
Abstract
Central apnea during sleep represents a manifestation of breathing instability in many clinical conditions of varied etiologies. Central apnea is the result of transient cessation of ventilatory motor output, which represents that inhibitory influences favoring instability predominate over excitatory influence favoring stable breathing. This article will review the determinants of central apnea, the specific features of CHF-related central apnea, and outline a management approach
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Determinants of central apnea during NREM sleep
The removal of the wakefulness “drive to breathe” renders respiration during sleep (specifically non-rapid eye movement or NREM sleep) critically dependent on chemical influences, especially PCO2. NREM sleep unmasks a highly sensitive, hypocapnic apneic threshold; accordingly, central apnea occurs if arterial PCO2 is lowered below the “apneic threshold” [1, 2]. This can be induced experimentally via passive nasal mechanical ventilation resulting in hypocapnia of varied degrees.
Hypocapnia is the most ubiquitous and potent influence favoring inhibition of ventilatory motor output in sleeping humans. However, some types of hyperventilation, including brief and active hyperventilation, may not result in apnea. Hypoxic hyperventilation, and possibly other forms of active hyperventilation, may be associated with activation of an excitatory neural mechanism referred to as short-term potentiation (STP) [3–5]. This neuronal phenomenon may preserve rhythmic respiration despite transient hypocapnia. In fact, termination of brief hypoxia or transient arousals from sleep rarely lead to central apnea despite hypocapnia at or below the apneic threshold [5, 6]. However, prolonged hypoxia may abolish STP, which may explain the development of periodic breathing after 20–25 min of hypoxia and the occurrence of central apnea upon termination of prolonged hypoxic exposure [5, 7]. Likewise, central apnea does not usually occur following brief episodes of hyperventilation in sleeping humans [8] or dogs [9] possibly due to insufficient reduction in PCO2 at the level of the central chemoreceptors.
Central apnea can also occur through non-chemical pathways; for example, application of negative pressure and subsequent deformation of the upper airway lead to central apnea in a canine model [10]. Likewise, central apnea is more frequent in the supine position [11–13] and may respond to nasal continuous positive airway pressure therapy (CPAP) [14]. In fact, the lateral position may lead to amelioration of severity of central apnea and Cheyne–Stokes respiration [11–13].
Apnea does not occur as an isolated event but in clusters of breaths separated by intervals of apnea or hypopnea. This has given rise to the maxim: apnea begets apnea. Patients with CHF and central apnea often display the typical crescendo-decrescendo pattern of Cheyne–Stokes respiration. This form of periodic breathing represents a cumulative effect of physiologic ventilatory responses aiming to minimize the fluctuation in arterial blood gases. Changes in PCO2 and PO2 produce proportional counteracting changes in ventilation; however, a delayed or exaggerated response produces periodic rather than stable breathing.
The properties of the ventilatory control during sleep underpin the notion that physiologic responses lead to pathologic instability. The ventilatory control system operates as a negative-feedback closed-loop cycle to maintain homeostasis of blood gas tensions within a physiologic range. Many authors have adopted the engineering concept of “loop gain” (GL) as a measure of ventilatory stability or susceptibility to central apnea and recurrent periodic breathing [15]. Loop gain represents the overall response of the plant (representing the lung and respiratory muscles), the controller (representing the ventilatory control centers and the chemoreceptors), and the delay, dilution, and diffusion inherent in transferring the signal between the plant and the controller. There are several excellent reviews discussing the dynamics of ventilatory control in humans [16–18].
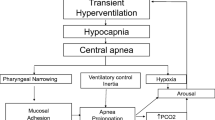
The occurrence of apnea leads to several consequences that conspire to promote further breathing instability. The first is the inertia of the ventilatory control system that prevents the return of rhythmic breathing until PaCO2 increases by 4–6 mmHg above eupneic arterial PCO2 [19]. In addition, central apnea is associated with narrowing or occlusion of the pharyngeal airway [20]. Thus, resumption of ventilation requires opening of a narrowed or occluded airway and overcoming tissue adhesion forces [21] and cranio-facial gravitational forces. Termination of central apnea is associated with variable asphyxia (hypoxia and hypercapnia) and transient arousal, resulting in ventilatory overshoot, subsequent hypocapnia, and further apnea/hypopnea. This sequence explains the overlap between central and obstructive apnea (upper airway obstruction often follows central apneas upon resumption of respiratory effort, i.e., mixed apnea).
Pathophysiologic classification of central sleep apnea
The classification of central apnea as hypercapnic or non-hypercapnic (based on the level of daytime PaCO2) does not capture the continuum of ventilatory abnormalities in clinical conditions. Physiologically, central apnea could be caused by reduced alveolar ventilation or by hypocapnia following alveolar hyperventilation.
Central sleep apnea, in its hypercapnic form, is caused by removal of the wakefulness stimulus to breathe in patients with neuromuscular hypoventilation disease or severe abnormalities in pulmonary mechanics [22]. Therefore, it is a form of nocturnal ventilatory failure in patients with marginal ventilatory status or worsening of existing chronic ventilatory failure. Arousal from sleep restores alveolar ventilation to a variable degree; resumption of sleep reduces ventilation in a cyclical fashion. This type of central apnea does not necessarily meet the strict criteria of “central” or of “apnea”. Consequently, the presenting clinical picture includes both features of the underlying ventilatory insufficiency and features of the sleep apnea/hypopnea syndrome. This type of central apnea is uncommon in patients with CHF unless a concomitant neuromuscular disease of chest wall deformity is present.
Hypocapnia secondary to hyperventilation is the most common underlying mechanism of central apnea and the most relevant mechanism in patients with CHF. This type of apnea occurs in the absence of daytime alveolar hypoventilation; in fact, common features include hyperventilation and hence hypocapnia, even during wakefulness [23] with no evidence of a neuromuscular disorder, abnormal lung mechanics, or impaired responses to chemical stimuli. Oscillating sleep state at sleep onset [24] or transient hypoxia possibly due to reduced lung volumes in obese supine patients may trigger the “first” apnea and set in motion the process of apnea-hyperapnea and leads to sustained breathing instability, manifested as periodic breathing (see above).
Central apnea risk factors
Several physiologic or pathologic factors influence the susceptibility to central apnea including sleep state, age, gender, sleep state, and several medical conditions.
Sleep state exerts a major influence on breathing during sleep. Breathing instability is likely to occur at sleep onset as sleep state oscillates [24–27], with reciprocal oscillation in PaCO2 around the hypocapnic apneic threshold. Consequently, central apnea may occur if sufficient hypocapnia occurred; recovery from apnea is associated with transient wakefulness and hyperventilation. The subsequent hypocapnia elicits apnea upon resumption of sleep. Consolidation of sleep stabilizes PaCO2 at a higher set point above the apneic threshold. Interestingly, central apnea may occur without preceding hyperventilation at the transition from alpha to theta sleep in normal subjects and is associated with prolongation of breath duration [28]. Post-hyperventilation central sleep apnea is uncommon during REM sleep, suggesting that REM sleep is impervious to chemical influences owing to increased ventilatory motor output [29, 30].
Aging is associated with increased prevalence of central sleep apnea [31–33]. Possible causes include age-related sleep state oscillations [24, 34]; co-morbid conditions such as thyroid disease [35], congestive heart failure [36], atrial fibrillation [37], and cerebrovascular disease [38], or that aging per se may contribute to increased susceptibility to develop central apnea in older adults [32].
Central sleep apnea is uncommon in pre-menopausal women [39]. There is evidence that women are less susceptible to the development of hypocapnic central apnea relative to men following mechanical ventilation. Physiologically, the hypocapnic central apneic threshold is higher in men relative to women [2]. Administration of testosterone to healthy pre-menopausal women elevates the apneic threshold [40]. Conversely, suppression of testosterone with leuprolide acetate in healthy males decreases the hypocapnic-apneic threshold and potentially stabilizing respiration [41]. Thus, male sex hormones are the most likely factor elevating the apneic threshold in men.
Sleep apnea is common after a cerebrovascular accident (CVA) [38, 42, 43], with central apnea being the predominant type in 40% of patients of sleep apnea after a CVA [43]. Likewise, central apnea occurs in 30% of patients stable methadone maintenance treatment [44]. Finally, several medical conditions predispose to the development of central apnea including hypothyroidism, acromegaly, and renal failure [45–49], all of which have an unexpectedly high prevalence of sleep apnea. Nocturnal hemodialysis is associated with improvement in sleep apnea indices [48].
Some patients may manifest central apnea with no apparent risk factor and are considered to have “idiopathic central apnea”, associated with increased chemo responsiveness and sleep state instability [50, 51]. Nevertheless, it is possible that these patients will have occult cardiac or metabolic disease. For example, idiopathic central sleep apnea is more prevalent in patients with atrial fibrillation.
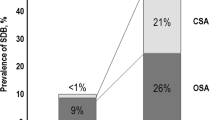
Sleep apnea is highly prevalent in patients with CHF [36, 52–54]. Javeheri et al. demonstrated that 51% of male patients with CHF had sleep-disordered breathing, 40% had central sleep apnea, and 11% obstructive apnea. Risk factors for CSA in this group of patients include male gender, atrial fibrillation, age >60 years, and daytime hypocapnia (PCO2 < 38 mmHg) [55]. Risk factors for OSA differed by gender; the only independent determinant in men was body mass index (BMI), whereas age over 60 was the only independent determinant in women.
The presence of pulmonary vascular congestion in patients with central apnea and CHF leads to hyperventilation and hypocapnia [36]. Paradoxically, steady-state hypocapnia is stabilizing by virtue of decreased plant gain [56–58]. In other words, steady-state reduction of PaCO2 is potentially stabilizing rather than destabilizing as is commonly thought. However, the apneic threshold in patients with central apnea and CHF is precariously close to the eupneic PCO2, because of increased chemoreflex sensitivity or controller gain.
Clinical features and diagnosis
Patients with central apnea may present with the usual symptoms of sleep apnea syndrome, or with insomnia and poor nocturnal sleep. These symptoms may be due to sleep fragmentation secondary to recurrent apnea. In fact, poor nocturnal sleep may be a presenting symptom in patients with central apnea due to CHF. Diagnosis of central sleep apnea requires nocturnal polysomnography and accurate detection of flow, measurement of oxyhemoglobin saturation, and detection of respiratory effort [59].
Accurate detection of respiratory effort is important to distinguish central from obstructive apnea. Cardiogenic oscillations (pulse artifacts) on the flow signal are often used as evidence of central etiology and a patent upper airway. The underlying rationale is the pulse artifacts represent transmission of a pulse waveform from the thorax, and hence indicates a patent upper airway that allows the transmission of cardiogenic oscillation. However, there is evidence that this index is invalid to ascertain upper airway patency. Morrell et al. [60] used fiber optic nasopharyngoscopy to evaluate upper airway patency during central apnea; cardiogenic oscillations were present even when the airway is completely occluded.
Other measurement of effort include measurement of the lapsed time for the pulse signal to reach the periphery, or pulse transit time (PTT) [59]. The technique measures the elapsed time between R wave on the ECG and the arrival of the pulse wave to the finger. The underlying physiologic principle is that blood pressure influences vessel stiffness and hence the speed of the pulse wave. When blood pressure increases during an obstructive apnea owing to increased intra-thoracic pressure swings, the pulse transit time displays parallel swings.
Management
Central apnea syndrome is a disorder with protean underlying conditions, therefore requiring a full panoply of therapeutic options. Management strategy incorporates underlying conditions, polysomnographic features, and individual factors. Optimal treatment of the underlying condition is a prerequisite component of the management plan in every patient. This is particularly important in patients with CHF where improvement of cardiac function may ameliorate central apnea. Likewise, diuresis may decrease pulmonary vascular congestion, improve oxygenation, and minimize overshoot. Specific therapeutic options include positive pressure therapy, pharmacologic therapy, and supplemental O2.
Positive pressure therapy
Central apnea may respond to nasal CPAP therapy, especially if it occurred in combination with episodes of obstructive or mixed apnea. Moreover, pure central sleep apnea may respond to nasal CPAP therapy for several reasons: (i) preventing pharyngeal narrowing during central apnea [14], (ii) improved oxygenation by increased lung volume, and (iii) improving cardiac function by decreasing pre-load and after load. The aggregate of these changes would dampen the ensuing ventilatory overshoot and perpetuation of ventilatory instability [20]. In fact, nasal CPAP is often effective in reversing central sleep apnea, even in the absence of concomitant obstructive apnea [14], possibly by preventing upper airway closure and subsequent ventilatory overshoot [20]. This may also explain the reported increase in PCO2 after CPAP [61]. Similarly, nasal CPAP therapy in patients with central sleep apnea and CHF produced increased left-ventricular ejection fraction (LVEF) and a reduction of combined mortality-cardiac transplantation risk by 81%, but only in patients with CSA [62].
The exuberance regarding nasal CPAP therapy in patients with central apnea and CHF did not withstand the rigors of controlled clinical trials. The Canadian Continuous Positive Airway Pressure trial, or CANPAP [63], tested the hypothesis that CPAP would improve the survival rate without heart transplantation in patients with heart failure and central sleep apnea. The study enrolled 258 patients who had heart failure and central sleep apnea; participants were randomly assigned to the nasal CPAP treatment group (n = 128) or no CPAP (130 patients). Duration of follow up was for a mean of 2 years. There was greater improvement in the CPAP group at 3 months relative to the placebo group as evidenced by greater reductions in apnea-hypopnea index, ejection fraction, mean nocturnal oxyhemoglobin saturation, plasma nor-epinephrine levels, and the distance walked in 6 min at 3 months. Nevertheless, there was no difference in the overall event rates (death and heart transplantation) between the two groups. Thus, nasal CPAP had no effect on survival, despite the effect on the “severity” of central apnea and several intermediate outcome variables. Nevertheless, a post-hoc analysis has shown decreased mortality if CPAP therapy resulted in amelioration of central sleep apnea. Therefore, current evidence does not support the use of CPAP to extend life in patients who have heart failure and central sleep apnea unless evidence of response is demonstrated by polysomnography.
The major finding of CANPAP was the absence of a survival benefit for nasal CPAP therapy. Nasal CPAP therapy had no effect on heart transplant-free survival; however, aggregate data showed only a modest reduction of AHI to 19 events per hour of sleep; a cutoff of 15 events/h was the inclusion threshold for the study and remains a commonly accepted threshold for pathologic AHI. Another limitation of the study was that nasal CPAP therapy was not titrated under polysomnographic monitoring but increased gradually to 10 cm/H2O or to the highest tolerated number. Optimal CPAP pressure was not determined. In addition, only 100 of the 128 randomized to CPAP underwent repeat polysomnography after 3 months of nasal CPAP therapy [64]. Fifty-seven patients responded to nasal CPAP as evidenced by decreased AHI below 15/h of sleep. Interestingly, the responders (CPAP-CSA-suppressed) group demonstrated a statistically significant increase in LVEF at 3 months (P = 0.001) and a higher transplant-free survival than control subjects. Conversely, the non-responders (CPAP-CSA-unsuppressed) group demonstrated no increase in either variable. These findings suggest that improvement in LVEF may be a marker of improved survival. In addition, this study highlights the need to conform optimal response to patients with CHF-CSA. Lowering AHI below 15 events/h of sleep may be suboptimal in terms of improved nocturnal respiration, cardiac function, or mortality.
Non-invasive positive pressure ventilation (NIPPV) using pressure support mode (bi-level nasal positive pressure) is a therapeutic option in patients with nocturnal ventilatory failure and central apnea secondary to hypoventilation. However, there is little evidence to support its use in non-hypercapnic central apnea including CHF. In fact, NIPPV in the pressure support, bi-level mode augments tidal volume, leading to hypocapnia and possible worsening of central apnea and periodic breathing during sleep [65]. Meza et al. [66] have shown that application of pressure support ventilation results in periodic breathing in most normal subjects when the pressure gradient is above 7 cm/H2O.
Recent technological advances allowed for variations in the mode of delivering positive pressure ventilation. One such method is adaptive servo ventilation (ASV) that provides a small but varying amount of ventilatory support against a background of low level of CPAP. Contrary to bi-level, pressure support devices, changes in respiratory effort result in reciprocal changes in the magnitude of ventilatory support. Thus, ventilation remains slightly below the baseline, eupneic average. There is evidence that ASV is more effective than CPAP, bi-level pressure support ventilation, or increased dead space in alleviating central sleep apnea [67].
Pharmacological therapy
Pharmacological therapy is of marginal role in central apnea. Small studies suggest acetazolamide and theophylline may be beneficial in the treatment of central apnea [68]. Acetazolamide is a carbonic anhydrase inhibitor and a weak diuretic that causes mild metabolic acidosis. Acetazolamide ameliorates central sleep apnea when administered as a single dose of 250 mg before bedtime [69]. Likewise, theophylline ameliorates the severity of Cheyne–Stokes respiration in patients with CHF [70], without adverse effect on sleep architecture. Pharmacologic therapy remains an unfulfilled opportunity that awaits further research.
Several pharmacologic agents may influence the propensity to develop central apnea; therefore, medication use is an important consideration in the management patients with central sleep apnea. For example, the use of beta-blockers in patients with central sleeps apnea and congestive heart failure is associated with reduced apnea index. Conversely, narcotics may worsen central apnea; a change in the pain control regimen may ameliorate the severity of central apnea [44, 71].
Supplemental O2 and CO2
Supplemental O2 therapy may be beneficial in patients with idiopathic central sleep apnea and patients with CHF-CSR [36, 72], most likely by ameliorating hypoxemia and minimizing the subsequent ventilatory overshoot. In addition, supplemental O2 may alleviate central apnea by increasing cerebral PCO2 through the Haldane effect. Likewise, supplemental CO2 abolishes central apnea in patients with pure central sleep apnea. The mechanism of action is by raising PCO2 above the apneic threshold. However, this therapy is not practical given the need for a closed circuit to deliver supplemental CO2.
Approach in selected clinical syndromes
-
1.
It is imperative to optimize treatment of the underlying condition such as CHF.
-
2.
Titration of CPAP in the sleep laboratory is beneficial to ascertain response to nasal positive airway pressure therapy. This may determine whether long-term treatment is of benefit.
-
3.
The optimal pressure settings have to be determined in a titration polysomnogram.
-
4.
The use of B-PAP in a pressure support mode may aggravate the severity of central apnea. ASV is a promising therapeutic modality for CHF-CSR.
-
5.
Supplemental O2 may be beneficial, particularly in patients with CHF-CSR.
-
6.
The use of pharmacologic agents remains uncommon. Additional studies are needed.
Summary
The pathogenesis of central sleep apnea varies depending on the clinical condition. Sleep-related withdrawal of the ventilatory drive to breathe is the common denominator among all cases of central apnea, whereas hypocapnia is the final common pathway leading to apnea in non-hypercapnic central apnea. The pathophysiologic heterogeneity may explain the protean clinical manifestations and the lack of a single effective therapy for all patients.
References
Skatrud JB, Dempsey JA (1983) Interaction of sleep state and chemical stimuli in sustaining rhythmic ventilation. J Appl Physiol 55:813–822
Zhou XS, Shahabuddin S, Zahn BK, Babcock MA, Badr MS (2000) Effect of gender on the development of hypocapnic apnea/hypopnea during NREM sleep. J Appl Physiol 89:192–199
Tawadrous FD, Eldridge FL (1974) Posthyperventilation breathing patterns after active hyperventilation in man. J Appl Physiol 37:353–356
Eldridge FL, Gill-Kumar P (1980) Central neural respiratory drive and afterdischarge. Respir Physiol 40:49–63. doi:10.1016/0034-5687(80)90004-3
Badr MS, Skatrud JB, Dempsey JA (1992) Determinants of poststimulus potentiation in humans during NREM sleep. J Appl Physiol 73:1958–1971
Badr MS, Morgan BJ, Finn L, Toiber FS, Crabtree DC, Puleo DS et al (1997) Ventilatory response to induced auditory arousals during NREM sleep. Sleep 20:707–714
Berssenbrugge A, Dempsey J, Iber C, Skatrud J, Wilson P (1983) Mechanisms of hypoxia-induced periodic breathing during sleep in humans. J Physiol 343:507–526
Badr MS, Kawak A (1996) Post-hyperventilation hypopnea in humans during NREM sleep. Respir Physiol 103:137–145. doi:10.1016/0034-5687(95)00083-6
Chow CM, Xi L, Smith CA, Saupe KW, Dempsey JA (1994) A volume-dependent apneic threshold during NREM sleep in the dog. J Appl Physiol 76:2315–2325
Harms CA, Zeng YJ, Smith CA, Vidruk EH, Dempsey JA (1996) Negative pressure-induced deformation of the upper airway causes central apnea in awake and sleeping dogs. J Appl Physiol 80:1528–1539
Sahlin C, Svanborg E, Stenlund H, Franklin KA (2005) Cheyne-Stokes respiration and supine dependency. Eur Respir J 25:829–833. doi:10.1183/09031936.05.00107904
Oksenberg A, Arons E, Snir D, Radwan H, Soroker N (2002) Cheyne-Stokes respiration during sleep: a possible effect of body position. Med Sci Monit 8:CS61–CS65
Szollosi I, Roebuck T, Thompson B, Naughton MT (2006) Lateral sleeping position reduces severity of central sleep apnea/Cheyne-Stokes respiration. Sleep 29:1045–1051
Issa FG, Sullivan CE (1986) Reversal of central sleep apnea using nasal CPAP. Chest 90:165–171. doi:10.1378/chest.90.2.165
Khoo MC, Kronauer RE, Strohl KP, Slutsky AS (1982) Factors inducing periodic breathing in humans: a general model. J Appl Physiol 53:644–659
Khoo MC (2000) Determinants of ventilatory instability and variability. Respir Physiol 122:167–182. doi:10.1016/S0034-5687(00)00157-2
Chapman KR, Bruce EN, Gothe B, Cherniack NS (1988) Possible mechanisms of periodic breathing during sleep. J Appl Physiol 64:1000–1008
Cherniack NS, Longobardo GS (2006) Mathematical models of periodic breathing and their usefulness in understanding cardiovascular and respiratory disorders. Exp Physiol 91:295–305. doi:10.1113/expphysiol.2005.032268
Leevers AM, Simon PM, Dempsey JA (1994) Apnea after normocapnic mechanical ventilation during NREM sleep. J Appl Physiol 77:2079–2085
Badr MS, Toiber F, Skatrud JB, Dempsey J (1995) Pharyngeal narrowing/occlusion during central sleep apnea. J Appl Physiol 78:1806–1815
Olson LG, Strohl KP (1988) Airway secretions influence upper airway patency in the rabbit. Am Rev Respir Dis 137:1379–1381
Mezon BL, West P, Israels J, Kryger M (1980) Sleep breathing abnormalities in kyphoscoliosis. Am Rev Respir Dis 122:617–621
Bradley TD, Phillipson EA (1992) Central sleep apnea. Clin Chest Med 13:493–505
Pack AI, Cola MF, Goldszmidt A, Ogilvie MD, Gottschalk A (1992) Correlation between oscillations in ventilation and frequency content of the electroencephalogram. J Appl Physiol 72:985–992
Trinder J, Whitworth F, Kay A, Wilkin P (1992) Respiratory instability during sleep onset. J Appl Physiol 73:2462–2469
Dunai J, Wilkinson M, Trinder J (1996) Interaction of chemical and state effects on ventilation during sleep onset. J Appl Physiol 81:2235–2243
Dunai J, Kleiman J, Trinder J (1999) Ventilatory instability during sleep onset in individuals with high peripheral chemosensitivity. J Appl Physiol 87:661–672
Thomson S, Morrell MJ, Cordingley JJ, Semple SJ (2005) Ventilation is unstable during drowsiness before sleep onset. J Appl Physiol 99:2036–2044
Orem J, Lovering AT, Dunin-Barkowski W, Vidruk EH (2002) Tonic activity in the respiratory system in wakefulness, NREM and REM sleep. Sleep 25:488–496
Orem J (1980) Neuronal mechanisms of respiration in REM sleep. Sleep 3:251–267
Phillips B, Cook Y, Schmitt F, Berry D (1989) Sleep apnea: prevalence of risk factors in a general population. South Med J 82:1090–1092
Phillips BA, Berry DT, Schmitt FA, Magan LK, Gerhardstein DC, Cook YR (1992) Sleep-disordered breathing in the healthy elderly. Clinically significant? Chest 101:345–349. doi:10.1378/chest.101.2.345
Ancoli-Israel S, Kripke DF, Klauber MR, Mason WJ, Fell R, Kaplan O (1991) Sleep-disordered breathing in community-dwelling elderly. Sleep 14:486–495
Pack AI, Silage DA, Millman RP, Knight H, Shore ET, Chung DC (1988) Spectral analysis of ventilation in elderly subjects awake and asleep. J Appl Physiol 64:1257–1267
Kapur VK, Koepsell TD, deMaine J, Hert R, Sandblom RE, Psaty BM (1998) Association of hypothyroidism and obstructive sleep apnea. Am J Respir Crit Care Med 158:1379–1383
Bradley TD, Floras JS (2003) Sleep apnea and heart failure: part II: central sleep apnea. Circulation 107:1822–1826. doi:10.1161/01.CIR.0000061758.05044.64
Leung RS, Huber MA, Rogge T, Maimon N, Chiu KL, Bradley TD (2005) Association between atrial fibrillation and central sleep apnea. Sleep 28:1543–1546
Bassetti C, Aldrich MS (1999) Sleep apnea in acute cerebrovascular diseases: final report on 128 patients. Sleep 22:217–223
Bixler EO, Vgontzas AN, Lin HM, Ten Have T, Rein J, Vela-Bueno A et al (2001) Prevalence of sleep-disordered breathing in women: effects of gender. Am J Respir Crit Care Med 163:608–613
Zhou XS, Rowley JA, Demirovic F, Diamond MP, Badr MS (2003) Effect of testosterone on the apneic threshold in women during NREM sleep. J Appl Physiol 94:101–107
Mateika JH, Omran Q, Rowley JA, Zhou XS, Diamond MP, Badr MS (2004) Treatment with leuprolide acetate decreases the threshold of the ventilatory response to carbon dioxide in healthy males. J Physiol 561:637–646. doi:10.1113/jphysiol.2004.071811
Bassetti C, Aldrich MS, Chervin RD, Quint D (1996) Sleep apnea in patients with transient ischemic attack and stroke: a prospective study of 59 patients. Neurology 47:1167–1173
Parra O, Arboix A, Bechich S, Garcia-Eroles L, Montserrat JM, Lopez JA, Ballester E, Guerra JM, Sopena JJ (2000) Time course of sleep-related breathing disorders in first-ever stroke or transient ischemic attack. Am J Respir Crit Care Med 161:375–380
Wang D, Teichtahl H, Drummer O, Goodman C, Cherry G, Cunnington D et al (2005) Central sleep apnea in stable methadone maintenance treatment patients. Chest 128:1348–1356. doi:10.1378/chest.128.3.1348
Grunstein RR, Ho KY, Berthon-Jones M, Stewart D, Sullivan CE (1994) Central sleep apnea is associated with increased ventilatory response to carbon dioxide and hypersecretion of growth hormone in patients with acromegaly. Am J Respir Crit Care Med 150:496–502
Grunstein RR, Ho KY, Sullivan CE (1991) Sleep apnea in acromegaly. Ann Intern Med 115:527–532
Grunstein RR, Sullivan CE (1988) Sleep apnea and hypothyroidism: mechanisms and management. Am J Med 85:775–779
Hanly PJ, Pierratos A (2001) Improvement of sleep apnea in patients with chronic renal failure who undergo nocturnal hemodialysis. N Engl J Med 344:102–107. doi:10.1056/NEJM200101113440204
Venmans BJW, van Kralingen KW, Chandi DD, de Vries PMJM, ter Wee PM, Postmus PE (1999) Sleep complaints and sleep disordered breathing in hemodialysis patients. Neth J Med 54:207–212. doi:10.1016/S0300-2977(99)00018-2
Xie A, Rutherford R, Rankin F, Wong B, Bradley TD (1995) Hypocapnia and increased ventilatory responsiveness in patients with idiopathic central sleep apnea. Am J Respir Crit Care Med 152:1950–1955
Xie A, Wong B, Phillipson EA, Slutsky AS, Bradley TD (1994) Interaction of hyperventilation and arousal in the pathogenesis of idiopathic central sleep apnea. Am J Respir Crit Care Med 150:489–495
Javaheri S, Parker TJ, Liming JD, Corbett WS, Nishiyama H, Wexler L et al (1998) Sleep apnea in 81 ambulatory male patients with stable heart failure. Types and their prevalences, consequences, and presentations. Circulation 97:2154–2159
Javaheri S, Parker TJ, Wexler L, Michaels SE, Stanberry E, Nishyama H et al (1995) Occult sleep-disordered breathing in stable congestive heart failure. Ann Intern Med 122:487–492
Javaheri S (1996) Central sleep apnea-hypopnea syndrome in heart failure: prevalence, impact, and treatment. Sleep 19:S229–S231
Sin DD, Fitzgerald F, Parker JD, Newton G, Floras JS, Bradley TD (1999) Risk factors for central and obstructive sleep apnea in 450 men and women with congestive heart failure. Am J Respir Crit Care Med 160:1101–1106
Xie A, Skatrud JB, Puleo DS, Rahko PS, Dempsey JA (2002) Apnea-hypopnea threshold for CO2 in patients with congestive heart failure. Am J Respir Crit Care Med 165:1245–1250. doi:10.1164/rccm.200110-022OC
Dempsey JA, Sheel AW, Haverkamp HC, Babcock MA, Harms CA (2003) [The John Sutton Lecture: CSEP, 2002]. Pulmonary system limitations to exercise in health. Can J Appl Physiol 28(Suppl):S2–S24
Dempsey JA, Smith CA, Przybylowski T, Chenuel B, Xie A, Nakayama H et al (2004) The ventilatory responsiveness to CO(2) below eupnoea as a determinant of ventilatory stability in sleep. J Physiol 560:1–11. doi:10.1113/jphysiol.2004.072371
Farre R, Montserrat JM, Navajas D (2004) Noninvasive monitoring of respiratory mechanics during sleep. Eur J Respir Dis 24:1052–1060. doi:10.1183/09031936.04.00072304
Morrell MJ, Badr MS, Harms CA, Dempsey JA (1995) The assessment of upper airway patency during apnea using cardiogenic oscillations in the airflow signal. Sleep 18:651–658
Naughton MT, Benard DC, Rutherford R, Bradley TD (1994) Effect of continuous positive airway pressure on central sleep apnea and nocturnal PCO2 in heart failure. Am J Respir Crit Care Med 150:1598–1604
Sin DD, Logan AG, Fitzgerald FS, Liu PP, Bradley TD (2000) Effects of continuous positive airway pressure on cardiovascular outcomes in heart failure patients with and without Cheyne-Stokes respiration. Circulation 102:61–66
Bradley TD, Logan AG, Kimoff RJ, Series F, Morrison D, Ferguson K et al (2005) Continuous positive airway pressure for central sleep apnea and heart failure. N Engl J Med 353:2025–2033. doi:10.1056/NEJMoa051001
Arzt M, Floras JS, Logan AG, Kimoff RJ, Series F, Morrison D et al (2007) Suppression of central sleep apnea by continuous positive airway pressure and transplant-free survival in heart failure: a post hoc analysis of the Canadian Continuous Positive Airway Pressure for Patients with Central Sleep Apnea and Heart Failure Trial (CANPAP). Circulation 115:3173–3180. doi:10.1161/CIRCULATIONAHA.106.683482
Johnson KG, Johnson DC (2005) Bilevel positive airway pressure worsens central apneas during sleep. Chest 128:2141–2150. doi:10.1378/chest.128.4.2141
Meza S, Mendez M, Ostrowski M, Younes M (1998) Susceptibility to periodic breathing with assisted ventilation during sleep in normal subjects. J Appl Physiol 85:1929–1940
Teschler H, Dohring J, Wang YM, Berthon-Jones M (2001) Adaptive pressure support servo-ventilation: a novel treatment for Cheyne-Stokes respiration in heart failure. Am J Respir Crit Care Med 164:614–619
Hudgel DW, Thanakitcharu S (1998) Pharmacologic treatment of sleep-disordered breathing. Am J Respir Crit Care Med 158:691–699
DeBacker WA, Verbraecken J, Willemen M, Wittesaele W, DeCock W, Van deHeyning P (1995) Central apnea index decreases after prolonged treatment with acetazolamide. Am J Respir Crit Care Med 151:87–91
Javaheri S, Parker TJ, Wexler L, Liming JD, Lindower P, Roselle GA (1996) Effect of theophylline on sleep-disordered breathing in heart failure. N Engl J Med 335:562–567. doi:10.1056/NEJM199608223350805
Sin DD, Man GC, Jones RL (2000) Central sleep apnea and heart failure. N Engl J Med 342:293–294. doi:10.1056/NEJM200001273420416
Javaheri S, Ahmed M, Parker TJ, Brown CR (1999) Effects of nasal O2 on sleep-related disordered breathing in ambulatory patients with stable heart failure. Sleep 22:1101–1106
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Badr, S. Central sleep apnea in patients with congestive heart failure. Heart Fail Rev 14, 135–141 (2009). https://doi.org/10.1007/s10741-008-9100-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-008-9100-3