
Abstract
During normal aging, the heart undergoes functional, morphological and cellular changes. Although aging per se does not lead to the expression of heart failure, it is likely that age-associated changes lower the threshold for the manifestation of signs and symptoms of heart failure. In patients, the susceptibility, age of onset and pace of progression of heart failure are highly variable. The presence of conventional risk factors cannot completely explain this variability. Accumulation of DNA damage and telomere attrition results in an increase in cellular senescence and apoptosis, resulting in a decrease in the number and function of cells, contributing to the overall tissue and organ dysfunction. Biological aging, characterized by reduced telomere length, provides an explanation for the highly interindividual variable threshold to express the clinical syndrome of heart failure at some stage during life. In this review, we will elaborate on the current knowledge of aging of the heart, telomere biology and its potential role in the development of heart failure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Congestive heart failure (CHF) is a highly prevalent condition affecting more than 15 million patients in Europe alone [1]. The incidence and prevalence of CHF increases steeply with age and is likely to rise in the next decade due to the increasing average age of the population [2]. The prognosis of CHF is poor with a 4-year survival of only ~50% [1]. Advanced age is one of the major risk factors for the development of CHF. Nevertheless, the susceptibility, age of onset and pace of progression are highly variable. The presence of conventional risk factors cannot completely explain this variability. The process of biological aging affects most cells, organisms and species. Although aging itself does not need to lead to CHF, it is likely that age-associated changes lower the threshold for the expression of this syndrome. Significant changes in diastolic function, hypertension, atherosclerosis, valve calcifications, senile cardiac amyloid depositions all are associated with aging and an increased risk of the clinical signs and symptoms of CHF [3]. The molecular mechanisms involved allow time for accumulated damage to occur and include free radicals, advance glycation end-products, apoptosis and senescence. Accumulation of DNA damage and telomere attrition can result in an increase in senescent cells in tissue and organs and can subsequently result in decreased function, providing an explanation for the lower threshold to express the clinical manifestation of heart failure. Here, we will briefly overview the current knowledge of the cardiac changes associated with aging. One aspect of aging in relation to CHF is of particular interest to us, telomere biology. Telomeres consist of an evolutionary conserved repetitive nucleotide sequence and are located at the terminal ends of the chromosomes [4, 5]. The process of telomere erosion has been put forward as an intracellular counting mechanism that runs parallel with the aging process and the susceptibility to develop CHF.
Functional changes of the aging heart
At the functional level, there are no age-related changes in cardiac output, end-diastolic or end-systolic volumes or ejection fraction at rest in healthy subjects [6]. However, the myocardial stiffness increases with advancing age, resulting in an elevated left ventricular end-diastolic pressure at rest and with exertion [7]. Early diastolic filling is decreased in elderly, a phenomenon seen on echocardiography as a change of the early to late filling velocity (E/A ratio) [8]. Important age-associated changes are particularly noticeable during exercise. Although the stroke-volume increase during exercise of the young and old is comparable, the old tend to augment stroke volume during exercise more through cardiac dilatation with an increase in end-diastolic volume, whereas the young rely more on increase in the ejection fraction with no cardiac dilatation [9]. During exercise, the older heart rate has a lesser increase in frequency and blood pressure has a greater increase [9]. The difference in heart rate response is associated with a decrease in response to catecholamines and axonal degeneration of sympathetic neurons innervating the atria [10, 11]. In addition, the number of pacemaker cells in the sinoatrial node diminishes significantly, from around 50% in youth to less than 30% in elderly [12]. One of the major vascular changes during aging is the decrease in aortic distensibility and decrease in the ‘Windkessel function’ explaining the frequently observed isolated systolic hypertension in elderly [7].
Morphological and cellular changes of the heart
The mature myocardium constitutes of cardiomyocytes and supporting connective tissue [13]. Healthy cardiac tissue is roughly composed of 20–25% cardiomyocytes, although this number varies among different species [13]. During aging, most components of the myocardium undergo structural changes. The change in morphology is characterized by a loss of myocytes number, with a subsequent hypertrophy of remaining viable myocytes [14]. The remaining myocytes frequently contain multiple nuclei (polyploidy). The mechanism responsible for loss of myocytes is likely to be apoptosis, or programmed cell death. In parallel, there is an increase in collagen content, fibrosis and depositions of ‘senile’ cardiac amyloid and lipofuscin [15–17]. Aging cardiomyocytes display prolonged contraction and relaxation caused by changes in calcium homeostasis [18]. Prolonged contraction with decreased force of the myofilaments is associated with down-regulation of genes that encode proteins that are involved in contractile activity, such as troponin and myosin forms in the aged heart [19, 20]. Age-related changes of the mitochondria include a tendency to become larger but less efficient, with a decreased ATP production per cell [21].
Cardiomyocyte turnover during aging
The paradigm that all cardiomyocytes are terminally differentiated has been challenged. Recent experiments using human left ventricular myocardial cells and carbon-dating techniques have established that DNA of cardiomyocytes continues to be synthesized many years after birth, indicating that cells in the human heart do renew well into adulthood [22]. Nevertheless, cardiomyocyte DNA synthesis decreases with age. Mathematical modeling predicts a ~1% cardiomyocyte renewal rate at the age of 25 and 0.45% at the age of 75. Considering this turnover rate, at the age of 50 years, 55% of the cardiomyocytes remain from the time around birth. There was no significant difference observed in 14C integration of DNA synthesis in cardiomyocytes from subjects with cardiac pathology. Interestingly, the 14C data also indicated a substantially higher renewal rate for non-cardiomyocytes, with a median annual turnover of 18%. Unfortunately, data derived from 14C integration do not allow the identification of the source of new cardiomyocytes. These could be derived either from cardiomyocyte duplication or from some sort of progenitor pool [22].
Advances in mouse genetic engineering allow cell tracking using ‘fate-mapping’ approach. Cells expressing the cardiomyocyte-specific alpha-myosin can be genetically labeled by an inducible recombination technique leading to permanent GFP expression of only cells that expressed alpha-myosin at time of induction [23]. If at a later stage, stem cells (which initially do not express alpha-myosin) contribute to cardiomyocyte renewal or regeneration, the percentage of GFP-positive myocytes will decrease. These experiments indicated that stem cells do not replace adult mouse cardiomyocytes during at least 1 year of aging. However, in the setting of myocardial infarction or pressure-overloaded hearts, a significant decrease in percentage of GFP-positive myocytes was observed, suggesting that precursor cells participate in the formation of new cardiomyocytes after injury [23]. The bone marrow is a pool for multiple types of progenitor cells, which are believed to contribute to cardiovascular repair [24]. Other possible sources of stem cells that can be stimulated to cardiogenic differentiation are located in adipose tissue or in the bone marrow [24].
One of the main determinants of functionality of bone marrow progenitor cells is age. A study in young and old bone marrow–derived endothelial progenitor cells (EPCs) showed that endothelial progenitor cells from 3-month-old donor mice augmented angiogenic capacity in the recipient 18-month-old mice, whereas EPCs from 18-month-old donor mice showed no effect [25]. Furthermore, young mesenchymal stem cells (MSCs) showed higher angiogenic response to anoxia than old MSCs in vitro, and were more resistant to apoptotic stimuli [26]. This was supported by a study in a myocardial infarction (MI) model. Infarct size of rats with experimental MI was attenuated after intramyocardial injections of young MSCs, whereas injection of old MSCs did not lead to any changes. In addition, cardiac function was preserved after MI in all rats that received MSCs—young, old or a mixture—but the beneficial effect on cardiac function was the largest in rats that received only young MSCs [26]. Age does not only determine functionality of progenitor cells but is also possibly associated with the number of progenitor cells [27–29].
Telomere biology
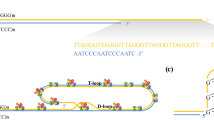
Telomeres are specialized DNA structures made up of tandem repeats (TTAGGG in humans) located at the end of chromosomes [4, 5]. Telomeres have a critical function as they serve as protective caps, preventing the chromosomal ends to be accidently recognized as DNA double strands by the DNA damage–repair system, and activation of the p53 or p16INK4a pathway, which eventually leads to senescence or apoptosis. The G-rich strand of the telomere, in conjunction with specialized proteins, form a so-called telomere loop (T-loop) concealing the terminal ends of the DNA strands (Fig. 1). These specialized proteins include telomeric repeat binding factor 1 (TRF1) and 2 (TRF2), which can both bind directly to double-stranded telomere DNA. Other telomere-associated proteins include the protein protection of telomeres 1 (POT1), binding directly to single-stranded telomere DNA, and repressor activator protein 1 (Rap1), TPP1 and TRF1-interacting nuclear factor 2 (TIN2) (Fig. 2). Telomeres lose 30–150 base pairs during each cell division (also known as the end replication problem) due to the inability of DNA polymerase to fully replicate the 3’ end of the DNA strand. Additional erosion occurs in presence of damaging environmental factors, e.g., oxidative stress [30]. Because telomere length marks the cumulative replicative history and cumulative exposure to environmental factors, it is strongly associated to date of birth (chronological) age and is even considered a marker of biological/cellular aging. When the telomere reaches a critical short length, the cell will no longer divide and can become dysfunctional or senescent. On average, cells are estimated to reach senescence after ~50 population doublings. Not only telomere length per se but also disruption of the associated telomere-binding proteins responsible for the three-dimensional loop structure can induce chromosomal instability, senescence or apoptosis.

Simplified scheme depicting the structure of the telomere and its location on the chromosome and in the cell. Reproduced with permission [68]

Simplified scheme depiction the terminal end of the telomere concealing the terminal single-stranded part with help of the shelterin complex. Reproduced with permission [68]
The function of the ribonucleoprotein enzyme telomerase is the elongation of the telomere sequence by addition of nucleotides to their ends. In most mature cells, telomerase activity is nearly undetectable, with exceptions including embryogenic stem cells, germline cells, malignantly transformed cells and some epithelial and lymphoid progenitor cells. Telomerase consists of two main components, telomerase RNA component (TERC) and telomerase reverse transcriptase (TERT). A third component (dyskerin) serves to stabilize the complex (Fig. 3). A less well-understood mechanism through which telomeres can be elongated has been named ‘alternative lengthening of telomeres’ (ALT). This mechanism is thought to be dependent on the cellular homologous recombination machinery, one of the systems for the reparation of DNA double-strand breaks. The ALT pathway is not an alternative to telomerase in cells that lack telomerase activity. Instead, the ALT pathway acts concurrently to telomerase [31].

Schematic overview of telomerase. Active telomerase is composed from 2 RNA-complexes (TERC; only 1 depicted) and 2 telomere-reverse-transcriptase (TERC; 1 depicted) stabilized by dyskerin. Reproduced with permission [68]
Recently, the paradigm that telomeres are transcriptional silent has been broken. Telomeric repeat-containing RNA (TERRA) is an in length heterogeneous non-coding RNA forming an integral component of the telomeric structure [32, 33]. As the sequence of TERRA is complementary to TERC, it might be involved in the regulation of telomerase activity or regulation of the DNA damage response at short telomeres. However, the exact function of TERRA remains to be discovered.
Telomere length is highly variable among individuals of the same age. Already at birth, remarkable differences in telomere length can be detected. Several studies have suggested that telomere length can be predicted by the telomere length of the parents. Heritability of telomere length has been estimated to be as high as 82% [34]. The effect of paternal telomere length is more obvious than that of maternal telomere length. The strength of the association with paternal telomere length is in addition larger for father–son than for father–daughter relationships [35]. Genome-wide scans have mapped loci associated with telomere length on chromosome 12[36], 14[37], 18[38] and 3 near TERC (component of the enzyme telomerase) [39].
Several environmental factors are also associated with telomere length and possible telomere attrition rate. Most important are oxidative stress [30] and factors related to oxidative stress such as smoking [40] and UV radiation [41].
Telomeres and heart failure
Telomere length has been related to factors predisposing to CHF, including hypertension [42], diabetes mellitus [43], premature myocardial infarction [44] and activation of the renin-angiotensin-aldosterone system (RAAS) [45]. The effect of RAAS inhibition, however, is not clear, since presumably all patients that suffer from CHF are taking RAAS-inhibiting medication, leaving little or no CHF patients without RAAS-inhibiting mediation to serve as valid controls. Of course, the effect of RAAS inhibition could very well be investigated in an experimental heart failure model, but to date no data on this matter have been reported.
Not surprisingly, patients with CHF also have shorter telomeres compared to healthy age- and gender-balanced controls [46]. Telomere length has even been associated with the severity of CHF symptoms and outcome [46, 47]. In addition, worse renal function (a powerful predictor of outcome in CHF and not directly related to vascular function [48–50]) has been associated with telomere length in subjects with CHF [51, 52]. It is tempting to speculate this is also due to higher senescence in the kidney and consequently nephron dropout. In apparent healthy elderly, one standard deviation of shorter leukocyte telomere length was associated with 5% reduced left ventricular ejection fraction and telomere length alone accounted for 12% of the observed variability [53]. Telomere length is usually measured in leukocytes but has also been evaluated in cardiac tissue from patients with CHF. Patients with dilated heart failure have ~25% shorter cardiac telomeres compared to healthy controls [54]. In aged diseased hearts characterized by mild hypertrophy and biventricular failure, shorter average telomere length was found [55]. Interestingly, cardiomyocytes with severely shortened telomeres (<2.5 kbp) were positive for p16INK4a, a marker for cellular senescence. The fraction of p16INK4a-positive cardiomyocytes was much larger in aged diseased heart compared to non-diseased controls. These findings are consistent with the idea that short telomere length is associated with increased levels of cellular senescence facilitating—if not causing—heart failure [55].
The most prominent question concerning the association between telomere length and heart failure is the causality of this association. Does short telomere length directly contribute to the development and progression of heart failure, or does short telomere length itself or the underlying cause of heart failure lead to accelerated telomere shortening? An attractive explanation could be that telomere dysfunction is a common pathway through which risk factors act and increase cardiomyocyte senescence and dysfunction. The diminished regenerative capacity might also include exhaustion of the progenitor pool with repair capacity [56–58]. Evidence for a causal role has been provided by telomerase knockout mice. Fifth-generation telomerase knockout mice have severely reduced telomere length and suffer from severe left ventricular failure, characterized by increased end-diastolic left ventricular pressure, decreased maximally developed left ventricular pressure and disturbed relaxation and contractility very similar to that observed in human dilated cardiomyopathies [59]. On the other hand, stabilizing telomeres by over-expression of TRF2 prevents doxorubicin-induced cardiac apoptosis in wild-type mice, but not in telomerase-deficient mice [60]. However, convincing evidence in humans for a causal role is lacking. Shorter telomeres could be a consequence or an epiphenomenon rather than a cause of CHF. Oxidative stress or increased inflammatory status, both related to outcome [61], could provide a true basis of the development of CHF and at the same time explain reduced telomere length.
Conclusions and future perspectives
Recent clinical CHF trials, for example with statins [62], have not fulfilled their promises in improving prognosis of CHF [63, 64]. Therapeutic strategies to improve myocardial function and outcome in CHF are urgently needed, and new medicines are rapidly being introduced [65–67]. Telomere biology might be involved in the biology of aging and age-associated pathology. Telomeres are connected to the basic biology of aging and trigger cellular senescence. It still needs to be established whether telomere biology is causally involved in the development of CHF in humans. Large, prospective, longitudinal studies are needed to provide us with more in-depth insights into the nature of the association between telomere length and CHF. These studies can clarify whether short telomere length predicts the development of CHF—which possibly supports a causal role—or telomere length merely shortens after manifestation of CHF. Of course, strongest evidence for a causal role would be delivered by interventional studies that demonstrate the cardiac effects of actively altered telomere length. At this time, however, this approach encounters not only several practical obstacles, but also moral objections, since our awareness of all the consequences of telomere length manipulation is surely incomplete. Nevertheless, answering the question on causality unambiguously in the near future is essential, as it will allow the development of novel strategies in the treatment and prevention of CHF, for example by beneficially modifying stem cells currently used in experimental trials.
References
Dickstein K, Cohen-Solal A, Filippatos G, McMurray JJ, Ponikowski P, Poole-Wilson PA, Stromberg A, van Veldhuisen DJ, Atar D, Hoes AW, Keren A, Mebazaa A, Nieminen M, Priori SG, Swedberg K, Vahanian A, Camm J, De CR, Dean V, Dickstein K, Filippatos G, Funck-Brentano C, Hellemans I, Kristensen SD, McGregor K, Sechtem U, Silber S, Tendera M, Widimsky P, Zamorano JL, Tendera M, Auricchio A, Bax J, Bohm M, Corra U, Della BP, Elliott PM, Follath F, Gheorghiade M, Hasin Y, Hernborg A, Jaarsma T, Komajda M, Kornowski R, Piepoli M, Prendergast B, Tavazzi L, Vachiery JL, Verheugt FW, Zamorano JL, Zannad F (2008) ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the task force for the diagnosis and treatment of acute and chronic heart failure 2008 of the European society of cardiology, developed in collaboration with the heart failure association of the ESC (HFA) and endorsed by the European society of intensive care medicine (ESICM). Eur J Heart Fail 10:933–989
Kannel WB (2000) Incidence and epidemiology of heart failure. Heart Fail Rev 5:167–173
Kitzman DW (2002) Diastolic heart failure in the elderly. Heart Fail Rev 7:17–27
Wong LS, de Boer RA, Samani NJ, van Veldhuisen DJ, van der Harst P (2008) Telomere biology in heart failure. Eur J Heart Fail 10:1049–1056
Samani NJ, van der Harst P (2008) Biological ageing and cardiovascular disease. Heart 94:537–539
Lakatta EG (2002) Age-associated cardiovascular changes in health: impact on cardiovascular disease in older persons. Heart Fail Rev 7:29–49
Pugh KG, Wei JY (2001) Clinical implications of physiological changes in the aging heart. Drugs Aging 18:263–276
Schulman SP, Lakatta EG, Fleg JL, Lakatta L, Becker LC, Gerstenblith G (1992) Age-related decline in left ventricular filling at rest and exercise. Am J Physiol 263:H1932–H1938
Stratton JR, Levy WC, Cerqueira MD, Schwartz RS, Abrass IB (1994) Cardiovascular responses to exercise. Effects of aging and exercise training in healthy men. Circulation 89:1648–1655
Lakatta EG, Gerstenblith G, Angell CS, Shock NW, Weisfeldt ML (1975) Diminished inotropic response of aged myocardium to catecholamines. Circ Res 36:262–269
McLean MR, Goldberg PB, Roberts J (1983) An ultrastructural study of the effects of age on sympathetic innervation and atrial tissue in the rat. J Mol Cell Cardiol 15:75–92
Rehman HU (1999) Age and the cardiovascular system. Hosp Med 60:645–652
Buja LM, Vela D (2008) Cardiomyocyte death and renewal in the normal and diseased heart. Cardiovasc Pathol 17:349–374
Olivetti G, Melissari M, Capasso JM, Anversa P (1991) Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ Res 68:1560–1568
Burgess ML, McCrea JC, Hedrick HL (2001) Age-associated changes in cardiac matrix and integrins. Mech Ageing Dev 122:1739–1756
Pandya K, Kim HS, Smithies O (2006) Fibrosis, not cell size, delineates beta-myosin heavy chain reexpression during cardiac hypertrophy and normal aging in vivo. Proc Natl Acad Sci U S A 103:16864–16869
Lie JT, Hammond PI (1988) Pathology of the senescent heart: anatomic observations on 237 autopsy studies of patients 90 to 105 years old. Mayo Clin Proc 63:552–564
Bernhard D, Laufer G (2008) The aging cardiomyocyte: a mini-review. Gerontology 54:24–31
Raizada V, Pathak D, Blomquist TM, Minser R, Woodfin B (1993) Alterations in cardiac myosin isozymes associated with aging and chronic hypertension: their modulation with nifedipine. Cardiovasc Res 27:1869–1872
Dhahbi JM, Tsuchiya T, Kim HJ, Mote PL, Spindler SR (2006) Gene expression and physiologic responses of the heart to the initiation and withdrawal of caloric restriction. J Gerontol A Biol Sci Med Sci 61:218–231
Muscari C, Finelli C, Stefanelli C, Flamigni F, Guarnieri C, Caldarera CM (1993) Age-dependent differences of ATP breakdown and ATP-catabolite release in ischemic and reperfused hearts. Mech Ageing Dev 67:1–11
Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J (2009) Evidence for cardiomyocyte renewal in humans. Science 324:98–102
Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT (2007) Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med 13:970–974
Dimmeler S, Leri A (2008) Aging and disease as modifiers of efficacy of cell therapy. Circ Res 102:1319–1330
Edelberg JM, Tang L, Hattori K, Lyden D, Rafii S (2002) Young adult bone marrow-derived endothelial precursor cells restore aging-impaired cardiac angiogenic function. Circ Res 90:E89–E93
Jiang S, Kh HH, Ahmed RP, Idris NM, Salim A, Ashraf M (2008) Transcriptional profiling of young and old mesenchymal stem cells in response to oxygen deprivation and reparability of the infarcted myocardium. J Mol Cell Cardiol 44:582–596
Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, Bohm M, Nickenig G (2005) Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med 353:999–1007
Kissel CK, Lehmann R, Assmus B, Aicher A, Honold J, Fischer-Rasokat U, Heeschen C, Spyridopoulos I, Dimmeler S, Zeiher AM (2007) Selective functional exhaustion of hematopoietic progenitor cells in the bone marrow of patients with post infarction heart failure. J Am Coll Cardiol 49:2341–2349
Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, Zeiher AM, Dimmeler S (2001) Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res 89:E1–E7
von Zglinicki T (2002) Oxidative stress shortens telomeres. Trends Biochem Sci 27:339–344
Grobelny JV, Kulp-McEliece M, Broccoli D (2001) Effects of reconstitution of telomerase activity on telomere maintenance by the alternative lengthening of telomeres (ALT) pathway. Hum Mol Genet 10:1953–1961
Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J (2007) Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 318:798–801
Schoeftner S, Blasco MA (2008) Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat Cell Biol 10:228–236
Slagboom PE, Droog S, Boomsma DI (1994) Genetic determination of telomere size in humans: a twin study of three age groups. Am J Hum Genet 55:876–882
Njajou OT, Cawthon RM, Damcott CM, Wu SH, Ott S, Garant MJ, Blackburn EH, Mitchell BD, Shuldiner AR, Hsueh WC (2007) Telomere length is paternally inherited and is associated with parental lifespan. Proc Natl Acad Sci U S A 104:12135–12139
Vasa-Nicotera M, Brouilette S, Mangino M, Thompson JR, Braund P, Clemitson JR, Mason A, Bodycote CL, Raleigh SM, Louis E, Samani NJ (2005) Mapping of a major locus that determines telomere length in humans. Am J Hum Genet 76:147–151
Andrew T, Aviv A, Falchi M, Surdulescu GL, Gardner JP, Lu X, Kimura M, Kato BS, Valdes AM, Spector TD (2006) Mapping genetic loci that determine leukocyte telomere length in a large sample of unselected female sibling pairs. Am J Hum Genet 78:480–486
Mangino M, Richards JB, Soranzo N, Zhai G, Aviv A, Valdes AM, Samani NJ, Deloukas P, Spector TD (2009) A genome-wide association study identifies a novel locus on chromosome 18q12.2 influencing white cell telomere length. J Med Genet 46:451–454
Codd V, Mangino M, van der Harst P, Braund PS, Kaiser M, Beveridge AJ, Rafelt S, Moore J, Nelson C, Soranzo N, Zhai G, Valdes AM, Blackburn H, Mateo LI, de Boer RA, Goodall AH, Ouwehand W, van Veldhuisen DJ, van Gilst WH, Navis G, Burton PR, Tobin MD, Hall AS, Thompson JR, Spector T, Samani NJ (2010) Common variants near TERC are associated with mean telomere length. Nat Genet 42:197–199
Valdes AM, Andrew T, Gardner JP, Kimura M, Oelsner E, Cherkas LF, Aviv A, Spector TD (2005) Obesity, cigarette smoking, and telomere length in women. Lancet 366:662–664
Oikawa S, Tada-Oikawa S, Kawanishi S (2001) Site-specific DNA damage at the GGG sequence by UVA involves acceleration of telomere shortening. Biochemistry 40:4763–4768
Jeanclos E, Schork NJ, Kyvik KO, Kimura M, Skurnick JH, Aviv A (2000) Telomere length inversely correlates with pulse pressure and is highly familial. Hypertension 36:195–200
Jeanclos E, Krolewski A, Skurnick J, Kimura M, Aviv H, Warram JH, Aviv A (1998) Shortened telomere length in white blood cells of patients with IDDM. Diabetes 47:482–486
Brouilette S, Singh RK, Thompson JR, Goodall AH, Samani NJ (2003) White cell telomere length and risk of premature myocardial infarction. Arterioscler Thromb Vasc Biol 23:842–846
Vasan RS, Demissie S, Kimura M, Cupples LA, Rifai N, White C, Wang TJ, Gardner JP, Cao X, Benjamin EJ, Levy D, Aviv A (2008) Association of leukocyte telomere length with circulating biomarkers of the renin-angiotensin-aldosterone system. The framingham heart study. Circulation 117:1138–1144
van der Harst P, van der Steege G, de Boer RA, Voors AA, Hall AS, Mulder MJ, van Gilst WH, van Veldhuisen DJ (2007) Telomere length of circulating leukocytes is decreased in patients with chronic heart failure. J Am Coll Cardiol 49:1459–1464
van der Harst P, de Boer RA, Samani NJ, Wong LS, Huzen J, Codd V, Hillege HL, Voors AA, van Gilst WH, Jaarsma T, van Veldhuisen DJ (2010) Telomere length and outcome in heart failure. Ann Med 42:36–44
Asselbergs FW, van der Harst P, Jessurun GA, Tio RA, van Gilst WH (2005) Clinical impact of vasomotor function assessment and the role of ACE-inhibitors and statins. Vascul Pharmacol 42:125–140
Smilde TD, Damman K, van der Harst P, Navis G, Daan WB, Voors AA, Boomsma F, van Veldhuisen DJ, Hillege HL (2009) Differential associations between renal function and “modifiable” risk factors in patients with chronic heart failure. Clin Res Cardiol 98:121–129
van der Harst P, Smilde TD, Buikema H, Voors AA, Navis G, van Veldhuisen DJ, van Gilst WH (2006) Vascular function and mild renal impairment in stable coronary artery disease. Arterioscler Thromb Vasc Biol 26:379–384
van der Harst P, Wong LS, de Boer RA, Brouilette SW, van der SG, Voors AA, Hall AS, Samani NJ, Wikstrand J, van Gilst WH, van Veldhuisen DJ (2008) Possible association between telomere length and renal dysfunction in patients with chronic heart failure. Am J Cardiol 102:207–210
Wong LS, van der Harst P, de Boer RA, Codd V, Huzen J, Samani NJ, Hillege HL, Voors AA, van Gilst WH, Jaarsma T, van Veldhuisen DJ (2008) Renal dysfunction is associated with shorter telomere length in patients with heart failure. Clin Res Cardiol 98:629–634
Collerton J, Martin-Ruiz C, Kenny A, Barrass K, von Zglinicki T, Kirkwood T, Keavney B (2007) Telomere length is associated with left ventricular function in the oldest old: the Newcastle 85 + study. Eur Heart J 28:172–176
Oh H, Wang SC, Prahash A, Sano M, Moravec CS, Taffet GE, Michael LH, Youker KA, Entman ML, Schneider MD (2003) Telomere attrition and Chk2 activation in human heart failure. Proc Natl Acad Sci U S A 100:5378–5383
Chimenti C, Kajstura J, Torella D, Urbanek K, Heleniak H, Colussi C, Di Meglio F, Nadal-Ginard B, Frustaci A, Leri A, Maseri A, Anversa P (2003) Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ Res 93:604–613
Spyridopoulos I, Erben Y, Brummendorf TH, Haendeler J, Dietz K, Seeger F, Kissel C, Martin H, Hoffmann J, Assmus B, Zeiher AM, Dimmeler S (2008) Telomere gap between granulocytes and lymphocytes is a determinant for haematopoetic progenitor cell impairment in patients with previous myocardial infarction. Arterioscler Thromb Vasc Biol 28:968–974
van der Harst P, van Veldhuisen DJ, Samani NJ (2008) Expanding the concept of telomere dysfunction in cardiovascular disease. Arterioscler Thromb Vasc Biol 28:807–808
Westenbrink BD, Voors AA, de Boer RA, Schuringa JJ, Klinkenberg T, van der Harst P, Vellenga E, van Veldhuisen DJ, van Gilst WH (2010) Bone marrow dysfunction in chronic heart failure patients. Eur J Heart Fail. In press
Leri A, Franco S, Zacheo A, Barlucchi L, Chimenti S, Limana F, Nadal-Ginard B, Kajstura J, Anversa P, Blasco MA (2003) Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation. EMBO J 22:131–139
Werner C, Hanhoun M, Widmann T, Kazakov A, Semenov A, Poss J, Bauersachs J, Thum T, Pfreundschuh M, Muller P, Haendeler J, Bohm M, Laufs U (2008) Effects of physical exercise on myocardial telomere-regulating proteins, survival pathways, and apoptosis. J Am Coll Cardiol 52:470–482
van der Harst P, Voors AA, Volbeda M, Buikema H, van Veldhuisen DJ, van Gilst WH (2006) Usefulness of preoperative C-reactive protein and soluble intercellular adhesion molecule-1 level for predicting future cardiovascular events after coronary artery bypass grafting. Am J Cardiol 97:1697–1701
van der Harst P, Voors AA, van Gilst WH, Bohm M, van Veldhuisen DJ (2006) Statins in the treatment of chronic heart failure: biological and clinical considerations. Cardiovasc Res 71:443–454
Kjekshus J, Apetrei E, Barrios V, Bohm M, Cleland JG, Cornel JH, Dunselman P, Fonseca C, Goudev A, Grande P, Gullestad L, Hjalmarson A, Hradec J, Janosi A, Kamensky G, Komajda M, Korewicki J, Kuusi T, Mach F, Mareev V, McMurray JJ, Ranjith N, Schaufelberger M, Vanhaecke J, van Veldhuisen DJ, Waagstein F, Wedel H, Wikstrand J (2007) Rosuvastatin in older patients with systolic heart failure. N Engl J Med 357:2248–2261
van der Harst P, Voors AA, van Gilst WH, Bohm M, van Veldhuisen DJ (2006) Statins in the treatment of chronic heart failure: a systematic review. PLoS Med 3:e333
Lipsic E, Westenbrink BD, van der Meer P, van der Harst P, Voors AA, van Veldhuisen DJ, Schoemaker RG, van Gilst WH (2008) Low-dose erythropoietin improves cardiac function in experimental heart failure without increasing haematocrit. Eur J Heart Fail 10:22–29
Westenbrink BD, Lipsic E, van der Meer P, van der Harst P, Oeseburg H, Du Marchie Sarvaas GJ, Koster J, Voors AA, van Veldhuisen DJ, van Gilst WH, Schoemaker RG (2007) Erythropoietin improves cardiac function through endothelial progenitor cell and vascular endothelial growth factor mediated neovascularization. Eur Heart J 28:2018–2027
Westenbrink BD, Oeseburg H, Kleijn L, van der Harst P, Belonje AM, Voors AA, Schoemaker RG, de Boer RA, van Veldhuisen DJ, van Gilst WH (2008) Erythropoietin stimulates normal endothelial progenitor cell-mediated endothelial turnover, but attributes to neovascularization only in the presence of local ischemia. Cardiovasc Drugs Ther 22:265–274
Huzen J, van Veldhuisen DJ, van Gilst WH, van der Harst P (2008) Telomeres and biological ageing in cardiovascular disease. Ned Tijdschr Geneeskd 152:1265–1270
Acknowledgments
This work was supported by Netherlands Heart Foundation (Grant 2006B140), the Innovational Research Incentives Scheme program of the Netherlands Organisation for Scientific Research (NWO VENI, grant 916.76.170 to P. van der Harst) and the Interuniversitair Cardiologisch Instituut Nederland (ICIN). L.S.M. Wong, P. van der Harst and R.A. de Boer are research fellows of the Netherlands Heart Foundation (grant 2008T028, 2006T003 and 2007T046, respectively).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wong, L.S.M., van der Harst, P., de Boer, R.A. et al. Aging, telomeres and heart failure. Heart Fail Rev 15, 479–486 (2010). https://doi.org/10.1007/s10741-010-9173-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-010-9173-7