
Abstract
Matricaria chamomilla L. (chamomile, Asteraceae) (GRIN, The Plant List 2013) has a long history of usage in traditional herbal medicine and is still today amongst the most important medicinal plants. Despite this importance, genetic diversity of cultivated and wild germplasm of M. chamomilla was rarely investigated so far. The objective of this study was to estimate the mitochondrial (mt) diversity of various cultivated M. chamomilla genotypes by determining point mutations in the mt genome. 89 SNPs (single nucleotide polymorphisms) were identified in the next generation sequencing data of 33 genotypes from 11 di- and tetraploid chamomile accessions representing a sequence diversity of 0.32 SNPs/kb. Based on the SNP analysis 19 mitochondrial haplotypes (mitotypes) could be specified with genetic distances ranging between 0.011 and 0.851. The examined mt variability within the accessions was higher than expected; only one monomorphic accession (variety ‘Camoflora’) was identified. Diploid accessions exhibited with 1.9 mitotypes per accession a higher variability than tetraploid accessions with a ratio of 1.3. Although some of the mitotypes were distributed over different accessions, identical mitotypes within di- and tetraploid accessions could not be determined. Furthermore, the mitotypes did not correspond to the geographical origin of the accessions. Although not the whole mt genome could be assembled in this study, the substitutions identified represent a valuable tool for further investigations of maternal phylogenetic relationships within M. chamomilla.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Matricariae flos (flowers from Matricaria chamomilla L., common name chamomile, Asteraceae) (GRIN, The Plant List 2013) is one of the most important medicinal plant raw materials with numerous applications in the pharmaceutical and cosmetic area. M. chamomilla is native to Europe and West Asia from where it gradually spread over many temperate zones worldwide (Franke and Schilcher 2007). The first documentation of cultivation of this plant dates back to 9000–7000 BC (Franke and Schilcher 2005). To meet the extensive demand on plant raw material, first breeding attempts started approximately 50 years ago (Franke and Schilcher 2007) with the centers being situated in the Czech and Slovak Republic, Poland, Hungary and Germany (Das 2014; Seidler-Lozykowska 2016). Breeding material was originally gained from wild collections, but information about the parental populations is available for only a few varieties (Das 2014). M. chamomilla is mainly outbreeding and originally diploid with tetraploid varieties induced by artificial polyploidisation to obtain higher crop yields due to larger flower heads or to obtain higher contents of essential oils or medicinally active compounds (Das 2014). Another aspect for the implementation of tetraploid varieties was to avoid crossings between cultivated and wild chamomile plants (Das 2014). However, accessions of both ploidy levels are cultivated and most of them still exhibit high phenotypic variability (Das 2014; Plescher and Sonnenschein 2013).
Genomic approaches to investigate the genetic diversity of cultivated and wild chamomile germplasm in order to improve breeding processes have already been made (Wagner et al. 2005; Solouki et al. 2008; Pirkhezri et al. 2010; Okon et al. 2013; Ahmadi et al. 2014; Otto et al. 2017). Nevertheless, the number of studies conducted on this topic is low compared to other crop plants and no effort has been made to investigate the diversity of the chloroplast (cp) or mitochondrial (mt) genome of chamomile lines.
One major problem in chamomile cultivation is the germination of chamomile (as weed) in the following crops because of self-seeding and seed shedding, which complicates regular crop rotation (Franke and Hannig 2012). The use of male sterility systems, already common in many crop species (Balk and Leaver 2001; Rao et al. 2018), for the prevention of self-fertilization would indeed improve the attractiveness of chamomile cultivation. Beneath others, the cytoplasmic male sterility (CMS) is one way to produce male sterile lines with preserved female fertility. Rearrangements or mutations within the mt genome, especially in the open reading frames (ORFs), often are responsible for the development of novel chimeric genes and thus the probability of male sterility in the progenies (Schnable and Wise 1998; Budar et al. 2003; Hanson and Bentolila 2004; Chase 2007). In contrast to animal and fungal mitochondrial DNA (mtDNA), the mt genome of plants differs greatly in size, gene content and gene order and alternative mitotypes coexisting with the main mtDNA are described (Gualberto et al. 2014; Kersten et al. 2016). The rare point mutations in the mitochondrion of plants compared to that of animals are believed to be explained by the occurrence of repair mechanisms because of an active DNA recombination (Gualberto et al. 2014). Due to these innate recombination and replication processes, the plant mt genome undergoes homologous recombination frequently, although only a low rate of point mutations is observed (Gualberto et al. 2014). The use of mt sequences for phylogenetic studies in plant genera therefore stands back behind the application of cp sequences although mtDNA can be rather informative for the study of maternal relationships, migration routes of populations in phylogeographic studies or the exploration of genetic backgrounds (Tomaru et al. 1998; Gugerli et al. 2001; Besnard et al. 2002; van de Paer et al. 2018). Another drawback of using mt markers is the low number of published universal primers. This is another reason why mt sequence information of plants is still underrepresented in public genebanks. Newer techniques like next generation sequencing (NGS) now enable the production of a high amount of sequences thus facilitating the exploitation of whole genome information and the identification of high quality SNPs in the resulting sequences.
Nevertheless, to date, the mt genome sequences of only seven Asteraceae species are deposited in Genbank with the total lengths of 363,342 bp in Lactuca sativa (Kozik et al. 2019), 363,328 bp in Lactuca serriola (Kozik et al. 2019), 208,097 bp in Chrysanthemum nankingense (Wang et al. 2018), 211,002 bp in Chrysanthemum boreale (Won et al. 2018), 277,718 bp in Diplostephium hartwegii, 300,945 bp in Helianthus annuus (Grassa et al. 2016) and 453,334 bp in Conyza canadensis (Peng et al. 2014). Sequence information of a total mt genome of M. chamomilla is not published yet.
In this study, we examine the mt diversity of different genotypes of M. chamomilla in order to determine their maternal relationships.
The obtained data can be useful for reconstructing the breeding history of the examined accessions and, eventually, to assign cultivated accessions to geographical origins. The identification of mitochondrial markers can further help organize genetically wide crossings increasing the probability of finding male sterile plants.
Material and method
Plant material
Based on the data of published GBS (genotyping by sequencing) analyses (Otto et al. 2017), eleven genetically diverse di- and tetraploid accessions of Matricaria chamomilla L. (Table 1) were selected and cultivated in a greenhouse. Three plants per accession were selected randomly for the extraction of mitochondrial DNA and further analysis.
Enrichment of mitochondria and extraction of mitochondrial DNA
Mitochondrial DNA was extracted according to the protocol of Triboush et al. (1998) with minor modifications. 1 g of very young leaves and apical shoot tips were ground in 10 ml of the recommended pre-chilled buffer STE with mortar and pistil using autoclaved sea sand. We did not filter the lysate but removed sea sand and cell debris by the first centrifugation step. Centrifugation steps for removal of nuclei and pelleting of mitochondria were done twice to optimize mt enrichment. All steps for isolation of mt organelles were performed on ice, centrifugation steps were done at 0 °C using the Hettich 320R centrifuge (Hettich, Bäch, Switzerland). After lysis of the organelles, centrifugation steps were done at room temperature using the microcentrifuge Hettich Mikro 200 (Hettich, Bäch, Switzerland). For the extraction of DNA phenol:chloroform:isoamylalcohol (25:24:1) and chloroform:isoamylalcohol (24:1) were consecutively used instead of phenol:chloroform 1:1 for removal of proteins. The second precipitation step of the protocol for a better removal of chloroplast DNA was skipped and the reaction was stopped after elution of the first DNA pellet in TE pH 8.0.
The quality and quantity of the DNA extracts were measured using a spectrophotometer (NanoDrop2000™, Thermofisher Scientific, Waltham, USA).
To evaluate residual amounts of nuclear and chloroplast DNA, samples were tested in a qPCR run by amplifying one mitochondrial (cox 1), chloroplast (trnL-F-IGS) and nuclear (ITS1) gene region. Concentrations of 1 ng/µl were compared to a predefined standard sample extracted with the same protocol and a two-fold dilution series (2–0.2 ng/µl) of a standard sample of total genomic DNA extracted using a modified CTAB-protocol (Schmiderer et al. 2013). Standard samples were set up by using a mixture of ‘Bona’ seedlings for both extractions. Implementing that the analysed gene regions are representatives of the whole plant genome, the standard sample showed a relation of 0.1%, 1.5% and 98.4% of the nuclear, chloroplast and mitochondrion amplicon, respectively. With the CTAB protocol without mt enrichment the relation of amplicons was 36.1%, 33.2% and 30.7%. The samples foreseen for sequencing were in the range of the standard sample.
For a 10 µl PCR reaction 1 ng of DNA was added to a master mix containing 1× HOT FIREPol® EvaGreen® HRM Mix (no ROX) (Solis BioDyne, Tartu, Estonia) and 150 nM forward and reverse primers (Life Technologies, Vienna, Austria) (Online Resource 1), respectively. The PCR cycle profile included a denaturation step at 95 °C for 14 min, followed by 50 cycles of 95 °C for 10 s, 55 °C for 20 s and 72 °C for 20 s. Samples and no-template controls were analyzed in duplicates. The qPCR was performed with the Rotor-Gene™ 6000 (Qiagen, Hilden, Germany) and analyses were conducted using the Rotor-Gene™ 6000 software.
NGS
Library construction, adapter ligation and sequencing (5 Mio. read pairs, 0.6 GB of an Illumina Miseq run) were outsourced (LGC Genomics GmbH, Berlin, Germany). After DNA fragmentation with a focused-ultrasonicator (Covaris, Woburn, Massachusetts, USA) libraries were prepared using the Ovation Rapid DR Multiplex System 1-96 (NuGEN) including the following steps: end repair, ligation, final repair, library purification and library amplification (14 cycles). Libraries were pooled, purified and size selected via preparative gel electrophoresis and quality controlled using the BioAnalyzer (Agilent, Santa Clara, CA 95051, USA) and Qubit fluorometer (Thermofisher Scientific, Waltham, USA).
One sample from the accession ‘Bona’ was previously sequenced in a separate run to test for sequence quality and mt enrichment. The Illumina bclfastq 1.8.4 software was used for demultiplexing and quality filtering of the reads under the allowance of one or two mismatches and removal of short reads (< 20 bases).
NGS data were deposited in Genbank (NCBI) under the following dataset: Temporary Submission ID: SUB5046906/BioProject ID: PRJNA515664.
Sequence assembly and SNP detection
After quality filtering, paired reads were mapped to Diplostephium hartwegii (KX063855), an Asteraceae species, which was used as reference sequence using the Geneious mapper [Geneious 9.1.5 (http://www.geneious.com, Kearse et al. 2012)] with low sensitivity under default parameters and choosing the function to find structural variants, short insertions and deletions of any size (Online Resource 2).
The resulting consensus sequence for mtDNA of all chamomile samples was used as new reference sequence to which paired reads of the samples were mapped separately under the same parameters. This resulted in individual consensus sequences for the samples, which were again aligned with Diplostephium hartwegii as reference using the LASTZ alignment as implemented in Geneious 9.1.5. Annotations were consecutively adopted from Diplostephium hartwegii. SNPs were detected with the SNP caller as implemented in Geneious 9.1.5 with default parameters and were used to set up the different mitochondrial haplotypes (= mitotypes). In order to compare the SNP frequency of chamomile with that of other species, the mitochondrial genomes of Lactuca sativa (NC_042756) and L. serriola (NC_042378) (Kozik et al. 2019) were downloaded from NCBI, aligned with MAFFT v7.308 (Katoh and Standley 2013) and SNPs detected as explained above.
Statistics
All genetic distances were calculated and visualized in R 3.5.2 (R Core Team 2018) with the R package poppr 2.8.1 (Kamvar et al. 2014, 2015) using Nei’s distance for distance calculation and the Davidson-Harel layout algorithm (Davidson and Harel 1996) for the minimum spanning network.
Results
Mitochondria enriched DNA of eleven accessions (32 plants) was sequenced on an Illumina Miseq run (5 Mio. read pairs). The run yielded 10,163,050 reads ranging between 87,428 and 741,788 raw sequences per sample (Online Resource 3). One sample (‘Bona5’) was previously sequenced in a separate run (30,806 raw sequences) to test for sequence quality and mitochondrial enrichment. An overall mitochondrial consensus sequence (162,685 bp) was established using paired reads of one sample per accession (about 30% of paired reads) and Diplostephium hartwegii (KX063855) as reference with a mean coverage of 5109 sequences. Paired reads of the samples were mapped separately to this consensus to generate individual mt consensus sequences. The mean coverage of individual samples ranged from 71 in ‘Promyk10’ to 479 in ‘Hun2_1’. Only two samples were outliers in the low range, the overall mean coverage was 201. The test sample ‘Bona5’ yielded a sequence coverage of 12. Consensus sequences were aligned with the reference D. hartwegii in a LASTZ alignment and SNPs were detected. Ninety-seven percent of the sequences (157,993 bp) could be aligned to the genome of D. hartwegii.
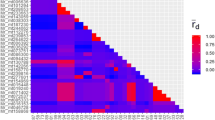
Twenty-three percent (36,383 bp) of the assembled sequences belonged to coding regions, 77% (121,610 bp) to non-coding regions. The area of highest diversity was found within the first 20,000 bp, in the region between the sequence coordinates 1 and 20,000 of D. hartwegii (Fig. 1).

The number of substitutions found within M. chamomilla in an LASTZ alignment; positions are corresponding to sequence coordinates of D. hartwegii. (The first column demonstrates mutations within bases 1–20,000, the second column shows polymorphic sites within positions 20,000–40,000 in the alignment, and so on.)
In total, 102 SNPs were detected in M. chamomilla in a 277,718 bp alignment, 89 SNPs (representing 0.03% of the aligned sequences) when sequence ambiguities within a single sample or mutations with low sequence coverage were omitted (Table 2). The sequence diversity between M. chamomilla and D. hartwegii was 0.45% or 1238 mutations within the D. hartwegii genome.
Out of the 89 SNPs 67 polymorphisms were substitutions from purine to pyrimidine bases or vice versa (Table 3). If the mutations would be evenly distributed between coding and non-coding regions, we would expect 20 SNPs in the coding regions based on the 23% coding regions found in our assembly. However, only 11% of the SNPs (10 mutations) were found in the genic regions, 89% (79 SNPs) were located in the intergenic regions. Three of the point mutations in the genic regions were found within the cox2 gene.
The SNP markers identified in this study distinguish nineteen haplotypes (mitotypes) in M. chamomilla (Online Resource 4). The mtDNA of only one accession (‘Camoflora’) was monomorphic for all three individuals, all other accessions were polymorphic. However, individuals of different accessions shared the same mitotype, although this was restricted to individuals of the same ploidy level (Table 4 and Fig. 2). Within the diploid accessions, fifteen mitotypes in twenty-four individuals were found, whereas four different mitotypes were identified in nine individuals of the tetraploid accessions. The ratio of mitotypes per accession was with 1.9 therefore slightly higher in diploid accessions than the ratio of 1.3 in tetraploid accessions. Genetic distances between the mitotypes ranged between 0.01 and 0.85. Three main clusters separating from each other with higher distances (0.43–0.45) could be identified in a minimum spanning network (Fig. 2). Cluster I comprising the tetraploid Mitotype 16 (‘Goral’, ‘Margaritar’) and several diploid accessions, cluster II is composed by the tetraploid Mitotype 19 and the diploid Mitotype 17 and cluster III is built by the rest of the tetraploid individuals (Mitotype 18) and the main group of diploid individuals (‘Promyk’, ‘Camoflora’, ‘Soroksári’, ‘Argenmilla’ and ‘Bona’). Clusters I and III could be further divided into several subclusters. Genetic distances between Mitotype 16 (4×) and Mitotype 10 (2×) (0.11) or Mitotype 16 (4×) and Mitotype 13 (2×) (0.07) for example are higher than between diploid mitotypes within the same cluster. The highest genetic distance (0.85) was calculated between Mitotype 1 (‘Bona’) and Mitotype 17 (‘Margaritar’). The accessions with the highest variability within were the Croatian provenance ‘PG029’ (2×), the Argentinean accession ‘Argenmilla’ (2×), ‘Bona’ (2×) from the Slovak republic and ‘Margaritar’ (4×) from Romania. The three mitotypes of the accession ‘Margaritar’ are so diverse that they are dispersed on the three main clusters of the minimum spanning network with genetic distances of 0.5–0.7. The three mitotypes of ‘Bona’ and ‘PG029’ respectively can be found in clusters I and III with one mitotype each that is more distantly related to the others (~ 0.6). Due to the high amount of polymorphic accessions it is impossible to determine which of the diploid accessions was used to attain a corresponding tetraploid accession or to determine a common ancestor of di- and tetraploid accessions. According to Das (2014), the Slovak accessions ‘Bona’ (2×) and ‘Goral’ (4×) e.g. resulted from breeding experiments using the same plant material. This fact, however, could based on this limited sample set be neither verified nor disproved. The analysed data also did not offer a clear connection between the mt lines and geographic origin of our samples. Anyhow, the high variability within the accessions shows that plants with different genetic background must have been used to establish distinct chamomile varieties.

Minimum spanning network of 33 individuals from eleven accessions and 89 polymorphic markers calculated with R. The different accessions are marked by different colours, the size of the circles corresponds to the sample number per node, genetic distances between the nodes are indicated by line thickness and shading
Discussion
The use of a closely related mt genome as a reference—although of limited use for the construction of the whole mt genome—was here a straightforward approach for the identification of point mutations enabling the evaluation of mt sequence data of a relatively large sample set in a short time.
Anyhow, the real number of mitotypes in M. chamomilla will be underestimated in this study because not the whole mt genome of M. chamomilla was assembled and indels, SSRs and rearrangements were excluded from the analyses.
As mtDNA is—like in most seed plants—in the closely related species Helianthus annuus solely maternally inherited (Wills et al. 2005), the probability that it is likewise in M. chamomilla is high. Anyhow, the mitochondrion e.g. is maternally inherited in Arabidopsis thaliana (Martínez et al. 1997) while paternal or biparental inheritance is specified in Brassica napus (Erickson and Kemble 1990) (both Brassicaceae). Different organelle inheritance modes are also described within Oleaceae (Van de Paer et al. 2018), which demonstrates that the way of inheritance of mitochondria is not always the same within plant families and it is not proven yet if the mt genome in M. chamomilla indeed is uniparentally inherited. As a slow mutation rate is expected for plant mitochondria (Gualberto et al. 2014), a relatively large number of SNPs would be contradictory in this case. However, an unexpected high variation of substitution rates of mt sequences across plants with some species exhibiting exceptionally high or low substitution rates were detected (Mower et al. 2007). Defects in mt repair mechanisms and/or DNA replication processes were beneath others discussed as possible reason for higher mt mutation rates in those cases (Mower et al. 2007).
Actually, reliable comparisons of our results to other populational studies concerning mt polymorphisms within Asteraceae are limited due to little comparable information. Mt sequence comparison of one cultivated and wild type of Helianthus annuus revealed a significantly lower amount of point mutations (0.027/kb) (Makarenko et al. 2016), the mitochondrions of Lactuca sativa and L. serriola even differed in 6 SNPs only (Kozik et al. 2019). Other publications examining inter- and intraspecific mt variability between related Asteraceae species did either not refer to mt data only (Peng et al. 2014) or used RFLP and no sequence data for the evaluation (Vermeulen et al. 1994). Two breeding lines of Capsicum revealed only one SNP (Wang et al. 2019). Kersten et al. (2016) found a mt sequence divergence of 0.065 SNPs/kb between Populus tremula and P. tremula x P. alba. Mitochondrial sequence comparisons in the subtribe Oleinae revealed 0.21 SNPs/kb between species of this subtribe (Van de Paer et al. 2018). In comparison to those studies we would at first glance assume a high amount of substitutions in our samples.
A biparental inheritance would explain a high variability but can almost be excluded as the number of substitutions we found in the mt genome was with 0.32 SNPs/kb very low compared to the 9.51 SNPs/kb identified in the GBS analysis of the same species (Otto et al. 2017). The high sequence diversity found in this study might also be a result of the preselection of the analysed accessions by GBS analysis (Otto et al. 2017) that clearly enhanced the probability to find as many mitotypes as possible. Otto et al. (2017) found a tendency towards lower genetic variability within tetraploid accessions and a lacking congruence of genetic structure and geographic origin of the analysed accessions. Our results, although focusing on mt variability, indeed concurred with the results of the GBS analysis.
Breeding of such small-scale crops is very pragmatically performed because of economic and time constraints. The high variability of mitotypes within accessions shows that plants collected for selection were not isolated from each other (so regarded as one big population) and selection was performed by mass selection. Our data gives reason to the assumption that active exchange of plant material between plant breeders resulted in the establishment of numerous haplotypes within one accession. The Romanian variety ‘Margaritar’ e.g., resulted from breeding experiments using the Polish ‘Zloty Lan’ and chamomile plants collected in the wild in Poland (Das 2014). Plants from the same populations were used to develop the Polish ‘Promyk’ via further selection (Das 2014). A clear connection between the two varieties ‘Margaritar’ and ‘Promyk’ could not be found in our data but the high variability within the variety ‘Margaritar’ could be explained by a combination of different chamomile germplasm. Further on, it is possible that the chamomile lines were developed by combining several plants of different natural populations thus enhancing mt variability of the progenies. The varieties ‘Bona’ and ‘Goral’ both were attained by breeding experiments using ‘Bohemia’ and a Spanish chamomile (Das 2014). The use of geographically distinct chamomile populations might have resulted in the establishment of several genetically diverse mitotypes in the accession ‘Bona’. As cultivated tetraploid chamomile varieties result from polyploidisation of diploid plants, it is not astonishing that tetraploid and diploid accessions both reflect the picture of high mt genetic diversity. Still it is remarkable that none of the mitotypes determined in diploid accessions could be found in tetraploid accessions as well. However, due to the unexpected high variability, three individuals per accession will not be enough to explain relationships between chamomile varieties and to clarify the breeding history of chamomile.
More samples should be examined in the intention to investigate the variability of the mt genome within the different chamomile origins more deeply. A bigger sample set and the additional analysis of natural populations would possibly provide an insight into the genetic background of the origins suitable for parentage analysis and population genetics.
Conclusion
To date, little is known about heritability, size, organisation or substitution rate of the mt genome of M. chamomilla. By means of this study a first step towards an exploration of mt genome variability in this medicinal plant was made. Promising regions for the development of phylogenetic markers were found which would enable further investigation of the distribution of different mitotypes in chamomile accessions, populations or varieties. Thus, the markers could be used for the development of a fast screening method of mt haplotypes, for evaluation of the organellar inheritance in M. chamomilla and for facilitation of phylogenetic relationships analyses between cytoplasmic lineages or chamomile populations.
References
Ahmadi H, Rahimmalek M, Zeinali H (2014) Assessment of the genetic variation of chamomile (Matricaria chamomilla L.) populations using phytochemical, morphological and ISSR markers. Biochem Syst Ecol 54:190–197. https://doi.org/10.1016/j.bse.2014.01.010
Balk J, Leaver CJ (2001) The PET1-CMS mitochondrial mutation in sunflower is associated with premature programmed cell death and cytochorome c release. Plant Cell 13(8):1803–1818
Besnard G, Khadari B, Baradat P, Bervillé A (2002) Combination of chloroplast and mitochondrial DNA polymorphisms to study cytoplasm genetic differentiation in the olive complex (Olea europaea L.). Theor Appl Genet 105(1):139–144. https://doi.org/10.1007/s00122-002-0868-6
Budar F, Touzet P, De Paepe R (2003) The nucleo-mitochondrial conflict in cytoplasmic male sterilities revisited. Genetica 117(1):3–16. https://doi.org/10.1023/A:1022381016145
Chase CD (2007) Cytoplasmic male sterility: a window to the world of plant mitochondrial-nuclear interactions. Trends Genet 23(2):81–90. https://doi.org/10.1016/j.tig.2006.12.004
Das M (2014) Chamomile. Medicinal, biochemical, and agricultural aspects, 1st edn. CRC Press, Boca Raton. https://doi.org/10.1201/b17160
Davidson R, Harel D (1996) Drawing graphs nicely using simulated annealing. ACM Trans Graph 15(4):301–331. https://doi.org/10.1145/234535.234538
Downie SR, Katz-Downie DS (1996) A phylogeny of Apiaceae subfamily Apioideae: evidence from nuclear ribosomal DNA internal transcribed spacer sequences. Am J Bot 83(2):234–251
Erickson L, Kemble R (1990) Paternal inheritance of mitochondria in rapeseed (Brassica napus). Mol Gen Genet 222(1):135–139. https://doi.org/10.1007/bf00283034
Faehnrich B, Otto L-G, Franz C, Mešić E, Cosendai A-C, Dobeš C (2019) Auxin application in interploidy crosses and genome stability: across-generation investigations on German chamomile (Matricaria recutita [L.] Rauschert) of various origins. Euphytica 215(2):237. https://doi.org/10.1007/s10681-019-2335-3
Franke R, Hannig H-J (2012) Kamille (Matricaria recutita L.). In: Hoppe B (ed) Handbuch des Arznei- und Gewürzpflanzenbaus, vol 4 Arznei- und Gewürzpflanzen A - K. Verein für Arznei- und Gewürzpflanzen SALUPLANTA e.V. Bernburg, pp 618–648
Franke R, Schilcher H (eds) (2005) Chamomile: industrial profiles. CRC Press Taylor and Francis Group, Boca Raton. https://doi.org/10.1201/9780203022382
Franke R, Schilcher H (2007) Relevance and use of chamomile (Matricaria recutita L.). ISHS Acta Horticulturae 749:29–43. https://doi.org/10.17660/ActaHortic.2007.749.2
Germplasm Resources Information Network (GRIN) Beltsville (MD): United States Department of Agriculture, Agricultural Research Service. https://npgsweb.ars-grin.gov/gringlobal/taxonomydetail.aspx?id=23475. Accessed 01 July 2019
Grassa CJ, Ebert DP, Kane NC, Rieseberg LH (2016) Complete mitochondrial genome sequence of sunflower (Helianthus annuus L.). Genome Announc. https://doi.org/10.1128/genomeA.00981-16
Gualberto JM, Mileshina D, Wallet C, Niazi AK, Weber-Lotfi F, Dietrich A (2014) The plant mitochondrial genome: dynamics and maintenance. Biochimie 100:107–120. https://doi.org/10.1016/j.biochi.2013.09.016
Gugerli F, Sperisen C, Buchler U, Magni F, Geburek T, Jeandroz S, Senn J (2001) Haplotype variation in a mitochondrial tandem repeat of Norway spruce (Picea abies) populations suggests a serious founder effect during postglacial re-colonization of the western Alps. Mol Ecol 10(5):1255–1263. https://doi.org/10.1046/j.1365-294X.2001.01279.x
Hanson MR, Bentolila S (2004) Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 16(Suppl):154–169. https://doi.org/10.1105/tpc.015966
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. https://doi.org/10.7717/peerj.281
Kamvar ZN, Brooks JC, Grünwald NJ (2015) Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front Genet 6:208. https://doi.org/10.3389/fgene.2015.00208
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30(4):772–780. https://doi.org/10.1093/molbev/mst010
Kersten B, Faivre Rampant P, Mader M, Le Paslier M-C, Bounon R, Berard A et al (2016) Genome sequences of Populus tremula chloroplast and mitochondrion: implications for holistic poplar breeding. PLoS ONE 11(1):e0147209. https://doi.org/10.1371/journal.pone.0147209
Kozik A, Rowan BA, Lavelle D, Berke L, Schranz ME, Michelmore RW, Christensen AC (2019) The alternative reality of plant mitochondrial DNA: one ring does not rule them all. PLoS Genet 15(8):e1008373. https://doi.org/10.1371/journal.pgen.1008373
Makarenko MS, Usatov AV, Markin NV, Azarin KV, Gorbachenko OF, Usatov NA (2016) Comparative genomics of domesticated and wild sunflower: complete chloroplast and mitochondrial genomes. Online J Biol Sci 16(1):71–75. https://doi.org/10.3844/ojbsci.2016.71.75
Martínez P, López C, Roldán M, Sabater B, Martín M (1997) Plastid DNA of five ecotypes of Arabidopsis thaliana: sequence of ndhG gene and maternal inheritance. Plant Sci 123(1–2):113–122. https://doi.org/10.1016/S0168-9452(96)04563-3
Mower JP, Touzet P, Gummow JS, Delph LF, Palmer JD (2007) Extensive variation in synonymous substitution rates in mitochondrial genes of seed plants. BMC Evol Biol 7:135. https://doi.org/10.1186/1471-2148-7-135
Okon S, Surmacz-Magdziak A, Paczos-Grzeda E (2013) Genetic diversity among cultivated and wild chamomile germplasm based on ISSR analysis. Acta Sci Pol Hortorum Cultus 12(2):43–50
Otto L-G, Mondal P, Brassac J, Preiss S, Degenhardt J, He S et al (2017) Use of genotyping-by-sequencing to determine the genetic structure in the medicinal plant chamomile, and to identify flowering time and alpha-bisabolol associated SNP-loci by genome-wide association mapping. BMC Genom 18(1):599. https://doi.org/10.1186/s12864-017-3991-0
Peng Y, Lai Z, Lane T, Nageswara-Rao M, Okada M, Jasieniuk M et al (2014) De novo genome assembly of the economically important weed horseweed using integrated data from multiple sequencing platforms. Plant Physiol 166(3):1241–1254. https://doi.org/10.1104/pp.114.247668
Pirkhezri M, Hassani ME, Hadian J (2010) Genetic diversity in different populations of Matricaria chamomilla L. growing in southwest of Iran, based on morphological and RAPD markers. Res J Med Plant 4(1):1–13. https://doi.org/10.3923/rjmp.2010.1.13
Plescher A, Sonnenschein M (2013) Züchtung einer Qualitätssorte von Kamille mit hoher Ertragsfähigkeit bei maschineller Ernte (Phase I). Abschlussbericht Demonstrationsprojekt Arzneipflanzen (KAMEL), FKZ08NR206 Fachagentur Nachwachsende Rohstoffe
R Core Team (2018) R: A language and environment for statistical computing. Hg. v. R Foundation for Statistical Computing, ViennaOnline. https://www.R-project.org/
Rao GS, Deveshwar P, Sharma M, Kapoor S, Rao KV (2018) Evolvement of transgenic male-sterility and fertility-restoration system in rice for production of hybrid varieties. Plant Mol Biol 96(1–2):35–51. https://doi.org/10.1007/s11103-017-0678-5
Sanchez-Puerta MV, Cho Y, Mower JP, Alverson AJ, Palmer JD (2008) Frequent, phylogenetically local horizontal transfer of the cox1 group I Intron in flowering plant mitochondria. Mol Biol Evol 25(8):1762–1777. https://doi.org/10.1093/molbev/msn129
Schmiderer C, Lukas B, Novak J (2013) Effect of different DNA extraction methods and DNA dilutions on the amplification success in the PCR of different medicinal and aromatic plants. Z Arznei- Gewurzpfla 18(2):65–72
Schnable P, Wise RP (1998) The molecular basis of cytoplasmic male sterility and fertility restoration. Trends Plant Sci 3(5):175–180. https://doi.org/10.1016/S1360-1385(98)01235-7
Seidler-Lozykowska K (2016) Medicinal plant breeding in Poland: history and nowadays, in Proceedings of the 6th International Symposium Breeding Research on Medicinal and Aromatic Plants, BREEDMAP 6, Quedlinburg, Germany. https://doi.org/10.5073/jka.2016.453.023
Solouki M, Mehdikhani H, Zeinali H, Emamjomeh AA (2008) Study of genetic diversity in chamomile (Matricaria chamomilla) based on morphological traits and molecular markers. Sci Hortic 117(3):281–287. https://doi.org/10.1016/j.scienta.2008.03.029
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
The Plant List (2013). Version 1.1. http://www.theplantlist.org/tpl1.1/record/gcc-103038. Accessed 01 July 2019
Tomaru N, Takahashi M, Tsumura Y, Takahashi M, Ohba K (1998) Intraspecific variation and phylogeographic patterns of Fagus crenata (Fagaceae) mitochondrial DNA. Am J Bot 85(5):629–636. https://doi.org/10.2307/2446531
Triboush SO, Danilenko NG, Davydenko OG (1998) A method for isolation of chloroplast DNA and mitochondrial DNA from sunflower. Plant Mol Biol Rep 16:183–189
Van de Paer C, Bouchez O, Besnard G (2018) Prospects on the evolutionary mitogenomics of plants: a case study on the olive family (Oleaceae). Mol Ecol Resour 18(3):407–423. https://doi.org/10.1111/1755-0998.12742
Vermeulen A, Desprez B, Lancelin D, Bannerot H (1994) Relationships among Cichorium species and related genera as determined by analysis of mitochondrial RFLPs. Theor Appl Genet 88(2):159–166. https://doi.org/10.1007/BF00225892
Wagner C, Friedt W, Marquard RA, Ordon F (2005) Molecular analyses on the genetic diversity and inheritance of (−)-α-bisabolol and chamazulene content in tetraploid chamomile (Chamomilla recutita (L.) Rausch.). Plant Science 169(5):917–927. https://doi.org/10.1016/j.plantsci.2005.06.010
Wang S, Song Q, Li S, Hu Z, Dong G, Song C et al (2018) Assembly of a complete mitogenome of Chrysanthemum nankingense using Oxford Nanopore Long Reads and the diversity and evolution of Asteraceae mitogenomes. Genes 9(11):547. https://doi.org/10.3390/genes9110547
Wang P, Lu Q, Ai Y, Wang Y, Li T, Wu L et al (2019) Candidate gene selection for cytoplasmic male sterility in pepper (Capsicum annuum L.) through whole mitochondrial genome sequencing. Int J Mol Sci. https://doi.org/10.3390/ijms20030578
White TJ, Bruns T, Lee S, Taylor J (1990) A guide to methods and applications. In: Innis M, Gelfand D, Sninsky J, White T (eds) PCR protocols. Academic Press, San Diego, pp 315–322
Wills DM, Hester ML, Liu A, Burke JM (2005) Chloroplast SSR polymorphisms in the Compositae and the mode of organellar inheritance in Helianthus annuus. Theor Appl Genet 110(5):941–947. https://doi.org/10.1007/s00122-004-1914-3
Won SY, Jung J-A, Kim JS (2018) The complete mitochondrial genome sequence of Chrysanthemum boreale (Asteraceae). Mitochondrial DNA Part B 3(2):529–530. https://doi.org/10.1080/23802359.2018.1468226
Acknowledgements
Open access funding provided by University of Veterinary Medicine Vienna. This work was initiated by a collaboration project with the Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), Department Plant Breeding Research, Apomixis Research Group, Gatersleben, Germany and the Pharmaplant GmbH, Artern, Germany. The authors would like to thank Dr. Lars-Gernot Otto (IPK) for his advice in the project and Marlis Sonnenschein (Pharmaplant GmbH) for providing seed material.
Funding
This work was financed based upon a resolution of the Federal Parliament of Germany (Deutscher Bundestag) by the Bundesministerium für Ernährung und Landwirtschaft (BMEL, support code 14NR063) via the Fachagentur Nachwachsende Rohstoffe e.V. (FNR) as project executing organisation for the funding programme ‘Renewable Resources’. The funding organisation did not execute any influence on the study nor the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ruzicka, J., Novak, J. Mitochondrial genome variation between different accessions of Matricaria chamomilla L. (Asteraceae) based on SNP mutation analysis. Genet Resour Crop Evol 67, 853–864 (2020). https://doi.org/10.1007/s10722-020-00881-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-020-00881-z