
Abstract
Capsicum species are of great economic, medicinal, culinary and cultural value worldwide, however their genetic potential is still underutilized. In this study, a set of 21 microsatellite markers showed utility for distinguishing a diverse group of 42 Capsicum genotypes representing 11 species. The level of polymorphism among these genotypes was evaluated by calculating the polymorphic information content (PIC) and discriminating power (D) for each locus. A total of 208 alleles were detected at the 21 loci evaluated. Markers detecting the highest number of alleles also exhibited the highest number of rare alleles. PIC values and D estimations showed a significant positive correlation (r = 0.98; P < 0.0000). PIC values ranged between 0.500 (marker NG20) and 0.899 (marker NG17), giving an average of 0.775. Genetic relationships among genotypes were evaluated using UPGMA clustering, Principal Coordinate Analysis (PCoA) and Bayesian model-based clustering, allowing genotypes to be discriminated into groups according to their degree of genetic similarity corresponding to distinct taxa. Based on these analyses, the 21 microsatellite included in this study now define a standard tool for efficient assessment of molecular variability of Capsicum germplasm collections conserved worldwide.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The fruits of the Capsicum (pepper) genus are valued worldwide as both a vegetable and spice. Pepper ranks fourth on the list of the most important vegetables in the global food economy, after tomatoes, cucurbits (pumpkins, squashes, cucumbers, and gherkins) and alliums (onions, shallots, and garlic) (Schreinemachers et al. 2018). In the United States, the 2017 fresh market value of bell and chili pepper crops was over $750 million dollars (National Agricultural Statistics Service 2018). The Capsicum genus is native to South and Central America (Bosland 1996; Perry et al. 2007; Walsh and Hoot 2001) and comprises approximately 35 species, of which five are domesticated and widely consumed worldwide as both vegetable foods and spices (Carrizo García et al. 2016; Moscone et al. 2007). This number, however, is only an estimate as new species are discovered and named on a regular basis (Barboza et al. 2019; Samuels 2015).
The five domesticated species are Capsicum annuum L., C. baccatum L., C. chinense Jacq., C. frutescens L., and C. pubescens Ruiz and Pav. Two centers of domestication of the genus have been identified: one in Mexico for C. annuum and the other one in South America for the other species (Eshbaugh 1993). One of pepper’s most notable characteristics is its pungency, attributed to a unique family of composite alkaloids produced in the fruit known as capsaicinoids (Bosland 1999). Most peppers grown commercially worldwide belong to the species C. annuum [including sweet varieties (e.g. bell peppers and paprika) and hot varieties like cayenne, jalapeños, and the chiltepin] with a few additional representatives from C. chinense (e.g. Habanero) and C. frutescens (e.g. Tabasco). In turn, C. baccatum and C. pubescens include the South American ají and rocoto peppers, respectively. Pepper fruits are an excellent source of micronutrient antioxidants (vitamins C and E as well as carotenoids), which confer to Capsicum a trending nutraceutical value due to their role in preventing or reducing chronic and age-related diseases. (Materska and Perucka 2005; Palevitch and Craker 1996; Soare et al. 2017).
Despite their broad presence in the world diet and global economic importance, the taxonomic classification of Capsicum within and between species is still confusing (Barboza 2011; Eshbaugh 1993). The terms pepper, chili, chile, and Capsicum are used interchangeably for different types and species within the genus (Andrews 1996). Additionally, there is no clear delimitation within two complexes of Capsicum species: (1) the C. annuum complex, formed by C. annuum, C. frutescens, and C. chinense, and (2) the C. eximium complex, formed by C. eximium Hunz. and C. cardenasii Heiser and P.G.Sm. (Moscone et al. 2007; Walsh and Hoot 2001). Ultimately, wild Capsicum germplasm represents an important source of genetic diversity but is underutilized and worthy of further research (van Zonneveld et al. 2015).
Currently, there exist several large germplasm ex situ collections of both wild and domesticated Capsicum species, particularly those maintained by the World Vegetable Center (AVRDC, Taiwan) with 8170 accessions, the United States Department of Agriculture (USDA) with 6067 entries, and the National Bureau of Plant Genetic Resources (NBPGR, India) with 2774 accessions (Chhapekar et al. 2018). Universities and other institutions from 21 countries in Latin America and the Caribbean also have genebanks that conserve smaller collections of Capsicum germplasm (Knudsen 2000). Nevertheless, only a few of these collections have been even partially characterized for morphological characters. Certain Capsicum species are not represented in the collections at all and perhaps will never be included because agricultural development and expanding human populations are reducing their natural habitats (Bosland and Gonzalez 2000).
Morphological characteristics have been traditionally used to classify species; however, they alone are not always enough to adequately categorize a specific entry to the species level in an ex situ collection. This type of characterization requires extensive resources, such as appropriate facilities and time (plants must be grown to the flowering or fruiting stage), as well as special expertise to determine characteristics such as pollen shape and flower morphology (IBPGR 1983; Singh et al. 2004). Alternatively, molecular assessment of germplasm collections may guide decisions about conservation activities, such as collecting, managing, identifying genes, and adding value to genetic resources (de Vicente et al. 2006).
Despite current advances in genome sequencing, microsatellite markers or simple sequence repeats (SSR) that have been converted into a DNA marker are still successfully used worldwide for routine genotyping in many crops, and for protection of ancient autochthonous olive trees in Italy (Rotondi et al. 2018), evaluation of genetic redundancy in ex situ apple collections in Norway (Gasi et al. 2016), evaluation of genetic structure in subspecies of black cherry from North America (Guzmán et al. 2018), estimation of natural outcrossing rate and genetic diversity in Lima bean landraces from Brazil (Penha et al. 2017), and in the analysis of the genetic diversity of rice cultivars in India (Dhama et al. 2018). This molecular technique is also ideal for designing conservation strategies for genetic resources, and for forming and validating core collections (Porta et al. 2018; de Vicente et al. 2006).
Capsicum germplasm collections are of actual and potential value in the breeding of the genus, and microsatellites are useful in the exploitation of these valuable sources of genetic diversity. Thus, the aims of this study were to (1) assess the utility of a collection of 54 microsatellite markers (Lee et al. 2004; Nagy et al. 2007) for distinguishing a diverse panel of 42 Capsicum genotypes, and (2) provide a rapid, accurate, and cost-effective set of SSR markers for assessment of genetic diversity in germplasm collections.
Materials and methods
Plant material and DNA extraction
This study included 42 genotypes representing 11 species of the Capsicum genus (Table 1). Germplasm was obtained from commercially available varieties or obtained from the USDA collection based on the USDA Genetic Resources Information Network (GRIN). At least three seeds per accession were planted under standard greenhouse conditions (16 h light and eight hours darkness with weekly fertilization). Young meristematic leaves were collected, and DNA was extracted according to Doyle and Doyle (1987). The leaf tissue of all individuals belonging to the same genotype was pooled to extract DNA. The quality of the extracted DNA was assessed by agarose gel electrophoresis (1% agarose) using a TBE 0.5X (Tris-borate-EDTA) buffer and staining with ethidium bromide. The gel was exposed to ultraviolet light and photographed. DNA was diluted to a final concentration of 2.0 ng/μL and stored at − 20 °C until use.
Microsatellite amplification
Fifty-four microsatellite markers were selected for preliminary evaluation. Thirty-three SSR were eliminated from the study because they exhibited poor band resolution. The remaining 21 microsatellite markers were then used to evaluate molecular diversity of the 42 genotypes included in this study (Table 2).
Fifteen SSRs have an NG designation, as they were initially reported by Nagy et al. (2007). Amplification was performed in a total volume of 15.0 μL containing 5.0 ng DNA, 0.5 µM of each primer, 0.125 mM of each dNTP, 1.5 µL 10X buffer (0.1 M Tris pH 8.3, 0.5 M KCl, 7.5 mM MgCl2, 0.1% gelatin), and 1 unit of Taq polymerase. The remaining six SSRs have a designation BD, as they were first reported by Lee et al. (2004), and their amplification was performed in a final volume of 10.0 μL containing 6.0 ng DNA, 0.3 µM of each primer, 0.1 mM of each dNTP, 1.0 µL 10X buffer (0.1 M Tris pH 8.3, 0.5 M KCl, 7.5 mM MgCl2, 0.1% gelatin), and 1 unit of Taq polymerase. Negative controls were run with every PCR to ensure no DNA contamination.
All amplifications were performed using a PTC-225 programmable thermocycler (MJ Research Inc., Waltham, MA, USA) under the conditions indicated in Table 2. Several amplifications were performed doing “Touchdown” PCR to reduce the amplification of non-specific products (Don et al. 1991). Touchdown amplification reactions began at a high hybridization temperature that gradually decreased by 1 °C per cycle until the respective hybridization temperatures of primers were reached.
The electrophoresis of amplified products was performed on 4% denaturing polyacrilamide gels, with 7 M urea and TBE 0.5X buffer. Six microliters of buffer [95% (v/v) formamide, 20 mM EDTA, pH 8.0, 0.05% (w/v) bromophenol blue and xylene cyanol FF] were added to each PCR product. This mixture was heated at 95 °C for 5 min and immediately cooled down on ice, then 5.0 µL of each sample were loaded into the wells of the gel.
The samples were run (2000 V, 75 W, 50 mA) and stained immediately afterwards with silver nitrate. Table 2 shows the electrophoresis run time for each marker. After staining with silver nitrate, discreet groups of two to five bands were observed (what is commonly called ‘shadow bands’ or ‘stuttering’) in most of the markers. ‘Stuttering’ refers to PCR products differing in the size of the principal band (allele), which makes each allele (band) observed in the gel appear to be more than one band (Litt et al. 1993).
Twenty-seven unique samples were run on each gel as a check to evaluate the reproducibility of patterns and compare bands between gels. Fifteen genotypes were also chosen at random and subsequently amplified separately and run several times on different gels.
The size of the most intensely amplified band of each microsatellite, measured as the number of nucleotides, was determined based on its migration in relation to that of molecular weight markers (50 bp and 10 bp DNA ladders, Roche Diagnostics Corporation, Indianapolis, IN, USA). Due to their codominance, the markers were scored as homozygous or heterozygous.
SSR data analysis
Each intensely amplified SSR band was manually scored as present (1) or absent (0), and a binary data matrix was generated. Two value measures for each marker were calculated on an Excel spreadsheet. First, the Polymorphism Information Content (PIC) value was estimated according to Botstein et al. (1980): \({\text{PIC}} = 1 - \left( {\sum\nolimits_{i = 1}^{n} {P_{i}^{2} } } \right) - \sum\nolimits_{i = 1}^{n - 1} {\sum\nolimits_{j = i + 1}^{n} {\left( {2P_{i}^{2} P_{j}^{2} } \right)} }\), where Pi and Pj are the relative frequencies of the ith and jth alleles in a given marker, and n is the total number of alleles detected by the marker. PIC value estimates the discriminatory power of an SSR by taking into account the number of alleles exhibited and their relative frequencies. Second, the discriminating power (D) per marker was calculated according to Tessier et al. (1999): \({\text{D}} = 1 - {\text{C}}_{j} = 1 - \sum\nolimits_{i = 1}^{I} {P_{i} \frac{{\left( {NP_{i} - 1} \right)}}{N - 1}}\), where I is the number of banding patterns (or genotypes) generated by the SSR, Pi is the frequency of the ith pattern of the given jth primer, N is the number of individuals evaluated and Cj is the confusion probability for the jth SSR, i.e. the probability that two randomly chosen individuals from the sample have identical banding patterns. In consequence, \({\text{D}} = 1 - {\text{C}}_{j}\) represents the probability that two randomly chosen individuals have different patterns and, therefore, are distinguishable from one another. Additionally, as N tends toward infinity, the limit of D, equals to DL, provides an estimate of discriminating power of each primer as follows: \({\text{D}}_{L} = 1 - {\text{C}} = 1 - \sum\nolimits_{i = 1}^{I} {P_{i}^{2} }\) (Tessier et al. 1999).
Theoretically, in a set of N individuals, it is possible to draw \(\left( {N\left( {N - 1} \right)/2} \right)\) different pairs. According to Tessier et al. (1999), the total number of non-differentiated pairs of genotypes for the jth SSR is given by \(x_{j} = \left( {N\left( {N - 1} \right)/2} \right){\text{C}}_{j} .\) Thus, for a given combination of K primers, \(X_{K} = \left( {N\left( {N - 1} \right)/2} \right)\varPi_{j = 1}^{k} C_{j} \) estimates the number of non-differentiated pairs of genotypes by the combination. The most efficient combination reduces the most the number of undifferentiated pairs, i.e., minimizes \(X_{K}\). To find this optimal combination, the primers were sorted in descending order of their discriminating power and, at each step, chosen one after the other to minimize \(X_{K}\).
GenAlEx 6.5b3 (Peakall and Smouse 2012) was used to assess the genetic diversity per marker by calculating allelic richness (number of alleles and number of private alleles) and observed (Ho) and expected heterozygosity (He). In GenAlEx, the genetic structure was studied by determining the number of private alleles for C. annuum, C. baccatum, C. chinense and C. frutescens.
Cluster analysis was used to evaluate the resolving power of the SSR markers. In PAST software (Hammer et al. 2001), the binary matrix was used to estimate a distance matrix using Nei’s unbiased measure of genetic distance (Nei 1987), which is based on the proportion of alleles shared between two genotypes for all possible pairwise combinations of samples. The resultant genetic distance matrix was subjected to a cluster analysis using the Unweighted Pair Group Method with Arithmetic Mean (UPGMA) to obtain a dendrogram that depicted the genetic relationships among genotypes. A bootstrap analysis (10,000 replicates) was performed to measure branch support by the data. In software GenAlEx, a Principal Coordinate Analysis (PCoA) was carried out using the genetic distance matrix with the goal to represent the distribution of the genotypes in a multidimensional metric space, which reflects the relationships among them based on their similarity in banding profiles, and to visualize their dispersion and possible structuring of the sample set studied. Three dimensions were chosen for illustration using PAST software.
The previous grouping analyses were complemented with Bayesian model-based clustering using the software STRUCTURE 2.3.4 (Pritchard et al. 2000), which differentiates groups of genotypes that have distinctive allele frequencies without a priori groupings and estimates the most likely number of genetic groups (K). STRUCTURE was run for K values ranging from 1 to 12. Each run was performed using the admixture model with 10,000 replicates for burn-in and 100,000 during the analysis (Falush et al. 2007; Pritchard et al. 2000), and 10 simulations per K value. The STRUCTURE output was summarized using STRUCTURE HARVESTER (Earl and von Holdt 2012). Both, the Evanno et al. (2005) delta K test and the method of Pritchard et al. (2000) [K with the highest Pr (X|K)] were performed to estimate the optimal value of K. CLUMPAK (Kopelman et al. 2015) was used for the graphical presentation of the STRUCTURE results.
Results
Molecular variation detected with the SSR markers
Table 3 presents the description of the 21 loci based on the number of alleles detected. A total of 208 alleles were identified, giving an average of 9.9 alleles per locus. The number of alleles ranged from five (markers NG20 and BD70) to 16 (marker NG17). The smallest allele (approx. 82 bp) was detected by SSR BD22 and the largest allele (approx. 800 bp) by NG15.
Alleles observed in only one or two of the 42 genotypes studied (frequency < 5%) were considered rare alleles (Table 3). Overall, 102 rare alleles were detected by 20 of the 21 markers evaluated. For each locus, the number of alleles and the number of rare alleles showed a significant positive correlation (r = 0.90; P < 0.0000). The highest percentages of rare alleles were detected by markers NG6 (78%) and NG7 (73%).
The absence of an amplification product for any given genotype-marker combination indicated the presence of genotypes with null alleles in the given locus. Null alleles are alleles that are not amplified during PCR, perhaps due to polymorphism at the hybridization sites of one or both primers (Dakin and Avis 2004). Genotype-marker combinations that exhibited null alleles were replicated at least twice to ensure that absence of amplification product was not due to experimental errors.
Thirteen of the 21 loci had null alleles, ranging from two (5%) to 10 (24%) genotypes exhibiting null alleles (Table 3). The two wild genotypes of C. rhomboideum (Dunal) Kuntze exhibited null alleles in all of these 13 SSR. In seven (54%) out of these 13 loci, null alleles were detected only for the two C. rhomboideum genotypes. Despite the known prevalence of null alleles, their evolutionary dynamics and model of variation in populations is still debated (Putman and Carbone 2014); as a result, little is known of their true impact on the estimation of population differences.
Any genotype-marker combination that produced two groups of bands was classified as heterogeneous (a mixture of alleles or multiple alleles). Genotypes with multiple alleles were detected by 62% of the 21 loci (Table 3). On average, 1.6 genotypes per locus presented multiple alleles. Markers NG4 (6 genotypes), NG7 (6), and NG12 (5) had the highest number of genotypes with multiple alleles.
There was a considerable variation in the frequency of the most common allele (Table 3). Among markers, a significant negative correlation was observed between the number of alleles and the frequency of the most common allele (r = − 0.66, P = 0.0011). On average, at any given locus, the common allele was shared by 33% of the genotypes evaluated.
The level of polymorphism among the 42 genotypes was evaluated by calculating a PIC value for each locus (Table 4). PIC values ranged between 0.5004 (NG20) and 0.8988 (NG17), with an average of 0.7748 per marker. Based on PIC values, 20 of the SSRs can be considered informative (PIC > 0.50), four of these being highly informative (PIC > 0.85). PIC values showed significant positive correlation with the number of alleles (r = 0.79; P < 0.0000), significant negative correlation with the frequency of the most common allele (r = − 0.95; P < 0.0000) and a non-significant very low correlation with the range of allele size (r = 0.17; P = 0.4627), which indicates that microsatellites detecting the highest number of alleles were the most informative, regardless of differences in allele size revealed.
Discriminatory power of the SSR markers
As seen with the PIC value, the estimate of the discriminating power (Dj) is based on the allele frequencies. Thus, they both correspond to nearly identical values (Tessier et al. 1999), and this was observed in the present study (Table 3 and 4). In fact, the PIC values and D estimations showed a significant high positive correlation (r = 0.98; P < 0.0000), which suggests that these two estimations give highly similar information for the SSR. Nonetheless, the discriminating power reveals that the efficiency of a given microsatellite marker does not depend only on the number of patterns it generates (Table 4). For instance, two SSRs producing a similar number of patterns can have very different discriminating powers, as illustrated by NG9 and BD33. In contrast, two markers that differ significantly with respect to the number of patterns observed may have similar discriminating powers (e.g. NG12 and NG34; 14 and 9 patterns, respectively). According to Tessier et al. (1999), the differences among the frequencies of the patterns generated by the markers help explain this result. When a marker generates patterns at the same frequency, then its discriminating power is maximal. Conversely, the farther the SSR is from this situation, the more its discriminating power diminishes.
Based on the values of the estimated total number of non-differentiated pairs, one optimal combination of three SSR markers would be sufficient to discriminate among the 42 genotypes studied (Table 5). In the present study, these three SSR markers, generated from the primers, NG17 + NG18 + BD12, were selected on the basis of their discriminating power (Table 4).
Resolving power of the SSR markers by cluster analysis
The 21 microsatellites evaluated made it possible to distinguish among the studied 42 genotypes. Figure 1 shows the UPGMA clustering pattern for the 42 genotypes with two main clusters. One cluster was formed by the two genotypes representing the wild species C. rhomboideum (Group A). These two genotypes showed virtually no similarity with the rest of the genotypes. This species presents a combination of morphological characteristics that contrasts with the other species of the Capsicum genus. The flowers of C. rhomboideum are yellow, unlike the other species that have white or purple flowers, and it is considered as a non-pungent and basal species of the Capsicum genus (Carrizo García et al. 2016; Egan et al. 2019; Walsh and Hoot 2001).

UPGMA dendrogram, based on Nei’s genetic distance, depicting the genetic relationships among the 42 genotypes studied. Numbers at the nodes provide branch support ≥ 75% based on 10,000 bootstrap replicates. Capital letters indicate the different genetic groups
The other main cluster grouped together genotypes from both domesticated and wild species, hampering a conclusive interpretation based on taxonomic status. Nonetheless, based on bootstrap values greater than or equal to 75%, it can be interpreted that, within this main cluster, there are six minor clusters that correspond to: (B) two genotypes of C. pubescens and one of C. minutiflorum Rusby (Hunz.); (C) two genotypes of C. chacoense Hunz.; (D) eleven genotypes of C. annuum (including the one from C. annuum var. abbreviatum Fingerh.) and one genotype of each species, C. baccatum, chinense x annuum, and C. luteum Lam.; (E) three genotypes of C. chinense and three of C. frutescens; (F) seven genotypes of C. baccatum and one of C. frutescens; and (G) four genotypes of C. chinense. Three genotypes did not belong to any of these groups but are still different from each other, making up unitary groups. These genotypes represented the domesticated species C. frutescens (16fru), and the wild species, C. cardenasii (24car) and C. flexuosum Sendtn. (25flex).
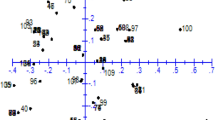
The cluster procedure applied to the coordinates obtained by PCoA revealed that the first three axes accounted together for 53.56% of the total variation, with 25.40, 15.54, and 12.62% explained, respectively, by PC axis 1, 2 and 3 (data not shown). The representation of the distribution of the genotypes in a tri-dimensional PCoA space suggested a structure of four main clusters (Fig. 2), corresponding to one group that included the 10 C. annuum genotypes (including var. abbreviatum) plus the chinense × annuum and C. luteum genotypes; a second group clustering three C. chinense genotypes and three of C. frutescens; a third group formed by six C. baccatum genotypes and one C. frutescens; and a fourth group composed of 14 genotypes representing species C. cardenasii, C. chacoense, C. flexuosum, C. pubescens and C. rhomboideum. Consistently in our clustering analyses, one C. baccatum genotype showed no evident close relation to other genotypes based on taxonomic species.

Spatial distribution of the groups of genotypes obtained with the three main axes of variation by PCoA
In our results from Bayesian model-based clustering, the delta K statistic in the Evanno test and median values of ln(Pr Data) showed that K = 10 was the optimal number of genetic groups in this study (Fig. 3). For K = 10, the clustering in different runs was almost identical (similarity coefficient 0.890). The run with the highest likelihood value (lnL = −1216.5) was selected to classify genotypes based on their posterior assignment probabilities (more than 50%) and to explore the possibility of a taxonomical explanation of these K = 10 genetic clusters (Table 1). These clusters mainly merged genotypes from the same species, thus a pattern of taxonomic structure can be deduced. For instance, clusters C1, C2, C3 and C4 grouped together genotypes of C. annuum, C. chinense, C. frutescens and C. baccatum, respectively; these species were represented by at least five genotypes.

Genetic groups formed by the 42 studied genotypes at K = 10
The split using a model-based Bayesian analysis was congruent with UPGMA clustering and PCoA analysis, although it was more discriminating in that it included nine of 10 genotypes from C. annuum in C1 (the other one clustered together with annuum × chinense, var. abbreviatum and C. luteum in C6) and four of seven genotypes from C. chinense in C2; whereas C. baccatum was contained in C4 and C. pubescens in C5, and still depicted an intermixed cluster (C3) of C. frutescens and C. chinense (Table 1; Fig. 3). This split also clearly grouped together the two genotypes of wild species C. rhomboideum (Cluster C10). In turn, C. minutiflorum (C8) and C. chacoense (C9) stood out in one cluster per species, and C. flexuosum and C. cardenasii made-up an admixed cluster (C7). One genotype of C. baccatum (20bac) had a posterior assignment probability value of less than 50%, which seems to be indicative of an admixed genotype (with genetic contributions from more than one cluster).
Markers to distinguish among same species genotypes
Table 6 summarizes the SSR description for species with five or more genotypes included in this evaluation. Due to their polymorphism, 15 SSRs were useful to differentiate the 10 genotypes of C. annuum (Table 6). None of these markers detected null alleles in any of these genotypes. The genotype C. annuum var. Chung yang showed multiple alleles in loci NG4, NG15, BD22 and BD70 (data not shown). The genotypes Jalapeño 3575, Jalapeño 1493, and PapriQueen exhibited multiple alleles in markers NG4, NG9, and NG15, respectively (data not shown).
Twenty microsatellites were polymorphic within the genotypes of C. baccatum (Table 6). PIC values indicate that loci NG7 and NG34 were the most informative markers in genotypes of this species. The SSR markers NG5 (genotype 51bacpen), NG34 (16bac and 60bac) and BD41 (16bac, 37bacbac, 43bacpen, 49bacpen, 51bacpen and 60bac) revealed nine genotypes with null alleles. Multiple alleles were detected by markers NG12, BD12 and BD22 in genotype C. baccatum var. praetermissum (Heiser and P.G.Sm.) Hunz., and by locus NG15 in genotype 56bac.
The seven genotypes of C. chinense were polymorphic when assayed with 20 microsatellite markers (Table 6). PIC values indicate that markers NG7, NG33 and NG34 were the most informative. Marker NG34 detected null alleles in genotype 14chi. Multiple alleles were determined at locus NG4 in genotypes 12chi and 53chi.
All 21 microsatellites evaluated were polymorphic in the set of five genotypes representing C. frutescens (Table 6). PIC values show that marker NG8 was the most informative microsatellite, as this SSR detected one different allele per every C. frutescens genotype evaluated. In turn, microsatellites NG17, NG18, BD22 and BD33 were the second most informative markers, each informative to an identical degree. Markers NG34 (genotypes 58fru, 59fru and 61fru) and BD41 (59fru) detected four genotypes with null alleles. One genotype with multiple alleles was detected by SSR NG5 (15fru).
Discussion
Polymorphism and usefulness of the studied markers
Microsatellite markers present several advantages over other types of molecular markers, because they are considered to be robust, more variable and informative than RFLP, RAPD and AFLP (He et al. 2003; Lee et al. 2004; Senan et al. 2014). In this study, the high level of polymorphism detected among the 42 genotypes assayed demonstrated the efficiency of SSR markers for discriminating both distinct taxa and variation within a species (Table 3).
The 21 microsatellite markers used in this study generated 208 alleles, with an average of 9.9 alleles per locus. The significant positive correlation (r = 0.79; P < 0.0000) between PIC value and number of alleles per SSR, discriminating power and number of alleles (r = 0.72; P < 0.0002), as well as the positive correlation between PIC values and discriminating power (r = 0.98; P < 0.0000), suggests that markers that detect higher number of alleles are more informative and therefore more useful in genotyping and diversity analysis of Capsicum germplasm. Our results confirm that any of these three estimators are potentially helpful in determining the informative value of an SSR marker used in diversity studies of the genus.
Botstein et al. (1980) reported that PIC values above 0.5 are consistent with usefulness of the marker. Based on PIC values, the level of polymorphism of the 21 microsatellites was very high, ranging from 0.500 to 0.899, with an average of 0.775. The polymorphism of these microsatellite markers could accordingly be used to discover duplicated entries in ex situ germplasm collections of Capsicum, as well as to resolve disputes related to intellectual property rights related to Capsicum germplasm. Furthermore, this high level of information was used to discriminate the genotypes in the present study, as shown by UPGMA clustering, PCoA analysis and Bayesian model-based clustering. The results from this study clearly confirm that microsatellites are a promising approach to definitive differentiation among cultivated varieties or different species of the genus Capsicum.
Rare alleles are highly informative for genotyping of varieties (Jain et al. 2004) and can serve as an indicator of the presence of unique genetic variants or differentiated populations. Genetic erosion, on the other hand, can rapidly lead to the loss of these alleles (Bijlsma and Loeschcke 2012). Nonetheless, this susceptibility makes them valuable tools for assessing losses of genetic diversity that occur during multiplication and regeneration of germplasm collections. The 20 microsatellites markers that detected rare alleles in this study are likely candidates for broad use within the genus for cultivar discrimination or identification of species of the genus Capsicum. They will also be helpful as baseline information in future decisions related to the most efficient way to disseminate and maintain ex situ collections of Capsicum germplasm.
Thirteen of the evaluated microsatellites detected genotypes with multiple alleles (Table 3). This level of polymorphism could be (1) the result of remnant heterozygosity in several genotypes, or (2) the consequence of the heterogeneity produced by involuntary mixture of seed. In turn, the residual heterozygosity could be the result of allogamy and would be a foreseeable characteristic in materials that have not been bred to develop pure lines. Capsicum is regarded as a primarily self-pollinating crop (Allard 1971); however, allogamy rates in Capsicum suggest that it should be considered as a facultative cross-pollination plant (Tanksley 1984). In the present study, heterozygosity cannot be clearly distinguished from heterogeneity because leaf tissue from three plants per genotype was pooled to extract the DNA. Nonetheless, the presence of allelic variants is highly valuable to find distinctive markers useful for discrimination of a given genotype.
Most of genotypes evaluated in this study (genotypes 1–30 in Table 1) have been multiplied by self-pollination under greenhouse conditions at Cornell University (former Jahn laboratory working group) over a period of several years to obtain seeds that are used in breeding programs. Although involuntary mistakes during handling cannot be discarded, it can be assumed that there was a very low likelihood of seed mixture events during the management of these genotypes. Therefore, genotypes provided by Cornell University that exhibited multiple alleles (e.g., Jalapeño 3575, Jalapeño 1493, PapriQueen and var. praetermissum) reinforce that the 21 microsatellites evaluated in this study are helpful markers to discover new molecular variability, or allelic variants, that can serve as distinctive markers for a given genotype.
The remaining genotypes were provided by the USDA (genotypes 31–42 in Table 1), but it is unclear the way these materials were managed during their conservation. Anecdotal reports suggest that any accession in the collection in the early 1980s was put through a round of open pollination in field increases early in that decade (Jahn, personal communication). This means that multiple alleles exhibited by genotypes of this subset (e.g. 53chi and 56bac in markers NG4 and NG15, respectively) may be attributable to allogamy in Capsicum. On the other hand, due to the lack of sound information about their management, it is also probably that these USDA genotypes showing multiple alleles reflect either unintentional mixture of seed that may have occurred during germplasm management, or inadvertent mistakes taken place while assigning introduction numbers (PI) in germplasm banks from which the genotypes were originally obtained. Accordingly, this study also identifies markers such as NG4 and NG15, that may be used by genebanks as an effective tool to detect mishandling that could have occurred during regeneration, multiplication and management of Capsicum germplasm.
Carrizo García et al. (2016) presented the most inclusive molecular phylogeny of Capsicum published to date using most of the recognized species of the genus. This study showed 11 well-supported clades, however the placements of some species and their closest relatives are not strongly resolved. In the present study, the Bayesian model-based analysis performed using software STRUCTURE to estimate the most probable K number of genetic groups, produced a most probable value of K = 10. This configuration of the 42 studied genotypes corresponded to some degree to the species included in the study. C. rhomboideum genotypes separate from the rest of the species (Cluster C10), as expected according to the classification indicated by cytogenetic and molecular data (Carrizo García et al. 2016; Moscone et al. 2007). According to the phylogenic analysis by Carrizo García et al. (2016), C. rhomboideum belongs to the Andean clade, the most ancient group of the genus which includes non-pungent species native to the Andes, western to north-western South and Central America. Further studies have indicated that this species is sister to the rest of the genus (Guzmán et al. 2009; Särkinen et al. 2013; Walsh and Hoot 2001).
As for the five domesticated species, the clustering of C. annuum genotypes together with one C. chinense genotype (C1) and also three genotypes of C. frutescens grouped with C. chinense (C3) reflects the strong relationship among these three species (Carrizo García et al. 2016). Together, these two clusters can be understood because these species form the C. annuum complex (C. annuum, C. chinense and C. frutescens). The difficulties in taxonomic differentiation within these three taxa have been previously reported (Baral and Bosland 2004; Moscone et al. 2007; Walsh and Hoot 2001), consistent with their high degree of genetic uniformity. In turn, C. baccatum genotypes are grouped in cluster C4. This species is characterized by white flowers, similar to those in the C. annuum complex (Carrizo García et al. 2016; Scaldaferro et al. 2018). It is remarkable that cluster C5 was composed of C. pubescens, the most distinct of any of the five domesticated species; characterized by strongly violet corollas, blackish brown seeds and pubescence on the entire plant (Carrizo García et al. 2016).
Owing to their codominance, multi allelism, and cross-species amplification, microsatellites are one of the most extensively harnessed markers in several areas of plant research (Senan et al. 2014). Microsatellite polymorphisms can be detected by agarose or polyacrylamide gel electrophoresis, and SSR alleles can be visualized using ethidium bromide or silver staining. Their reproducibility allows compare data among laboratories and their protocols imply the use of relative low-cost reagents and equipment. As a result, microsatellites can be very helpful in developing countries or any context where limited funding may constrain the scientific community working toward long term conservation of genetic diversity within Capsicum and use of Capsicum genetic resources.
With the aim to contribute to safeguarding ex situ collections of Capsicum spp., the molecular resources presented in this study define a major advance for the conservation of Capsicum genetic resources, especially those from developing countries, allowing much more accurate and efficient assessment of Capsicum spp. germplasm to support better decisions in day-to-day conservation activities with highly constrained resources.
Enabling conservation and use of Capsicum genetic diversity
The conservation of genetic resources of Capsicum sp. in germplasm banks is justified by the need and ongoing, even intensified use of this germplasm by both public and private sector plant breeders. In turn, use of Capsicum germplasm among researchers and conservation activities requires reliable information about the genetic makeup of the conserved accessions. Thus, ex situ collections are fundamental for the present and future utilization of Capsicum sp. genetic resources.
This study characterizes a set of 21 polymorphic microsatellite markers that successfully amplify across 11 Capsicum species, establishing their utility for broad molecular characterization of genetic diversity of Capsicum species. These results spotlights the relevance of this set of markers for both enhanced characterization of Capsicum germplasm and potentially improved efficiencies in breeding programs. Due to their polymorphism, these microsatellites can further be used to estimate changes in the genetic structure of ex situ collections of Capsicum germplasm. In addition, available information on allelic frequencies can serve as a baseline to make sound decisions about conservation of Capsicum germplasm. Allelic variants that occur at low frequency can be identified and maintained, and sources of genetic variability can be ascertained through genotyping of local varieties. Finally, these markers can also be used to trace the recent genetic history of a given modern commercial variety or landrace endemic to a geographical region.
The use of these microsatellites to characterize Capsicum germplasm conserved ex situ will promote the utilization of more collections which are the foundation for the development of improved varieties. These markers will also be helpful in assessing to what extent any Capsicum ex situ collection is representative of the genetic diversity present in a specific country or region, and in identifying gaps within collections, thus providing guidelines to define complementary conservation strategies.
The overall results of this study highlight that identification and establishment of useful, efficient microsatellites can offer consistent results and enable preservation of genetic distinctness, uniformity, and discrimination of promising genotypes for Capsicum breeding. Morphological markers, in conjunction with molecular genetic evidence provided by this set of microsatellites, would be beneficial for definition of strategies for complementary conservation of Capsicum, as well as for selection of germplasm to be included in breeding programs.
References
Allard RW (1971) Princípios do melhoramento genético das plantas. Edgard Blucher, São Paulo
Andrews J (1996) Chili or chile is silly, when it should be chilli: why I call pungent Capsicums chilies (Capsicum). In: Proceedings of the national pepper conference. American Society for Horticultural Science, Alexandria, VA, p 8
Baral JB, Bosland PW (2004) Unravelling the species dilemma in Capsicum frutescens and C. chinense (Solanaceae): a multiple evidence approach using morphology, molecular analysis, and sexual compatibility. J Am Soc Hortic Sci 129:826–832
Barboza G (2011) Lectotypifications, synonymy, and a new name in Capsicum (Solanoideae, Solanaceae). PhytoKeys 2:23–38. https://doi.org/10.3897/phytokeys.2.730
Barboza GE, Carrizo García C, Leiva González S, Scaldaferro M, Reyes X (2019) Four new species of Capsicum (Solanaceae) from the tropical Andes and an update on the phylogeny of the genus. PLoS ONE 14(1):e0209792. https://doi.org/10.1371/journal.pone.0209792
Bijlsma R, Loeschcke V (2012) Genetic erosion impedes adaptive responses to stressful environments. Evol Appl 5:117–129. https://doi.org/10.1111/j.1752-4571.2011.00214.x
Bosland PW (1996) Capsicums: innovative uses of an ancient crop. In: Janick J (ed) Progress in new crops. ASHS Press, Arlington, pp 479–487
Bosland PW (1999) Chiles: a gift from fiery god. HortScience 34:809–811
Bosland PW, Gonzalez MM (2000) The rediscovery of Capsicum lanceolatum (Solanaceae), and the importance of nature reserves in preserving cryptic biodiversity. Biodivers Conserv 9:1391–1397. https://doi.org/10.1023/A:1008930931976
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32(3):314–331
Carrizo García C, Barfuss MH, Sehr EM, Barboza GE, Samuel R, Moscone EA, Ehrendorfer F (2016) Phylogenetic relationships, diversification and expansion of chili peppers (Capsicum, Solanaceae). Ann Bot 118(1):35–51. https://doi.org/10.1093/aob/mcw079
Chhapekar SS, Jaiswal V, Ahmad I, Gaur R, Ramchiary N (2018) Progress and prospects in Capsicum breeding for biotic and abiotic stresses. In: Vats S (ed) Biotic and abiotic stress tolerance in plants. Springer, Singapore. https://doi.org/10.1007/978-981-10-9029-5_11. ISBN 978-981-10-9029-5
Dakin EE, Avis JC (2004) Microsatellite null alleles in parentage analysis. Heredity 93:504–509. https://doi.org/10.1038/sj.hdy.6800545
de Vicente MC, Guzmán FA, Engels J, Ramanatha Rao V (2006) Genetic characterization and its use in decision-making for the conservation of crop germplasm. In: Ruane J, Sonnino A (eds) The role of biotechnology in exploring and protecting agricultural genetic resources, Chap. 12. FAO, Rome, pp 129–138. ISBN 978-92-5-105480-2
Dhama N, Saini RK, Kumar R, Chaudhary DP, Maurya BK, Sharma M, Sen R, Kumar D, Malik P, Kumar P (2018) Analysis of genetic diversity in rice (Oryza Sativa L.) cultivars using SSR markers. Bull Environ Pharmacol Life Sci 7(3):1–7
Don RH, Cox PT, Wainwright BJ, Baker K, Mattick JS (1991) ‘Touchdown’ PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res 19:4008
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Earl DA, von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Egan AN, Moore S, Stellari GM, Kang BC, Jahn MM (2019) Tandem gene duplication and recombination at the AT3 locus in the Solanaceae, a gene essential for capsaicinoid biosynthesis in Capsicum. PLoS ONE 14(1):e0210510. https://doi.org/10.1371/journal.pone.0210510
Eshbaugh WH (1993) History and exploitation of a serendipitous new crop discovery. In: Janick J, Simon JE (eds) New crops. Wiley, New York, pp 132–139
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Falush D, Stephens M, Pritchard JK (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7(4):574–578. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Gasi F, Kanlić K, Kalamujić Stroil B, Pojskić N, Asdal Å, Rasmussen M, Kaiser C, Meland M (2016) Redundancies and genetic structure among ex situ apple collections in Norway examined with microsatellite markers. HortScience 51(12):1458–1462. https://doi.org/10.21273/HORTSCI11212-16
Guzmán FA, Dean E, Bohs L (2009) Hot or not so hot: phylogenetic relationships in Capsicum and Lycianthes (Solanaceae). Snowbird: botany and mycology 2009 meeting, Botanical Society of America. http://bohs.biology.utah.edu/PDFs/Poster-LycCap.jpg. Accessed 02 Feb 2019
Guzmán FA, Segura S, Aradhya M, Potter D (2018) Evaluation of the genetic structure present in natural populations of four subspecies of black cherry (Prunus serotina Ehrh) from North America using SSR markers. Sci Hortic 232:206–215. https://doi.org/10.1016/j.scienta.2018.01.013
Hammer Ø, Harper DAT, Ryan PD (2001) PAST: paleontological statistics software package for education and data analysis. Palaeontol Electron 4(1):1–9
He GH, Meng RH, Newman M, Gao GQ, Pittman RN, Prakash CS (2003) Microsatellites as DNA markers in cultivated peanut (Arachis hypogaea L.). BMC Plant Biol. https://doi.org/10.1186/1471-2229-3-3
IBPGR (1983) Genetic resources of Capsicum. International Board for Plant Genetic Resources, IBPGR, Rome
Jain S, Jain RK, McCouch S (2004) Genetic analysis of Indian aromatic and quality rice (Oryza sativa L.) germplasm using panels of fluorescently-labeled microsatellite markers. Theor Appl Genet 109(5):965–977. https://doi.org/10.1007/s00122-004-1700-2
Knudsen H (2000) Directorio de colecciones de germoplasma en América Latina y el Caribe, 1st edn. International Plant Genetic Resources Institute (IPGRI), Roma
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15(5):1179–1191. https://doi.org/10.1111/1755-0998.12387
Lee JM, Nahm SH, Kim YM, Kim BD (2004) Characterization and molecular genetic mapping of microsatellite loci in pepper. Theor Appl Genet 108(4):619–627. https://doi.org/10.1007/s00122-003-1467-x
Litt M, Hauge X, Sharma V (1993) Shadow bands seen when typing polymorphic repeats: some causes and cures. Biotechniques 15:280–284
Materska M, Perucka I (2005) Antioxidant activity of the main phenolic compounds isolated from hot pepper fruit (Capsicum annuum L.). J Agric Food Chem 53(5):1750–1756. https://doi.org/10.1021/jf035331k
Moscone EA, Scaldaferro MA, Grabiele M, Cecchini NM, Sánchez García Y, Jarret R, Daviña JR, Ducasse DA, Barboza GE, Ehrendorfer F (2007) The evolution of chili peppers (Capsicum—solanaceae): a cytogenetic perspective. Acta Hortic 745:137–170. https://doi.org/10.17660/ActaHortic.2007.745.5
Nagy I, Stágel A, Sasvári Z, Röder M, Ganal M (2007) Development, characterization, and transferability to other Solanaceae of microsatellite markers in pepper (Capsicum annuum L.). Genome 50(7):668–688. https://doi.org/10.1139/g07-047
National Agricultural Statistics Service (2018) Crop values summary 2017. United States Department of Agriculture. ISSN 1949-0372. http://www.nass.usda.gov. Accessed 21 Sept 2018
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Palevitch D, Craker LE (1996) Nutritional and medical importance of red pepper (Capsicum spp.). J Herbs Spices Med Plants 3(2):55–83. https://doi.org/10.1300/J044v03n02_08
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28(19):2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Penha JS, Lopes ACA, Gomes RLF, Pinheiro JB, Assunção Filho JR, Silvestre EA, Viana JPG, Martínez-Castillo J (2017) Estimation of natural outcrossing rate and genetic diversity in Lima bean (Phaseolus lunatus L. var. lunatus) from Brazil using SSR markers: implications for conservation and breeding. Genet Resour Crop Evol 64:1355–1364. https://doi.org/10.1007/s10722-016-0441-9
Perry L, Dickau R, Zarrillo S, Holst I, Pearsall DM, Piperno DR, Berman MJ, Cooke RG, Rademaker K, Ranere AJ, Raymond JS, Sandweiss DH, Scaramelli F, Tarble K, Zeidler JA (2007) Starch fossils and the domestication and dispersal of chili peppers (Capsicum spp.) in the Americas. Science 315:986–988. https://doi.org/10.1126/science.1136914
Porta B, Condón F, Franco J, Iriarte W, Bonnecarrère V, Guimaraens-Moreira M, Vidal R, Galván GA (2018) Genetic structure, core collection, and regeneration quality in white dent corn landraces. Crop Sci 58:1644–1658. https://doi.org/10.2135/cropsci2017.12.0752
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Putman AI, Carbone I (2014) Challenges in analysis and interpretation of microsatellite data for population genetic studies. Ecol Evol 4(22):4399–4428. https://doi.org/10.1002/ece3.1305
Rotondi A, Ganino T, Beghè D, Di Virgilio N, Morrone L, Fabbri A, Neri L (2018) Genetic and landscape characterization of ancient autochthonous olive trees in northern Italy. Plant Biosyst Int J Dealing Asp Plant Biol 152(5):1067–1074. https://doi.org/10.1080/11263504.2017.1415993
Samuels J (2015) Biodiversity of food species of the Solanaceae family: a preliminary taxonomic inventory of subfamily Solanoideae. Resources 4(2):277–322. https://doi.org/10.3390/resources4020277
Särkinen T, Bohs L, Olmstead R, Knapp SD (2013) A phylogenetic framework for evolutionary study of the nightshades (Solanaceae): a dated 1000-tip tree. BMC Evol Biol 13:214. https://doi.org/10.1186/1471-2148-13-214
Scaldaferro M, Barboza GE, Acosta MC (2018) Evolutionary history of the chili pepper Capsicum baccatum L. (Solanaceae): domestication in South America and natural diversification in the Seasonally Dry Tropical Forests. Biol J Linnean Soc 124(3):466–478. https://doi.org/10.1093/biolinnean/bly062
Schreinemachers P, Simmons EB, Wopereis MCS (2018) Tapping the economic and nutritional power of vegetables. Glob Food Secur 16:36–45. https://doi.org/10.1016/j.gfs.2017.09.005
Senan S, Kizhakayil D, Sasikumar B, Sheeja TE (2014) Methods for development of microsatellite markers: an overview. Notulae Scientia Biologicae 6(1):1–13. https://doi.org/10.15835/nsb619199
Singh RK, Sharma RK, Singh AK, Singh VP, Singh NK, Tiwari SP, Mohapatra T (2004) Suitability of mapped sequence tagged microsatellite site markers for establishing distinctness, uniformity and stability in aromatic rice. Euphytica 135:135–143. https://doi.org/10.1023/B:EUPH.0000014905.10397.08
Soare R, Dinu M, Băbeanu C, Popescu M, Popescu A (2017) Nutritional value and antioxidant activities in fruit of some cultivars of pepper (Capsicum annuum L.). J Agroaliment Process Technol 23(4):217–222
Tanksley SD (1984) High rates of cross-pollination in chile pepper. HortScience 19:580–582
Tessier C, David J, This P, Boursiquot JM, Charrier A (1999) Optimization of the choice of molecular markers for varietal identification in Vitis vinifera L. Theor Appl Genet 98:171–177. https://doi.org/10.1007/s001220051054
van Zonneveld M, Ramírez M, Williams DE, Petz M, Meckelmann S, Avila T, Bejarano C, Ll Ríos, Peña K, Jäger M, Libreros D, Amaya K, Scheldeman X (2015) Screening genetic resources of Capsicum peppers in their primary center of diversity in Bolivia and Peru. PLoS ONE 10(9):e0134663. https://doi.org/10.1371/journal.pone.0134663
Walsh BM, Hoot SB (2001) Phylogenetic relationships of Capsicum (Solanaceae) using DNA sequences from two noncoding regions: the chloroplast atpB-rbcL spacer region and nuclear waxy introns. Int J Plant Sci 162(6):1409–1418. https://doi.org/10.1086/323273
Acknowledgements
This work was conducted under a cooperative agreement between Bioversity International and Cornell University. The authors want to thank to the members of the Jahn Lab at Cornell University, particularly to Mary Kreitinger, Sherry Roof and Michael Mazourek for their assistance in the lab, and Liam Hutchison for assistance with manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

{kind=link}
Cite this article
Guzmán, F.A., Moore, S., de Vicente, M.C. et al. Microsatellites to enhance characterization, conservation and breeding value of Capsicum germplasm. Genet Resour Crop Evol 67, 569–585 (2020). https://doi.org/10.1007/s10722-019-00801-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10722-019-00801-w