
Abstract
Pistacia genus is botanically classified in the Sapindales order and the Anacardiaceae family, and the member of this genus P. eurycarpa is used as a rootstock for pistachio. Due to its rich oil content, various soaps are produced from the green peels of P. eurycarpa nuts and are used in the cosmetic industry as soap. In the current study, we aimed to release the complete chloroplast genome of P. eurycarpa to detect the relationship of the members of the Sapindales and also Pistacia genus. The length of the chloroplast genome of P. eurycarpa was identified 160,693 (bp) and consisted of the distinct parts of the genomic fragments such as LSC, SSC, and IR regions. The length of the LSC was 88,410 (bp), and SSC genomic range represented a total of 19,087 (bp). Inverted regions length was 26,598 (bp). A total of 125 genes were determined within the whole chloroplast genome of P. eurycarpa. The number of simple sequence repeats (SSRs) in the chloroplast genome was computed as 74. Three main clusters defined the dendrogram of all accessions. The closest pairs of species were identified among the P. eurycarpa and P. vera species. The farthest genetic relationship was calculated among the members of the Sapindales species Mangifera indica and the Fagales species Castanea mollissima. The sequences of the chloroplast genome of P. eurycarpa species can be evaluated for the construction of the basis of the complete evolution of the order, genera, and the development of the SSRs for species-specific markers for revealing the evolution of Pistacia species for future breeding research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pistacia L. genus takes place within the Sapindales order, and it is a member of the Anacardiaceae family. Horticultural important species such as mango, pepper tree, and sumac are also members of the Anacardiaceae (Kafkas et al. 2006). The genus Pistacia contains at least eleven species with more sub-species (Kafkas 2006). Pistacia species are generally dioecious flowering structure with a few exceptions (Kafkas et al. 2000). P. eurycarpa is one of the most significant species within the Pistacia, and is believed to be derived from ancestral species of P. vera (Kafkas 2019).
Taxonomic investigation of the genus Pistacia was first performed by Zohary (1952) in several morphological traits of the accessions. The researcher stated that P. eurycarpa has not evaluated a novel species, and it could be a subspecies of P. atlantica (var. kurdica); despite the detection of the distinct species from other wild Pistacia species by Yaltirik (1967). He reported that six different species were recorded in Türkiye, especially in southeastern regions such as P. terebinthus, P. eurycarpa, P. khinjuk, P. vera, P. atlantica and P. lentiscus. Yaltırık added a new species to the literature used as a rootstock for pistachios in Türkiye, and reported that the P. eurycarpa species is a different species from others. However, since this is different from Zohary's classification, Yaltırık (1967) distinguished P. eurycarpa from P. atlantica in terms of two features. Firstly, P. atlantica has a petiole blade that continues from the terminal leaflet, but the petiole blade of P. eurycarpa is narrower, and sometimes present and sometimes empty. Another feature is that its nuts have a more flattened shape.
The first transcriptomic paper related to P. eurycarpa was carried out using the samples from a location obtained from Yaltırık (1967) mentioned regions in Göbek village of Gaziantep province of Türkiye (Karcι et al. 2020). The latter authors studied the genetic background of the Pistacia species using simple sequence repeats (SSRs) developed from transcriptomes of P. vera. The results of the paper quoted that P. eurycarpa could be a separate species and closer to P. atlantica than P. khinjuk. Although a few phylogenetic papers included P. eurycarpa species were reported using different molecular marker systems with dominant or codominant inheritance, the ambiguity is still ongoing about the closeness or distance of P. eurycarpa to other species or subspecies (Kafkas and Perl-Treves 2001; Karcι et al. 2020). However, the results of the molecular characterization papers supported by Yaltırık (1967) and by Kafkas and Perl-Treves (2001) that P. eurycarpa was a separate species from the others.
Pistacia eurycarpa is cultivated naturally in Eastern Anatolia and Southeastern Anatolia regions in Türkiye, and it is grown naturally at altitudes between 1100 and 1800 m (AL-Saghir and Porter 2012). The natural distribution of P. eurycarpa from Afghanistan, Armenia, Iran, Iraq, Pakistan, Syria (desert) to Türkiye (Eastern Anatolia) and temperate Asia. And, it is distributed in the Eastern and Southeastern Anatolia regions of Türkiye (Yaltirik 1967).
Pistacia eurycarpa is used as a rootstock for pistachios since it adapts to clayey, calcareous, and hillside lands. Its nuts are used for different purposes. In Türkiye, its nuts are used as snacks in Southeastern Anatolia and especially in Siirt province. Due to its rich oil content, various soaps are produced from the green peels of P. eurycarpa nuts and are used in the cosmetic industry as soap. In addition, Demirci et al. (2001) reported that P. eurycarpa resin had different levels of antibacterial activity against negative and positive gram bacteria. Thus, P. eurycarpa has not only importance in horticulture but also has potential in the cosmetic industry.
To date, the previous research on P. eurycarpa mainly focused on the development of the markers and the detection of the rootstock potential or power for P. vera (Kafkas and Perl-Treves 2001; Kafkas 2006; Karcι et al. 2020). The discussion related to P. eurycarpa is a separate species from other wild Pistacia species or is a subspecies of the closely related Pistacia species is still ongoing within the Pistacia breeders since there are not enough papers related to chloroplast genomes of the genus Pistacia (Kafkas 2019; Karcι et al. 2020). Recently, several complete chloroplast genome sequences belonging to Pistacia species were released in P. weinmannifolia (Zheng et al. 2018), P. chinensis (Xu et al. 2019, 2020), and P. vera (Mao et al. 2019) for completing of the relationship among Pistacia species. However, the genomic information of the P. eurycarpa species was an extremely limited number, although it is quite a significant species as a horticulturally.
Here, the main objectives of the current paper are to (i) construct the complete chloroplast genome of the P. eurycarpa for the first time, (ii) reveal the genomic location of the P. eurycarpa within the Pistacia and Sapindales species and (iii) consist of the basis for completing the evolution of the Pistacia genus. Revealing complexity within Pistacia sup-species/species using the other wild Pistacia chloroplast genome sequences will be the next step, after this paper.
Material and methods
Plant material and DNA extraction
Pistacia eurycarpa genotype was used as the plant material of the present study. The fresh plant leaves of the genotype were gathered from its wide distribution area, the province of Siirt in Türkiye (Fig. 1).

The distribution fields of P. eurycarpa all over the world and the region of the sampled of P. eurycarpa genotype in Türkiye
Genomic DNA was extracted utilizing the CTAB (cetyltrimethylammonium bromide) technique (Doyle and Doyle 1987) with minor alterations (Kafkas et al. 2006). The quality of the extracted genomic DNA was controlled by 0.8% agarose gel. The measurement of concentration was performed using both the gel electrophoresis method and Qubit Fluorimeter (Invitrogen) by following the standard procedures.
Whole genome re-sequencing (WGRS) and chloroplast genome assembly
The high-quality paired-end sequencing was performed by BGI (Beijing Genomic Institute) using an Illumina (Hi-Seq 2500) NGS platform. The sequencing coverage was set to 20× coverage. The clean reads (150 bp) of P. eurycarpa sample were mapped to the reference genome of P. vera (Kafkas et al. 2022) assembled with the chloroplast genome of P. vera (KY549635) using BWA (v0.7.12) (Li and Durbin 2009). Then the Sequence Alignment/Map (SAM/BAM) file were sorted using the SAMTOOLS (Li et al. 2009) software. The duplicated reads of the sample were marked using the PICARD software (http://picard.sourceforge.net). The final output of reads (BAM) was saved for extraction of the fragment of the chloroplast. The extracted (BAM) file was converted to (SAM), and PICARD was used for converting to fastq files with forward and reverse paired in the following step. The new paired reads were assembled with SPADES and scaffold.contigs were produced for generating reference-based merged sequences using Geneious software (Grant and Stothard 2008).
Gene annotation and sequence analysis
The constructed P. eurycarpa chloroplast genome fasta file was loaded to annotate the chloroplast genome Genbank file to CHLOROBOX GeSeq software (Tillich et al. 2017). Then, the chloroplast genome map was created using the fasta and Genbank file with OGDRAW v1.2 (Greiner et al. 2019). CpGAVAS was used to annotate the sequences for detailed results (Liu et al. 2012).
Detection of the simple sequence repeats (SSRs)
MIcroSAtellite (MISA) was used to detect simple sequence repeats in the chloroplast genome of P. eurycarpa (Beier et al. 2017). The parameters for SSRs were set as follows: a minimum number of 10, 5, 4, 3, 3, and 3 repeat units was adjusted for the identification of perfect mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides motifs, respectively. In addition to the whole chloroplast genome, SSRs were separately analyzed in different regions of genomes. Tandem repeat finder (https://tandem.bu.edu/trf/trf.submit.options.html, accessed on September 2nd 2023) was used to identify tandem repeats in the chloroplast genomes. Palindromic and forward repeats were detected by REPuter software (Kurtz et al. 2001). The minimum sizes for repeat elements and the sequence identity were considered to be 30 bp and more than 90% (hamming distance of 3), respectively.
Phylogenetic analysis
The analysis of the relationship was carried out using a total of 18 chloroplast genomes. Pistacia species such as P. eurycarpa, P. vera, P. chinensis, and P. weinmanniifolia and the species of members of the Sapindales order within other genera were used to detect genetic dissimilarities of the genomes. In addition, hard-shelled nuts such as Castanea mollissima Blume and Junglans regia L. were included in the phylogenetic analysis (Table 1). The dendrogram of chloroplast genomes was generated with a neighbor-joining tree and genetic dissimilarity coefficients of the chloroplast genomes were calculated in Geneious software (Grant and Stothard 2008).
Results
Pistacia eurycarpa chloroplast genome structure and features
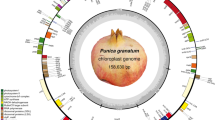
The first complete P. eurycarpa chloroplast genome was sequenced using the samples from Siirt province of the Türkiye. The statistics of the genomic positions and lengths of the chloroplast genome were mined in several open or commercial and web-based programs. The length of the chloroplast genome of P. eurycarpa was identified 160,693 (bp) and consisted of the distinct parts of the genomic fragments such as LSC, SSC, and IR regions. The length of the LSC was 88,410 (bp), and SSC genomic range represented a total of 19,087 (bp) (Fig. 2, Table 2). The covering values of the genome of these regions were 55.02% and 11.88%, respectively. Two inverted regions covered the genome with 33.10%, this length was 26,598 (bp). A total of 125 genes were determined within the whole chloroplast genome. They consisted of four rRNA, 38 tRNA, and 83 protein-coding genes. The most abounded (49 genes) were photosynthesis-related genes, followed by proteins for subunits of ribosomal proteins (29 genes) and RNA polymerase-coding genes (4 genes) among the 87 protein-coding genes. In total, six protein coding genes were responsible for subunit acetyl-CoA-carboxylase, maturase, c-type cytochrome synthesis, and envelope membrane protein genes (Table 3). The highest proportion of the predicted genes were localized into the LSC region with 90 genes. The high amount of the rRNA-related genes distributed in LSC and SSC sections, except ccsA were located in LSC. tRNA genes were taken place into IR regions, although many NADH dehydrogenase genes were defined into SSC genomic part (Table 4).

Circle map of Pistacia eurycarpa chloroplast genome drawn using CPGview (Liu et al. 2023). The first circle represents the whole chloroplast genome size of P. eurycarpa and its GC content. LSC, SSC, and inverted repeat (IRA and IRB) regions were shown on the second track. The third track belongs to the GC contents of the parts of the genome, and the outside circle represents the genes
The percentage of the chloroplast genome GC content in all of the fragments was calculated, and the highest rate of the GC content was defined in rRNA regions as 55.64%. The high variability in nucleotide distribution was exhibited in P. eurycarpa chloroplast genome. And, IR regions attracted attention being second with the highest GC content proportion (42.97%). LSC and SSC regions produced 35.91 and 32.39% GC contents, respectively, although the GC content of tRNA genes was close to the whole genome GC content of the P. eurycarpa (37.92%) (Table 2).
A total of ten genes owing to intron were recognized into P. eurycarpa chloroplast genome. Among them, nine intron-containing genes had one intron while pafI gene contained in Intron II. Moreover, two intron-owing genes exhibited the duplication into IR genomic regions. The biggest intron sequence was calculated as 1120 (bp) within ndhA gene, although only one intron-owing gene was recorded within the SSC region (Table 4).
Frequencies of the amino acids were computed into the chloroplast genome of P. eurycarpa. In total, 52,637 codons were recognized within P. eurycarpa chloroplast genes. The leucine amino acid was the most frequent and consisted of a total of 5459 different codon types (10.37%), in the following, isoleucine with 4398 (8.36%). On the other hand, cysteine (690, 1.31%) and tryptophan (984, 1.87%) amino acids were identified as the least prevalent amino acids in protein-coding regions of P. eurycarpa chloroplast genome. The type of the ATT codon derived from isoleucine represented the most prevalent codon (1072) within P. eurycarpa chloroplast genome (Table 5).
Comparison of P. eurycarpa chloroplast genome with Pistacia species
The P. eurycarpa chloroplast genome size and parts of the genomes were compared with other genus Pistacia species (Table 6). To date, the reported four Pistacia species including P. eurycarpa of chloroplast genome sizes were detected similar to each other. The range of the whole genomes varied from 160,598 to 160,767 (bp), and the largest chloroplast genome size was detected in P. weinmaniifolia. However, P. eurycarpa genome size (160,693 bp) was almost similar to P. chinensis and P. weinmaniifolia. The whole chloroplast genome size variation was calculated as 169 (bp) among Pistacia species. The GC contents of the chloroplast genomes had close values, GC contents of the P. eurycarpa was 37.83%. The biggest genomic fragment of P. eurycarpa was a large single-copy (LSC = 88,410 bp), and the size of the LSC of variation of all genomes was 236 (bp). In addition, although P. eurycarpa had 19,087 (bp) SSC genomic length, the smallest and largest SSC lengths were calculated as 19,330 bp (P. vera) and 19,057 bp (P. chinensis), respectively. The lengths of IR regions of Pistacia species ranged from 26,547 (P. vera) to 26,618 (bp) (P. weinmaniifolia). The numbers of genes in the P. eurycarpa genome were detected as 125 genes, and the total genes were ordered from highest to lowest values, P. chinensis (132), P. weinmaniifolia (131 genes) P. eurycarpa (125) and P. vera (121 genes). The lowest tRNA-related genes were found in P. vera, while the highest tRNA genes were exhibited in P. eurycarpa. On the other hand, the number of rRNA genes was not recognized variability among Pistacia species.
Detection of the SSRs in P. eurycarpa chloroplast genome
The whole chloroplast genome sequences of P. eurycarpa were screened for detection of the SSRs. The number of SSRs in the chloroplast genome was computed as a total of 74 SSRs, and the distribution of the SSRs in the whole genome fragments was carried out between three and 54 SSRs (Table 2). The most abounded SSRs were mined from the largest genomic regions (LSC) as a total of 54 SSRs, while the region of the rRNA was not identified in the length of the 4520 (bp). The rest of the detected SSRs were distributed according to counts from SSC to tRNA coding sequences. Mononucleotide repeats were the most frequent motifs, while the repetition of the di-nucleotides was found in quite low numbers. The most common type of simple sequence repeats was T/A repeats in P. eurycarpa. The other motif types were not found in P. eurycarpa chloroplast genome.
The long repeat sequences within the P. eurycarpa genome were recognized, and tandem repeats distribution was found frequently in the chloroplast genome. A total of 42 tandem repeats were identified in P. eurycarpa chloroplast genome. The most tandem repeats were ranged from 18 to 20 (bp). Most of the forward (93.75%) and palindromic (52.94%) repeat lengths varied from 22 to 52 (bp). In total, 16 forward and 34 palindromic and 42 tandem repeats were revealed in the chloroplast genome of P. eurycarpa, respectively.
Identification of genetic relationship of the species
Cluster analysis of a total of 18 species belonging to Sapindales and Fagales orders was carried out using the neighbor-joining method. Three main clusters consisted in the dendrogram, and the accessions belonging to Sapindales order were grouped within two sub-groups. The first main Sapindales cluster covered all species between P. eurycarpa and L. chinensis species. The second sub-cluster consisted of the C. sinensis, Z. bungeanum, L. floridana and K. senegalensis species. The members of the Fagales order species J. regia and C. mollissima were separated sharply and defined as outgroups. The genus Pistacia samples were separated from the other members of the Sapindales. The closest pairs of species were identified among the P. eurycarpa and P. vera species. In the following, P. chinensis joined these species, and it was the second close species within Pistacia genus according to coefficients. Although P. vera and P. eurycarpa species had the closest relationship, the most distant of them was exhibited with P. weinmannifolia species into Pistacia species. The closest Pistacia species with the rest of the Sapindales species was identified R. chinensis, belonging to the same tribe but a different genus species (Rhus). On the other hand, the species of the members of the Anacardieae tribe such as A. occidentale and M. indica were clustered the another sub-group, they took place relatively closer Pistacia species. The species of the Spondias genus and Anacardiaceae family members, S. tuberosa and S. mombin had the lowest genetic dissimilarity coefficient, these species were recognized the closer relationships with other Anacardiaceae family species such as P. eurycarpa, P. vera, P. chinensis, P. weinmannifolia, R. chinensis, A. occidentale, M. indica (Fig. 3). The highest genetic distance or the farthest genetic relationship was calculated among the member of the Sapindales species M. indica and the member of the Fagales species C. mollissima. The detected genetic dissimilarity coefficient between them was 0.144899 (Supplementary File-1).

Phylogenetic tree obtained from the chloroplast genome sequences of the members of the Sapindales and Fagales orders
Discussion
This study focused on the significance of understanding the organization and evolution of chloroplast genomes, highlighting its dual benefits in enhancing plant yield and gaining more precise insights into plant phylogeny based on evolution. To achieve this, the researchers utilized high-throughput next-generation sequencing techniques to assemble the chloroplast genome of P. eurycarpa, revealing its complete size at 160,693 (bp), a range commonly observed in chloroplast genomes of other angiosperm plants as previously documented by Daniell et al. (2016). Furthermore, it was observed that the chloroplast genome of P. eurycarpa exhibits a high degree of conservation and maintains a consistent four-section circular DNA structure with similar gene content and order to other angiosperms, corroborating findings from various research (Zuo et al. 2017; Li et al. 2019; Song et al. 2019; Zhou et al. 2019). Notably, the research highlights that the inverted repeat sections within the chloroplast genome display significant size variation among the members of the Pistacia species, with a wide variation of 71 bp. This size variability is attributed to the expansion and contraction of the inverted repeat sections, serving as a primary factor contributing to the variations in chloroplast genome length, as elucidated by He et al. (2017).
The GC content serves as a crucial metric in discerning genetic relationships among distinct species, as emphasized by previous research conducted by Asaf et al. (2017) and Xu et al. (2017). In the case of the P. eurycarpa chloroplast genome, its GC content closely resembled that of within the Pistacia genus and demonstrated the highest similarity to P. vera.
In comparison to closely related species, P. eurycarpa exhibited a higher count of genes containing introns, unlike P. vera. Notably, pafI was found to contain more than one intron in the current paper like the previous findings in distinct chloroplast genomes (Jang et al. 2023; Lee et al. 2023), while this discovery of multiple introns was carried out by clP and ycf3 aligns with findings from previous research on various chloroplast genomes (Guo et al. 2018; Li et al. 2019; Song et al. 2019; Zhou et al. 2019). Interestingly, it has been reported that pafI gene was responsible for Photosystem I assembly protein Ycf3, since it was a synonym of the Ycf3. Thus, it plays a crucial role in the stable accumulation of the photosystem I complex (Naver et al. 2001). The presence of additional introns within ycf3 may hold significance for research focusing on the evolution of photosynthesis (Guo et al. 2018).
Similar to the pattern observed in other plant chloroplast genomes, the regions with the highest GC content were found within the inverted repeat (IR) regions, a phenomenon corroborated by Guo et al. (2018). This elevated GC content in the IR regions can be attributed to the accumulation of a substantial number of rRNA and tRNA genes within these specific regions (He et al. 2017; Shen et al. 2017).
The amino acid codons within the P. eurycarpa chloroplast genome sequences displayed a pronounced bias toward a specific codon. This phenomenon underscores the significance of codon degeneracy in higher organisms, a factor that can mitigate the adverse impacts of point mutations (Freeland and Hurst 1998; Błażej et al. 2018; Gonzalez et al. 2019).
Conversely, the uneven distribution of certain amino acid codons in the genome suggests that nucleotide mutations do not occur randomly; rather, they reveal a preference for mutation and exert selective pressure, thereby giving rise to synonymous codon usage bias. This pattern aligns with observations in the nucleic acid composition of other angiosperm plants (Zuo et al. 2017; Li et al. 2019; Song et al. 2019).
Repetitive sequences hold a pivotal role in shaping the structure and genetic diversity of chloroplast genomes. They facilitate genetic variation through mechanisms such as slipped strand mispairing during DNA replication and illegitimate recombination, as extensively discussed in the scientific literature (Cavalier-Smith 2002; Zrzavy et al. 2009; Weng et al. 2014; Zhou et al. 2019).
Among these repetitive sequences, SSRs emerge as valuable tools with a wide range of applications. They are instrumental in phylogenetic analysis, ecological research, species differentiation, and plant breeding efforts (Karcı et al. 2022; Karcı 2023). Their utility extends to serving as potent molecular markers for exploring genetic diversity, conducting phylogenetic analyses, and delving into population genetics (Hu et al. 2017; Zhou et al. 2019; Zarei et al. 2022).
Historically, many phylogenetic research in plant species relied on a limited number of genetic loci, which can be insufficient for gaining a precise understanding of evolutionary relationships, particularly when studying taxa at lower taxonomic levels or contentious plant species (Welch et al. 2016; Song et al. 2019). In the current investigation, researchers identified a noteworthy count of 74 SSRs within the chloroplast genome of P. eurycarpa, surpassing the numbers found in closely related taxa. Interestingly, the most prevalent repeat units were A/T repeats. This observation aligns with previous reports, which consistently noted the predominance of mononucleotide SSRs in chloroplast genomes across diverse plant lineages, often characterized by a strong bias towards A/T nucleotide composition (Guo et al. 2018; Li et al. 2019; Song et al. 2019; Zhou et al. 2019). Furthermore, it’s worth noting that the absence of six-nucleotide repeats in three members of the Rhoeae tribe suggests the presence of shared pattern similarities among the repeat units of closely related species (Zarei et al. 2022), while mono- and dinucleotides repeats were detected in P. eurycarpa species. The low or high repetitions into chloroplast genomes will make it easier to understand the evolutionary connections within each classification. The identification of chloroplast SSRs is of great significance as these are valuable molecular markers that can greatly enhance our understanding of genetic relationships within Pictacia genus and its closely related species. SSR loci hold immense potential for genetic diversity research in P. eurycarpa, thus offering a powerful tool to evaluate and quantify the genetic variability within this species.
Moreover, the utilization of SSRs has the potential to significantly improve the precision of discrimination among controversial taxonomic classifications of P. eurycarpa and its close relatives, including P. khinjuk, P. atlantica, P. atlantica var. mutica and P. atlantica var kurdica (Kafkas 2006). By harnessing these SSRs in combination with genomic or genic SSRs in nuclear DNA, researchers can augment the capacity for distinguishing between different sub-species, thereby enhancing our ability to discern and classify these closely related taxa accurately. This integrated approach may prove invaluable in shedding light on the intricate relationships and evolutionary dynamics among these plant interspecies or genera.
Chloroplast genomes have emerged as a valuable platform for resolving contentious phylogenetic relationships among diverse species, even at lower taxonomic levels, as demonstrated in previous research (Dong et al. 2018; Zhao et al. 2018). Traditional approaches such as morphological resemblance, molecular markers, and various barcode systems have been employed to classify uncertain genera into their correct families. Nevertheless, many earlier phylogenetic analyses relied on a limited number of genetic loci, which often fell short of providing precise insights into phylogenetic relationships, particularly within closely related species (Song et al. 2019). However, the advent of next-generation sequencing technologies has revolutionized the sequencing and assembly of plastid genomes across a wide spectrum of plant species, surpassing the capabilities of traditional sequencing methods. These technological advancements have empowered researchers to more accurately assign controversial species to their appropriate genera and species, thus significantly enhancing our ability to unravel and delineate accurate taxonomic relationships among these plants.
In the current study, the members of the Sapindales order available in NCBI were divided into main three clusters. Pistacia species such as P. eurycarpa, P. vera, P. chinensis, and P. weinmannifolia were clustered together. However, P. eurycarpa and P. vera were the closest species in Pistacia genus with chloroplast genomes. Consequently, the closest species to P. vera have been mentioned as P. khinjuk Stocks in previous papers. In this study, the Pistacia species was only four species. Thus, the expected result was exhibited, and the pairs of species P. eurycarpa and P. vera were grouped. As genetic distance increased, each species was gradually separated from the others. The first separated species was P. chinensis, followed by P. weinmannifolia. The separation was carried out like expectation and found compatible with previous papers (Kafkas and Perl-Treves 2001; Kafkas 2006). In summary, R. chinensis, which is in the same Tribe as Pistacia species, was clustered closely with the Pistacia group. On the other hand, species with tribes Anacardieae and Spondieae members were grouped in two distinct groups. Similarly, close relationships between species in the Sapindaceae family and the Rutaceae family were identified as pairs. J. regia, known as the Persian walnut, and C. mollissima, known as the Chinese chestnut, were separated from the members of the Sapindales order since they were the members of the same family. Thus, the results of this paper were verified by previous reported by Zarei et al. (2022).
Although molecular research has been carried out in recent years to prove that P. eurycarpa is a separate species, a clear distinction based on genome degree between P. eurycarpa and its closeness to P. khinjuk and P. atlantica has not yet been explained. There have been investigations that there are also subspecies of P. atlantica var. mutica and var. kurdica subspecies that were confused with this species (AL-Saghir and Porter 2012; Karcι et al. 2020). This imprecise information has caused a lot of controversy and the spread of misinformation. To clarify this complex information, it is necessary to perform chloroplast genome research on P. khinjuk and P. atlantica, P. terebinthus, P. palaestina, P. lentiscus, P. integerrima, and their sub-species. The next aim should be revealing relationships among Pistacia sup-species/species using the other wild Pistacia chloroplast genome sequences, after this paper. Consequently, the evolution of Pistacia species will be clearly explained with the correct information. Despite the high heterozygosity of Pistacia species in breeding research, thanks to species-specific genetic markers, marker-assisted selection can be applied at an early stage, and breeding time can be seriously saved.
Conclusion
Pistacia eurycarpa species has quite significant horticultural and industrial value, it is evaluated as rootstock for P. vera and cosmetic industrial products. And, its importance has been revealed day by day. The results of the study have been discussed in detail its lack of genomic information and ongoing debate about the place within the genus. The first chloroplast genome report of P. eurycarpa was released, with a relationship within the Sapindales order. It is considered that the current study will contribute to revealing taxonomic classifications within the genus Pistacia by sequencing P. eurycarpa species.
References
AL-Saghir MG, Porter DM (2012) Taxonomic revision of the genus Pistacia L. (Anacardiaceae). Am J Plant Sci. https://doi.org/10.4236/ajps.2012.31002
Asaf S, Khan AL, Khan MA et al (2017) Chloroplast genomes of Arabidopsis halleri ssp. gemmifera and Arabidopsis lyrata ssp. petraea: Structures and comparative analysis. Sci Rep. https://doi.org/10.1038/s41598-017-07891-5
Beier S, Thiel T, Münch T et al (2017) MISA-web: A web server for microsatellite prediction. Bioinformatics. https://doi.org/10.1093/bioinformatics/btx198
Błażej P, Wnętrzak M, Mackiewicz D, Mackiewicz P (2018) Optimization of the standard genetic code according to three codon positions using an evolutionary algorithm. PLoS ONE. https://doi.org/10.1371/journal.pone.0201715
Cavalier-Smith T (2002) Chloroplast evolution: secondary symbiogenesis and multiple losses. Curr Biol 12(2):R62–R64
Daniell H, Lin CS, Yu M, Chang WJ (2016) Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol 17:1–29
Demirci F, Baser KHC, Calis I, Gokhan E (2001) Essential oil and antimicrobial evaluation of the Pistacia eurycarpa. Chem Nat Compd. https://doi.org/10.1023/A:1013766400932
Dong W, Xu C, Wu P et al (2018) Resolving the systematic positions of enigmatic taxa: manipulating the chloroplast genome data of Saxifragales. Mol Phylogenet Evol. https://doi.org/10.1016/j.ympev.2018.04.033
Doyle J, Doyle J (1987) A rapid isolation procedure for small amounts of leaf tissue. Phytochem Bull. https://doi.org/10.2307/4119796
Freeland SJ, Hurst LD (1998) Load minimization of the genetic code: history does not explain the pattern. Proc R Soc B Biol Sci. https://doi.org/10.1098/rspb.1998.0547
Gonzalez DL, Giannerini S, Rosa R (2019) On the origin of degeneracy in the genetic code. Interface Focus. https://doi.org/10.1098/rsfs.2019.0038
Grant JR, Stothard P (2008) The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. https://doi.org/10.1093/nar/gkn179
Greiner S, Lehwark P, Bock R (2019) OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucl Acids Res. https://doi.org/10.1093/nar/gkz238
Guo S, Guo L, Zhao W et al (2018) Complete chloroplast genome sequence and phylogenetic analysis of paeonia ostii. Molecules. https://doi.org/10.3390/molecules23020246
He L, Qian J, Li X et al (2017) Complete chloroplast genome of medicinal plant Lonicera japonica: Genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules. https://doi.org/10.3390/molecules22020249
Hu Y, Woeste KE, Zhao P (2017) Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Front Plant Sci. https://doi.org/10.3389/fpls.2016.01955
Jang JE, Kim SC, Kim JE et al (2023) The complete chloroplast genome sequence of Ligustrum foliosum (Oleaceae), a species endemic to the Ulleungdo Island Korea. J Asia-Pac Biodiv. https://doi.org/10.1016/j.japb.2022.11.003
Kafkas S (2006) Phylogenetic analysis of the genus Pistacia by AFLP markers. Plant Syst Evol. https://doi.org/10.1007/s00606-006-0460-7
Kafkas S (2019) Advances in breeding of pistachio. In: Serdar Ü, Fulbright D (eds) Achieving sustainable cultivation of tree nuts. Burleigh Dodds Science Publising, Cambridge, p 42
Kafkas S, Perl-Treves R (2001) Morphological and molecular phylogeny of Pistacia species in Turkey. Theor Appl Genet. https://doi.org/10.1007/s001220000526
Kafkas S, Perl-Treves R, Kaska N (2000) Unusual Pistacia atlantica Desf. (Anacardiaceae) monoecious sex type in the yunt mountains of the Manisa province of Turkey. Isr J Plant Sci. https://doi.org/10.1092/UFCU-7LF6-T0A3-UXWY
Kafkas S, Ozkan H, Ak BE et al (2006) Detecting DNA polymorphism and genetic diversity in a wide pistachio germplasm: comparison of AFLP, ISSR and RAPD markers. J Am Soc Hortic Sci. https://doi.org/10.21273/jashs.131.4.522
Kafkas S, Ma X, Zhang X, et al (2022) The pistachio genomes provide insights into nut tree domestication and ZW sex chromosome evolution. Plant Commun 100497
Karcı H (2023) QTL-seq for the identification of candidate genes responsible for double seeds in almond. Turk J Agric For 47:633–644
Karcı H, Paizila A, Güney M et al (2022) Revealing genetic diversity and population structure in Pistachio (Pistacia vera L.) by SSR markers. Genet Resour Crop Evol 69:1–13
Karcι H, Paizila A, Topçu H et al (2020) Transcriptome sequencing and development of novel genic SSR markers from Pistacia vera L. Front Genet. https://doi.org/10.3389/fgene.2020.01021
Kurtz S, Choudhuri JV, Ohlebusch E et al (2001) REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. https://doi.org/10.1093/nar/29.22.4633
Lee SR, Ha YH, Son DC, Kim SC (2023) First record of the complete chloroplast genome of Polygonatum infundiflorum (Asparagaceae), a Korean endemic species. Mitochondrial DNA Part B Resour. https://doi.org/10.1080/23802359.2023.2215349
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. https://doi.org/10.1093/bioinformatics/btp324
Li H, Handsaker B, Wysoker A et al (2009) The 0sequence alignment/map format and SAMtools. Bioinformatics. https://doi.org/10.1093/bioinformatics/btp352
Li DM, Zhao CY, Liu XF (2019) Complete chloroplast genome sequences of Kaempferia galanga and Kaempferia elegans: Molecular structures and comparative analysis. Molecules. https://doi.org/10.3390/molecules24030474
Liu C, Shi L, Zhu Y et al (2012) CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genom. https://doi.org/10.1186/1471-2164-13-715
Liu S, Jingling N, Xinyi L, Heyu Z et al (2023) CPGView : A package for visualizing detailed chloroplast genome structures. https://doi.org/10.1111/1755-0998.13729
Mao W, Yao G, Chen B et al (2019) Characterization of the complete chloroplast genome of the Pistacia vera L. Mitochondrial DNA Part B 4:4140–4141
Naver H, Boudreau E, Rochaix JD (2001) Functional studies of Ycf 3: its role in assembly of photosystem I and interactions with some of its subunits. Plant Cell. https://doi.org/10.1105/tpc.13.12.2731
Shen X, Wu M, Liao B et al (2017) Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant artemisia annua. Molecules. https://doi.org/10.3390/molecules22081330
Socias R (1990) Breeding self-compatible almonds. Plant Breed Rev 8:313–338
Song Y, Chen Y, Lv J et al (2019) Comparative chloroplast genomes of sorghum species: sequence divergence and phylogenetic relationships. Biomed Res Int. https://doi.org/10.1155/2019/5046958
Tillich M, Lehwark P, Pellizzer T et al (2017) GeSeq - Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx391
Welch AJ, Collins K, Ratan A et al (2016) The quest to resolve recent radiations: plastid phylogenomics of extinct and endangered Hawaiian endemic mints (Lamiaceae). Mol Phylogenet Evol. https://doi.org/10.1016/j.ympev.2016.02.024
Weng ML, Blazier JC, Govindu M, Jansen RK (2014) Reconstruction of the ancestral plastid genome in geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol Biol Evol. https://doi.org/10.1093/molbev/mst257
Xu C, Dong W, Li W et al (2017) Comparative analysis of six lagerstroemia complete chloroplast genomes. Front Plant Sci. https://doi.org/10.3389/fpls.2017.00015
Xu J-H, Zhang D-X, Sun K et al (2019) The complete chloroplast genome sequences of Pistacia chinensis Bunge, a potential bioenergy tree. Mitochondrial DNA Part B 4:1774–1775
Xu Y, Zhang Y, Ren Z (2020) Complete chloroplast genome of Pistacia chinensis Bunge (Anacardiaceae: Rhoideae), an important economical and ornamental plant. Mitochondrial DNA Part B 5:1931–1932
Yaltirik F (1967) Anacardiaceae. Flora Turk 2:544–548
Zarei A, Ebrahimi A, Mathur S, Lawson S (2022) The first complete chloroplast genome sequence and phylogenetic analysis of pistachio (Pistacia vera). Diversity. https://doi.org/10.3390/d14070577
Zhao Z, Wang X, Yu Y et al (2018) Complete chloroplast genome sequences of dioscorea: characterization, genomic resources, and phylogenetic analyses. PeerJ. https://doi.org/10.7717/peerj.6032
Zheng W, Li K, Wang W, Xu X (2018) The complete chloroplast genome of the threatened Pistacia weinmannifolia, an economically and horticulturally important evergreen plant. Conserv Genet Resour 10:535–538
Zhou T, Ruhsam M, Wang J et al (2019) The complete chloroplast genome of Euphrasia regelii, Pseudogenization of ndh genes and the phylogenetic relationships within orobanchaceae. Front Genet. https://doi.org/10.3389/fgene.2019.00444
Zohary M (1952) A monographical study of the genus Pistacia. Palest J Bot Jerusalem 5:187–228
Zrzavy J, Mihulka S, Kepka P, et al (2009) Artificial life & complex systems. Bioinformatics
Zuo LH, Shang AQ, Zhang S et al (2017) The first complete chloroplast genome sequences of Ulmus species by de novo sequencing: genome comparative and taxonomic position analysis. PLoS ONE. https://doi.org/10.1371/journal.pone.0171264
Acknowledgements
All data generated or analyzed during this study are included in this published article and its supplementary information file. The genetic dissimilarity of the chloroplast genomes obtained from Geneious was given in Supplementary File-1.
Funding
Open access funding provided by the Scientific and Technological Research Council of Türkiye (TÜBİTAK). This research received no external funding.
Author information
Authors and Affiliations
Contributions
HK and SK conceived and designed the experiments. HK performed most of the experiments and analyzed the data. HK and SK wrote the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The author declares no conflict of interest.
Consent to participate
Not applicable.
Ethical approval
This article does not contain any research with human participants or animals performed by the author.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Karcı, H., Kafkas, S. The first complete chloroplast genome sequences of Pistacia eurycarpa Yalt, a horticultural important rootstock and industrial tree. Genet Resour Crop Evol (2024). https://doi.org/10.1007/s10722-024-01877-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10722-024-01877-9