–1351
Rahuel J, Rasetti V, Maibaum J, Rueger H, Goschke R, Cohen NC et al (2000) Structure-based drug design: the discovery of novel nonpeptide orally active inhibitors of human renin. Chem Biol 7(7):493–504
Schweda F, Friis U, Wagner C, Skott O, Kurtz A (2007) Renin release. Physiology (Bethesda) 22:310–319
Alund M (1980) Juxtaglomerular cell activity during hemorrhage and ischemia as revealed by quinacrine histofluorescence. Acta Physiol Scand 110(2):113–121
Buhrle CP, Nobiling R, Mannek E, Schneider D, Hackenthal E, Taugner R (1984) The afferent glomerular arteriole: immunocytochemical and electrophysiological investigations. J Cardiovasc Pharmacol 6(Suppl 2):S383–S393
Pratt RE, Carleton JE, Richie JP, Heusser C, Dzau VJ (1987) Human renin biosynthesis and secretion in normal and ischemic kidneys. Proc Natl Acad Sci USA 84(22):7837–7840
Danser AH, Deinum J (2005) Renin, prorenin and the putative (pro)renin receptor. Hypertension 46(5):1069–1076
Toffelmire EB, Slater K, Corvol P, Menard J, Schambelan M (1989) Response of plasma prorenin and active renin to chronic and acute alterations of renin secretion in normal humans. Studies using a direct immunoradiometric assay. J Clin Invest 83(2):679–687
Luetscher JA, Kraemer FB, Wilson DM, Schwartz HC, Bryer-Ash M (1985) Increased plasma inactive renin in diabetes mellitus. A marker of microvascular complications. N Engl J Med 312(22):1412–1417
Nguyen G, Danser AH (2008) Prorenin and (pro)renin receptor: a review of available data from in vitro studies and experimental models in rodents. Exp Physiol 93(5):557–563
Franken AA, Derkx FH, Man in’t Veld AJ, Hop WC, van Rens GH, Peperkamp E et al (1990) High plasma prorenin in diabetes mellitus and its correlation with some complications. J Clin Endocrinol Metab 71(4):1008–1015
Skott O (1986) Episodic release of renin from single isolated superfused rat afferent arterioles. Pflugers Arch 407(1):41–45
Harrison-Bernard LM (2009) The renal renin-angiotensin system. Adv Physiol Educ 33(4):270–274
Bader M, Ganten D (2000) Regulation of renin: new evidence from cultured cells and genetically modified mice. J Mol Med 78(3):130–139
Pan L, Gross KW (2005) Transcriptional regulation of renin: an update. Hypertension 45(1):3–8
Castrop H, Hocherl K, Kurtz A, Schweda F, Todorov V, Wagner C (2010) Physiology of kidney renin. Physiol Rev 90(2):607–673
Chen L, Kim SM, Oppermann M, Faulhaber-Walter R, Huang Y, Mizel D et al (2007) Regulation of renin in mice with Cre recombinase-mediated deletion of G protein Gs{alpha} in juxtaglomerular cells. Am J Physiol Renal Physiol 292(1):F27–F37
Friis UG, Stubbe J, Uhrenholt TR, Svenningsen P, Nusing RM, Skott O et al (2005) Prostaglandin E2 EP2 and EP4 receptor activation mediates cAMP-dependent hyperpolarization and exocytosis of renin in juxtaglomerular cells. Am J Physiol Renal Physiol 289(5):F989–F997
Jensen BL, Schmid C, Kurtz A (1996) Prostaglandins stimulate renin secretion and renin mRNA in mouse renal juxtaglomerular cells. Am J Physiol Renal Physiol 271(3):F659–F669
Jensen BL, Kramer BK, Kurtz A (1997) Adrenomedullin stimulates renin release and renin mRNA in mouse Juxtaglomerular granular cells. Hypertension 29(5):1148–1155
Kurtz A, Bruna RD, Pratz J, Cavero I (1998) Rat Juxtaglomerular cells are endowed with Da-1 dopamine-receptors mediating renin release. J Cardiovasc Pharmacol 12(6):658–663
Kurtz A, Muff R, Born W, Lundberg JM, Millberg BI, Gnadinger MP et al (1988) Calcitonin gene-related peptide is a stimulator of renin secretion. J Clin Invest 82(2):538–543
Hautmann M, Friis UG, Desch M, Todorov V, Castrop H, Segerer F et al (2007) Pituitary adenylate cyclase-activating polypeptide stimulates renin secretion via activation of PAC1 receptors. J Am Soc Nephrol 18(4):1150–1156
Hackenthal E, Paul M, Ganten D, Taugner R (1990) Morphology, physiology, and molecular biology of renin secretion. Physiol Rev 70(4):1067–1116
Grunberger C, Obermayer B, Klar J, Kurtz A, Schweda F (2006) The calcium paradoxon of renin release: calcium suppresses renin exocytosis by inhibition of calcium-dependent adenylate cyclases AC5 and AC6. Circ Res 99(11):1197–1206
Ortiz-Capisano MC, Ortiz PA, Harding P, Garvin JL, Beierwaltes WH (2007) Decreased intracellular calcium stimulates renin release via calcium-inhibitable adenylyl cyclase. Hypertension 49(1):162–169
Kurtz A, Pfeilschifter J, Hutter A, Buhrle C, Nobiling R, Taugner R et al (1986) Role of protein kinase C in inhibition of renin release caused by vasoconstrictors. Am J Physiol Cell Physiol 250(4):C563–C571
Ritthaler T, DellaBruna R, Kramer BK, Kurtz A (1996) Endothelins inhibit cyclic-AMP induced renin gene expression in cultured mouse juxtaglomerular cells. Kidney Int 50(1):108–115
Vandonge R, Peart WS (1974) Calcium dependence of inhibitory effect of angiotensin on renin secretion in isolated perfused kidney of rat. Br J Pharmacol 50(1):125–129
Kuipers I, van der Harst P, Navis G, van Genne L, Morello F, van Gilst WH et al (2008) Nuclear hormone receptors as regulators of the renin-angiotensin-aldosterone system. Hypertension 51(6):1442–1448
Resnick LM, Muller FB, Laragh JH (1986) Calcium-regulating hormones in essential hypertension. Relation to plasma renin activity and sodium metabolism. Ann Intern Med 105(5):649–654
Burgess ED, Hawkins RG, Watanabe M (1990) Interaction of 1, 25-dihydroxyvitamin D and plasma renin activity in high renin essential hypertension. Am J Hypertens 3(12 Pt 1):903–905
Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP (2002) 1, 25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 110(2):229–238
Kimura Y, Kawamura M, Owada M, Oshima T, Murooka M, Fujiwara T et al (1999) Effectiveness of 1, 25-dihydroxyvitamin D supplementation on blood pressure reduction in a pseudohypoparathyroidism patient with high renin activity. Intern Med 38(1):31–35
Sigmund CD (2002) Regulation of renin expression and blood pressure by vitamin D(3). J Clin Invest 110(2):155–156
Kuipers I, van der Harst P, Kuipers F, van Genne L, Goris M, Lehtonen JY et al (2010) Activation of liver X receptor-alpha reduces activation of the renal and cardiac renin-angiotensin-aldosterone system. Lab Invest 90(4):630–636
Newton-Cheh C, Guo CY, Gona P, Larson MG, Benjamin EJ, Wang TJ et al (2007) Clinical and genetic correlates of aldosterone-to-renin ratio and relations to blood pressure in a community sample. Hypertension 49(4):846–856
Castrop H, Lorenz JN, Hansen PB, Friis U, Mizel D, Oppermann M et al (2005) Contribution of the basolateral isoform of the Na-K-2Cl- cotransporter (NKCC1/BSC2) to renin secretion. Am J Physiol Renal Physiol 289(6):F1185–F1192
Holmer SR, Hengstenberg C, Mayer B, Engel S, Lowel H, Riegger GA et al (2001) Marked suppression of renin levels by beta-receptor blocker in patients treated with standard heart failure therapy: a potential mechanism of benefit from beta-blockade. J Intern Med 249(2):167–172
Zhou A, Carrell RW, Murphy MP, Wei Z, Yan Y, Stanley PL et al (2010) A redox switch in angiotensinogen modulates angiotensin release. Nature 468(7320):108–111
Huang Y, Border WA, Noble NA (2007) Functional renin receptors in renal mesangial cells. Curr Hypertens Rep 9(2):133–139
Huang Y, Noble NA, Zhang J, Xu C, Border WA (2007) Renin-stimulated TGF-beta1 expression is regulated by a mitogen-activated protein kinase in mesangial cells. Kidney Int 72(1):45–52
Huang Y, Wongamorntham S, Kasting J, McQuillan D, Owens RT, Yu L et al (2006) Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms. Kidney Int 69(1):105–113
Hartman D, Sagnella GA, Chesters CA, Macgregor GA (2004) Direct renin assay and plasma renin activity assay compared. Clin Chem 50(11):2159–2161
Schalekamp MA, Derkx FH, Deinum J, Danser AJ (2008) Newly developed renin and prorenin assays and the clinical evaluation of renin inhibitors. J Hypertens 26(5):928–937
Tsutamoto T, Sakai H, Tanaka T, Fujii M, Yamamoto T, Wada A et al (2007) Comparison of active renin concentration and plasma renin activity as a prognostic predictor in patients with heart failure. Circ J 71(6):915–921
Alderman MH, Madhavan S, Ooi WL, Cohen H, Sealey JE, Laragh JH (1991) Association of the renin-sodium profile with the risk of myocardial infarction in patients with hypertension. N Engl J Med 324(16):1098–1104
Parikh NI, Gona P, Larson MG, Wang TJ, Newton-Cheh C, Levy D et al (2007) Plasma renin and risk of cardiovascular disease and mortality: the Framingham Heart Study. Eur Heart J 28(21):2644–2652
Meade TW, Cooper JA, Peart WS (1993) Plasma renin activity and ischemic heart disease. N Engl J Med 329(9):616–619
Muhlestein JB, May HT, Bair TL, Prescott MF, Horne BD, White R et al (2010) Relation of elevated plasma renin activity at baseline to cardiac events in patients with angiographically proven coronary artery disease. Am J Cardiol 106(6):764–769
Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L (1990) Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS trial study group. Circulation 82(5):1730–1736
Voors AA, Pinto YM, Buikema H, Urata H, Oosterga M, Rooks G et al (1998) Dual pathway for angiotensin II formation in human internal mammary arteries. Br J Pharmacol 125(5):1028–1032
Oosterga M, Voors AA, Buikema H, Pinto YM, Haber HE, Ebels T et al (2000) Angiotensin II formation in human vasculature after chronic ACE inhibition: a prospective, randomized, placebo-controlled study QUO VADIS investigators. Cardiovasc Drugs Ther 14(1):55–60
Hillege HL, Girbes AR, de Kam PJ, Boomsma F, de Zeeuw D, Charlesworth A et al (2000) Renal function, neurohormonal activation, and survival in patients with chronic heart failure. Circulation 102(2):203–210
Damman K, Navis G, Smilde TD, Voors AA, van der Bij W, van Veldhuisen DJ et al (2007) Decreased cardiac output, venous congestion and the association with renal impairment in patients with cardiac dysfunction. Eur J Heart Fail 9(9):872–878
Packer M (1990) Why do the kidneys release renin in patients with congestive heart failure? A nephrocentric view of converting-enzyme inhibition. Eur Heart J 11(Suppl D):44–52
Cohn JN, Tognoni G (2001) Valsartan heart failure trial investigators. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med 345(23):1667–1675
Szymanski MK, Damman K, van Veldhuisen DJ, van Gilst WH, Hillege HL, de Boer RA (2011). Prognostic value of renin and prorenin in heart failure patients with decreased kidney function. Am Heart J. doi:10.1016/j.ahj.2011.06.001
Pfeffer MA, McMurray JJ, Velazquez EJ, Rouleau JL, Kober L, Maggioni AP et al (2003) Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 349(20):1893–1906
Mann JF, Schmieder RE, McQueen M, Dyal L, Schumacher H, Pogue J et al (2008) Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 372(9638):547–553
Kaneshiro Y, Ichihara A, Sakoda M, Takemitsu T, Nabi AH, Uddin MN et al (2007) Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats. J Am Soc Nephrol 18(6):1789–1795
Burckle CA, Jan Danser AH, Muller DN, Garrelds IM, Gasc JM, Popova E et al (2006) Elevated blood pressure and heart rate in human renin receptor transgenic rats. Hypertension 47(3):552–556
Kinouchi K, Ichihara A, Sano M, Sun-Wada GH, Wada Y, Kurauchi-Mito A et al (2010) The (pro)renin receptor/ATP6AP2 is essential for vacuolar H + -ATPase assembly in murine cardiomyocytes. Circ Res 107(1):30–34
Cruciat CM, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D et al (2010) Requirement of prorenin receptor and vacuolar H + -ATPase-mediated acidification for Wnt signaling. Science 327(5964):459–463
Suzuki F, Hayakawa M, Nakagawa T, Nasir UM, Ebihara A, Iwasawa A et al (2003) Human prorenin has “gate and handle” regions for its non-proteolytic activation. J Biol Chem 278(25):22217–22222
Ichihara A, Kaneshiro Y, Takemitsu T, Sakoda M, Suzuki F, Nakagawa T et al (2006) Nonproteolytic activation of prorenin contributes to development of cardiac fibrosis in genetic hypertension. Hypertension 47(5):894–900
Takahashi H, Ichihara A, Kaneshiro Y, Inomata K, Sakoda M, Takemitsu T et al (2007) Regression of nephropathy developed in diabetes by (Pro)renin receptor blockade. J Am Soc Nephrol 18(7):2054–2061
Muller DN, Klanke B, Feldt S, Cordasic N, Hartner A, Schmieder RE et al (2008) (Pro)renin receptor peptide inhibitor “handle-region” peptide does not affect hypertensive nephrosclerosis in Goldblatt rats. Hypertension 51(3):676–681
Mercure C, Prescott G, Lacombe MJ, Silversides DW, Reudelhuber TL (2009) Chronic increases in circulating prorenin are not associated with renal or cardiac pathologies. Hypertension 53(6):1062–1069
Feldt S, Batenburg WW, Mazak I, Maschke U, Wellner M, Kvakan H et al (2008) Prorenin and renin-induced extracellular signal-regulated kinase 1/2 activation in monocytes is not blocked by aliskiren or the handle-region peptide. Hypertension 51(3):682–688
Burton J, Cody RJ Jr, Herd JA, Haber E (1980) Specific inhibition of renin by an angiotensinogen analog: studies in sodium depletion and renin-dependent hypertension. Proc Natl Acad Sci USA 77(9):5476–5479
Fisher ND, Hollenberg NK (2001) Is there a future for renin inhibitors? Expert Opin Investig Drugs 10(3):417–426
Seed A, Gardner R, McMurray J, Hillier C, Murdoch D, MacFadyen R et al (2007) Neurohumoral effects of the new orally active renin inhibitor, aliskiren, in chronic heart failure. Eur J Heart Fail 9(11):1120–1127
van Esch JH, Moltzer E, van Veghel R, Garrelds IM, Leijten F, Bouhuizen AM et al (2010) Beneficial cardiac effects of the renin inhibitor aliskiren in spontaneously hypertensive rats. J Hypertens 28(10):2145–2155
O’Brien E, Barton J, Nussberger J, Mulcahy D, Jensen C, Dicker P et al (2007) Aliskiren reduces blood pressure and suppresses plasma renin activity in combination with a thiazide diuretic, an angiotensin-converting enzyme inhibitor, or an angiotensin receptor blocker. Hypertension 49(2):276–284
McMurray JJ, Pitt B, Latini R, Maggioni AP, Solomon SD, Keefe DL et al (2008) Effects of the oral direct renin inhibitor aliskiren in patients with symptomatic heart failure. Circ Heart Fail 1(1):17–24
Solomon SD, Appelbaum E, Manning WJ, Verma A, Berglund T, Lukashevich V et al (2009) Effect of the direct Renin inhibitor aliskiren, the Angiotensin receptor blocker losartan, or both on left ventricular mass in patients with hypertension and left ventricular hypertrophy. Circulation 119(4):530–537
Solomon SD, Hee Shin S, Shah A, Skali H, Desai A, Kober L et al (2011) Effect of the direct renin inhibitor aliskiren on left ventricular remodelling following myocardial infarction with systolic dysfunction. Eur Heart J 32(10):1227–1234
Feldman DL, Jin L, Xuan H, Contrepas A, Zhou Y, Webb RL et al (2008) Effects of aliskiren on blood pressure, albuminuria, and (pro)renin receptor expression in diabetic TG(mRen-2)27 rats. Hypertension 52(1):130–136
Danser AH (2010) The increase in renin during renin inhibition: does it result in harmful effects by the (pro)renin receptor? Hypertens Res 33(1):4–10
Krum H, Massie B, Abraham WT, Dickstein K, Kober L, McMurray JJ et al (2011) Direct renin inhibition in addition to or as an alternative to angiotensin converting enzyme inhibition in patients with chronic systolic heart failure: rationale and design of the Aliskiren Trial to Minimize OutcomeS in Patients with HEart failuRE (ATMOSPHERE) study. Eur J Heart Fail 13(1):107–114
Gheorghiade M, Albaghdadi M, Zannad F, Fonarow GC, Bohm M, Gimpelewicz C et al (2011) Rationale and design of the multicentre, randomized, double-blind, placebo-controlled Aliskiren trial on acute heart failure outcomes (ASTRONAUT). Eur J Heart Fail 13(1):100–106
Hogg K, McMurray J (2005) Neurohumoral pathways in heart failure with preserved systolic function. Prog Cardiovasc Dis 47(6):357–366
Cleland JG, Tendera M, Adamus J, Freemantle N, Polonski L, Taylor J et al (2006) The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur Heart J 27(19):2338–2345
Yusuf S, Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJ et al (2003) Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 362(9386):777–781
Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR et al (2008) Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 359(23):2456–2467
Fisher ND, Jan Danser AH, Nussberger J, Dole WP, Hollenberg NK (2008) Renal and hormonal responses to direct renin inhibition with aliskiren in healthy humans. Circulation 117(25):3199–3205
Cherney DZ, Lai V, Scholey JW, Miller JA, Zinman B, Reich HN (2010) Effect of direct renin inhibition on renal hemodynamic function, arterial stiffness, and endothelial function in humans with uncomplicated type 1 diabetes: a pilot study. Diabetes Care 33(2):361–365
Dietz R, Dechend R, Yu CM, Bheda M, Ford J, Prescott MF et al (2008) Effects of the direct renin inhibitor aliskiren and atenolol alone or in combination in patients with hypertension. J Renin Angiotensin Aldosterone Syst 9(3):163–175
Teisman AC, van Veldhuisen DJ, Boomsma F, de Kam PJ, Tjeerdsma G, Pinto YM, et al. (2000) Chronic beta-blocker treatment in patients with advanced heart failure. Effects on neurohormones. Int J Cardiol 73(1):7–12; discussion 13–4
Fryer RM, Rakestraw PA, Nakane M, Dixon D, Banfor PN, Koch KA et al (2007) Differential inhibition of renin mRNA expression by paricalcitol and calcitriol in C57/BL6 mice. Nephron Physiol 106(4):p76–p81
Bodyak N, Ayus JC, Achinger S, Shivalingappa V, Ke Q, Chen YS et al (2007) Activated vitamin D attenuates left ventricular abnormalities induced by dietary sodium in Dahl salt-sensitive animals. Proc Natl Acad Sci USA 104(43):16810–16815
Kong J, Kim GH, Wei M, Sun T, Li G, Liu SQ et al (2010) Therapeutic effects of vitamin D analogs on cardiac hypertrophy in spontaneously hypertensive rats. Am J Pathol 177(2):622–631
Freundlich M, Quiroz Y, Zhang Z, Zhang Y, Bravo Y, Weisinger JR et al (2008) Suppression of renin-angiotensin gene expression in the kidney by paricalcitol. Kidney Int 74(11):1394–1402
Park CW, Oh YS, Shin YS, Kim CM, Kim YS, Kim SY et al (1999) Intravenous calcitriol regresses myocardial hypertrophy in hemodialysis patients with secondary hyperparathyroidism. Am J Kidney Dis 33(1):73–81
de Zeeuw D, Agarwal R, Amdahl M, Audhya P, Coyne D, Garimella T et al (2010) Selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL study): a randomised controlled trial. Lancet 376(9752):1543–1551
Krum H, Gilbert RE (2007) Novel therapies blocking the renin-angiotensin-aldosterone system in the management of hypertension and related disorders. J Hypertens 25(1):25–35
Parving HH, Brenner BM, McMurray JJ, de Zeeuw D, Haffner SM, Solomon SD et al (2009) Aliskiren trial in type 2 diabetes using cardio-renal endpoints (ALTITUDE): rationale and study design. Nephrol Dial Transplant 24(5):1663–1671
Acknowledgments
Supported by the Netherlands Heart Foundation grant 2007T046 (Renin regulation in heart failure) and the Innovational Research Incentives Scheme program of the Netherlands Organization for Scientific Research (NWO VENI, grant 916.10.117), both to Dr. de Boer.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Schroten, N.F., Gaillard, C.A.J.M., van Veldhuisen, D.J. et al. New roles for renin and prorenin in heart failure and cardiorenal crosstalk. Heart Fail Rev 17, 191–201 (2012). https://doi.org/10.1007/s10741-011-9262-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-011-9262-2