
Abstract
The glyoxalase system in the cytoplasm of cells provides the primary defence against glycation by methylglyoxal catalysing its metabolism to D-lactate. Methylglyoxal is the precursor of the major quantitative advanced glycation endproducts in physiological systems - arginine-derived hydroimidazolones and deoxyguanosine-derived imidazopurinones. Glyoxalase 1 of the glyoxalase system was linked to anthropometric measurements of obesity in human subjects and to body weight in strains of mice. Recent conference reports described increased weight gain on high fat diet-fed mouse with lifelong deficiency of glyoxalase 1 deficiency, compared to wild-type controls, and decreased weight gain in glyoxalase 1-overexpressing transgenic mice, suggesting a functional role of glyoxalase 1 and dicarbonyl stress in obesity. Increased methylglyoxal, dicarbonyl stress, in white adipose tissue and liver may be a mediator of obesity and insulin resistance and thereby a risk factor for development of type 2 diabetes and non-alcoholic fatty liver disease. Increased methylglyoxal formation from glyceroneogenesis on adipose tissue and liver and decreased glyoxalase 1 activity in obesity likely drives dicarbonyl stress in white adipose tissue increasing the dicarbonyl proteome and related dysfunction. The clinical significance will likely emerge from on-going clinical evaluation of inducers of glyoxalase 1 expression in overweight and obese subjects. Increased transcapillary escape rate of albumin and increased total body interstitial fluid volume in obesity likely makes levels of glycation of plasma protein unreliable indicators of glycation status in obesity as there is a shift of albumin dwell time from plasma to interstitial fluid, which decreases overall glycation for a given glycemic exposure.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
The obesity epidemic and related complications
In the last 30 years the causes of global premature death and loss of productive life, as assessed in disability adjusted life years reported by the World Health Organization, has changed from communicable diseases in children towards non-communicable diseases in adults. Impaired metabolic health – development of insulin resistance leading to type 2 diabetes (T2DM) is now a leading cause of disability-adjusted life years. A major risk factor for this is being overweight and obese (body mass index >25 kg/m2) in which insulin resistance is a common feature and contributor to development of T2DM [1]. The number of people who are overweight and obese worldwide is now >2.1 billion, a prevalence of ca. 37 % in the adult population and 13 % in adolescents and children in developing countries and 23 % in developed countries [2]. In 2010, overweight and obesity were estimated to cause 3.4 million deaths worldwide. Most deaths attributable to overweight and obesity are cardiovascular deaths; 36 % of the increased risk of coronary heart disease (CHD) mortality and 59 % of the increased risk of stroke mortality risk associated with obesity was linked to increased blood pressure and cholesterol, 14 % and 25 % of CHD and stroke excess mortality were directly linked to glucose and the remaining excess risk is unexplained [3]. A further complication of overweight and obesity is non-alcoholic fatty liver disease (NAFLD) leading to hepatic steatosis (NASH), cirrhosis, liver failure and cancer [4]. Obesity-induced insulin resistance is a major driver of development of T2DM and NAFLD. NAFLD affects 20–40 % of the population in Westernised countries and will likely increase direct and indirect medical costs by 25 % in the next 5 years. There is an urgent requirement for improved understanding of the risk factors of insulin resistance, obesity and NAFLD and to guide interventions to decrease incidence and health impact.
Obesity and the glyoxalase system
Recent reviews have described the impact of obesity on the glyoxalase system [5–8]. Herein consideration of the subject is advanced with description of recent experimental studies and findings with discussion of new underlying concepts, interpretation and inferences.
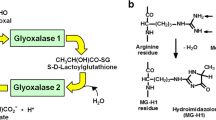
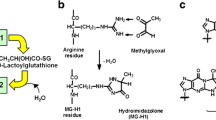
Genetic factors, dietary factors and early-life nutrition influence risk of insulin resistance and obesity. A gene functionally linked to obesity and diabetes is the glyoxalase 1 (Glo-1) gene, GLO-1 [9]. Glo-1 is part of the cytosolic glyoxalase system present in the cytoplasm of all mammalian cells – Fig. 1a. The glyoxalase system catalyses the metabolism of methylglyoxal (MG) to D-lactate via the intermediate S-D-lactoylglutathione and thereby suppresses the spontaneous modification of proteins and DNA by MG forming advanced glycation endproducts (AGEs). MG is the major precursor of AGEs in vivo, modifying mainly arginine residues in proteins to form hydroimidazolone MG-H1 - Fig. 1b, and deoxyguanosine residues in DNA to form imidazopurinones MGdG [10, 11] - Fig. 1c. Accumulation of MG adducts would otherwise cause protein dysfunction and mutagenesis. The function of the glyoxalase system is the enzymatic defence against MG glycation where Glo-1 catalyses the key step of removing the potentially damaging MG. Periods of increased MG concentration are called “dicarbonyl stress” produced by increased MG formation and/or decrease Glo-1 activity.

Biochemistry of dicarbonyl stress. a Metabolism of MG by the glyoxalase system. b Formation of hydroimidazolone MG-H1 from arginine residues. c Formation of imidazopurinone MGdG in DNA. Adduct residue is shown with guanyl base only
In human subjects, GLO-1 was linked to anthropometric measurements of obesity - upper-arm circumference and supra-iliac skinfold thickness [12]. In mice, meta-analysis of 34 mouse cross-breeding experiments linked GLO-1 to body weight [13]. Mice with a preference for a high energy-rich diet without marked health impairment have a relatively high expression of Glo-1 [5]. In the mouse overeating model of obesity, leptin mutant (ob/ob) mice, Glo-1 protein was decreased 80 % in the liver [14]. Recent conference reports described increased weight gain on high fat diet (HFD)-fed mouse with through-life expression of GLO-1 siRNA and mild Glo-1 deficiency, compared to wild-type controls [15], and decreased weight gain in Glo-1 overexpressing transgenic mice [16], suggesting a functional role of Glo-1 and dicarbonyl stress in obesity. We found increased MG concentration in hepatocyte-like hepatoma G2 cells in vitro incubated with saturated fatty acid and mono-unsaturated fatty acid, palmitic acid and oleic acid, respectively, suggesting that fatty acid metabolism may drive increased MG formation [17] – see below. HFD-fed wild-type mice had increased MG-H1 content of heart and liver, as judged by immunoassay [18]. Dicarbonyl stress may be a mediator of obesity and insulin resistance and thereby a risk factor for development of T2DM and NAFLD. Moreover, in a mouse model of hepatocellular carcinoma, Glo-1 was a tumour suppressor protein [19]. Hence, decrease of Glo-1 activity and hepatic dicarbonyl stress in NAFLD with progression to NASH may also increase risk of hepatocellular carcinoma.
Obesity and dicarbonyl stress
Several studies have attempted to model dicarbonyl stress in obesity by administration of exogenous MG. Difficulties performing such studies are: (i) lack of commercial availability of suitable high purity MG, (ii) interference-free assay of MG, and (iii) and judgement of an appropriate dose to administer. Commercial 40 % MG contains 9–17 mol% formaldehyde and many other compounds that potentially interfere in studies of dicarbonyl stress [20]. Methods for preparation of high purity MG and interference-free assay of MG have been described [21, 22]. The flux of endogenous formation of MG has been estimated at ca. 3–6 mg/kg (ca. 0.05 % glycolytic rate, which we find relatively constant in many cell types) [23]. Experimental studies have often used 10–20 fold higher than this – which is likely similar to and exceeds the upper limit of severe dicarbonyl stress of poorly-controlled clinical diabetes and end stage renal disease [24, 25]. MG formation of cells with GLUT1 glucose transport increased only 2–3 fold in the high glucose concentration characteristic of T2DM and MG concentration in blood of patients with T2DM showed a similar 2–3 fold increase [24, 26].
Infusion of MG (60 mg/kg/day) into healthy rats induced impaired glucose tolerance, decreased glucose transporter GLUT-4, phosphoinositide-3-kinase activity, and insulin-stimulated glucose uptake in adipose tissue [27]. Administration of exogenous MG (50–75 mg/kg, daily, i.p.) induced insulin resistance in mice [28], inhibited insulin-stimulated phosphorylation of protein kinase B and extracellularly-regulated kinase, contributing to insulin resistance in muscle cells [29]. It also inhibited insulin-induced insulin receptor substrate tyrosine phosphorylation and phosphatidylinositol 3-kinase/protein kinase B pathway activation in pancreatic beta-cells [30], increased free fatty acids, hypoadiponectinemia, hypoxia and macrophage recruitment of adipose tissue [31]. These levels of MG exposure also arrested growth of rats, impaired renal function, induced hypercholesterolaemia and impaired vasodilation. There were also degenerative changes in cutaneous capillaries with loss of endothelial cells, basement membrane thickening, luminal occlusion and inflammatory response – increased receptor for AGE (RAGE), interleukin-1ß, tumour necrosis factor-α and connective tissue growth factor in medial layers of arteries, and transforming growth factor-ß in glomerular tufts, tubular epithelial cells and interstitial endothelial cells [32]. These MG administration models to date, therefore, explore features of MG intoxication. Some of the features produced may be similar to those developing in obesity – although they are likely markedly less severe.
Moderate dicarbonyl stress in clinical obesity
To investigate dicarbonyl stress in clinical obesity we recruited obese and non-obese healthy human subjects and placed them on an isocaloric diet for 2 weeks. Blood samples were collected after overnight fasting and plasma prepared. Plasma MG was determined by stable isotopic dilution analysis liquid chromatography-tandem mass spectrometry (LC-MS/MS) [22] and plasma D-lactate concentration by endpoint enzymatic assay [33]. Plasma MG was increased 35 % in obese subjects and D-lactate was increased ca. 2-fold – Table 1. Plasma MG levels were intermediate between those found in non-obese healthy subjects and patients with type 1 or type 2 diabetes [22, 34, 35], suggesting clinical obesity is a state of moderate dicarbonyl stress. Plasma D-lactate levels, a surrogate and qualitative indicator of MG flux, also supported this [24, 35, 36] and suggest the flux of formation of MG is increased in obesity. (D-Lactate is metabolised in human subjects [37]). The MG-H1 residue content of plasma protein was determined in obese and non-obese subjects by exhaustive enzymatic hydrolysis of plasma protein and MG-H1 detection and quantitation by and stable isotopic dilution analysis LC-MS/MS [38] and no significant difference was found.
Estimates of MG and in white adipose tissue (WAT) in experimental models of overfeeding, HFD fed mice, were ca. 5000 nmol per g tissue versus ca. 2000 nmol per g tissue in controls – equivalent to ca. 5 mM and 2 mM MG, respectively [39]. Our estimates of MG in WAT of the same experimental model by the reference protocol [22] are markedly lower (mean ± SD): 2.91 ± 0.98 (n = 7) versus 1.47 ± 0.65 (n = 8, P < 0.01); equivalent to ca. 3 and 1.5 μM, respectively. We also found markedly lower levels of glyoxal in WAT but similar levels of 3-deoxyglucosone (Masania, J., Rabbani, N., Rossmeisl, M., Kopecky, J. and Thornalley, P.J.; unpublished observations). Mathematical modelling of MG formation and protein glycation in mouse tissue predicted MG concentrations in the 1–2 μM range in normal metabolism and are likely increased 2–3 fold in obesity [22]. Hence markedly higher estimates are not sustainable metabolically and may have been caused by interference – MG and glyoxal formation in pre-analytic processing. Trichloroacetic acid deproteinization and azide blocking of acid-stable peroxidase is required to avoid overestimation of MG and glyoxal in mouse tissues [40]. This requires further investigation.
Plasma protein glycation adducts as reporters of glycation exposure in obesity
Glycated albumin
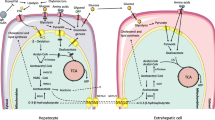
In clinical obesity as insulin resistance develops, there is progressive decline in glycemic control with impaired fasting and postprandial hyperglycemia – as detected by continuous glucose monitoring. There is a moderate increase in glycated hemoglobin (A1C) as prediabetes develops [41] but surprisingly there is often a decrease in glycated albumin with increased BMI although A1C is increased. This is found both in obese adults and children [42, 43] and lower than expected glycated albumin in obesity also extends into the patients with T2DM [43]. The steady-state level of albumin glycation in plasma depends on the increased glucose concentration in plasma and duration over which it occurs, and also the residence time of albumin in the plasma compartment – as judged by the albumin transcapillary escape rate (TER). Until degraded with a half-life of ca. 20 days [44], albumin cycles from plasma into interstitial fluid, lymph and returns to plasma – with some leakage through renal glomeruli and return to venous circulation by the renal albumin retrieval pathway [45]. The rate of glycation by glucose normally is 4-fold higher in the plasma compared to interstitial fluid so that decrease in dwell time in plasma by increased TER may decrease glycation without change in plasma glucose concentration [45, 46] – Fig. 2. The rate of glycation of albumin by glucose, rGlycation, is directly proportional to the concentration of glucose and the concentration of albumin; rGlycation = kGlycation [Glucose][Albumin], where kGlycation is the rate constant for glycation of albumin by glucose. The concentrations of albumin and glucose are 2.7-fold and 1.4 fold higher in plasma than interstitial fluid, which multiplied together indicate that the rate of glycation by glucose normally is 3.8 fold or ca. 4-fold higher in the plasma compared to interstitial fluid. Albumin TER is increased by hypertension (from 5.6 % per hour to 7.6 % per h) and plasma volume decreases by up to 10 % [47]. It is also increased in overweight/obese subjects with metabolic syndrome [48]. Obesity also increases the total body interstitial fluid volume [49]. This suggests that the explanation for decreased plasma glycated albumin in obesity with increased glucose exposure, as indicated by decreased glycated albumin/A1C ratio, may be due to a shift of albumin residence time from plasma to interstitial fluid in favour of the latter. This suggests glycated albumin is an unreliable marker of glycemic control in obesity.

Glycation of albumin by glucose – glycation kinetics and dynamics in vascular and extravascular compartments. Physiological data from [45, 46]. Relative glycation kinetics deduced from: rGlycation = k [Glucose][albumin], k is the glycation rate constant and assuming rGlycation in the plasma compartment =100 %
The rate of degradation of albumin in obesity and diabetes may also influence the level of glycated albumin if changed. In obese subjects and patients with diabetes, with normal renal function, the synthesis of albumin and plasma concentration of albumin are unchanged from those of lean healthy controls, suggesting that the rate of degradation of albumin is also normal [50, 51]. In experimental diabetes early studies of Baynes and co-workers and others showed that the rate of degradation of albumin was not increased but slightly lower in streptozotocin-induced diabetic rats than in healthy controls [52, 53]. So there is no evidence that an increased rate of albumin degradation that contributes to decreased glycated albumin or AGE-modified albumin in obesity and diabetes. This may change, however, with impaired renal function with increased urinary loss of albumin and compensatory increased albumin synthesis [54].
AGEs in plasma/serum protein in obesity
A shift of albumin residence time from plasma to interstitial fluid may also influence AGEs. Nε-Carboxymethyl-lysine (CML) residue content of serum protein was inversely linked to BMI and body fat mass [55, 56]. In studies where plasma early glycated (glycated albumin), CML and florescent AGE content were determined, all were decreased in obese subjects compared to controls [57]. Decrease of CML residue content of plasma protein in obesity was confirmed in an independent study and association with central obesity and inflammation [58]. Decreased CML residue content of plasma protein in obesity has been explained as due to enlargement of adipose tissue mass in obesity [59] and a contributory feature to this is likely decreased glycation of albumin due to the shift of albumin from plasma to interstitial fluid. An association with inflammation is also expected as albumin TER increases with increased capillary permeability in vascular inflammation [60]. Decreased residence time of albumin in the vascular lumen in obese subjects may also explain herein how plasma MG is increased without increase in MG-H1 residue content of plasma protein in our study – Table 1. This is supported by a recent study with a Glo-1 inducer where plasma MG concentration and whole body endogenous formation of MG-H1 adduct flux was decreased without decrease in plasma protein MG-H1 residue content. This was attributed to improved vascular function, decreased albumin TER and increased plasma dwell time of albumin with Glo-1 inducer intervention [61]. Given this, it is also likely that plasma protein AGE content in obesity is not a reliable indicator of AGE tissue exposure – as found previously [59].
Source of dicarbonyl stress in obesity and its likely effects
Our finding of increased plasma MG and D-lactate concentrations in obese human subjects compared to non-obese subjects on an isocaloric diet suggests the source of dicarbonyl stress in obesity is not of dietary origin. Indeed, recent studies of metabolic transit of MG indicate that dietary MG is metabolised and/or reacts with protein in the intestinal lumen and has limited bioavailability [62]. Moreover the insulin resistance in obesity suggests the usually dominant source of MG formation from increased flux through anaerobic glycolysis may not be increased. Triosephosphates, however, are not limited to intermediates of anaerobic glycolysis but are also intermediates of glyceroneogenesis and gluconeogenesis. Increased glyceroneogenesis is associated with adipocyte expansion in obesity supporting increased fatty acid esterification for triglyceride deposition [63]. Most triglyceride synthesis involves glyceroneogenesis via triosephosphate intermediates [64]. Glyceroneogenesis is not limited, however, because it uses pyruvate as the carbon source, which has unrestricted entry and metabolism into major cell types experiencing insulin resistance – such as adipocytes, hepatocytes and skeletal muscle cells. As flux of glyceroneogenesis increases to support triglyceride synthesis, there is proportionate increase in MG formation by related increase in flux of triosephosphate formation. Most triosephosphate intermediates go on to form glycero-l-3-phosphate but with 0.05 % of triosephosphate degrading to MG, when the flux through glyceroneogenesis increases then concomitantly the flux of MG formation also increases. Increased glyceroneogenesis, therefore, is likely a major driver of increased MG formation in obesity – Fig. 3.

Increased formation of methylglyoxal in the triglyceride/free fatty acid cycle. Percentage flux of glyceroneogenesis in triglyceride formation in liver and adipose tissue is from [64]
There is no inconsistency with previous findings of increased MG from red blood cells in high glucose concentration as therein MG is formed from increased concentrations of triosephosphates driven by increased uptake and metabolism of glucose in anaerobic glycolysis. Red blood cells do not suffer impaired glucose metabolism in insulin resistance. MG is mainly formed non-enzymatically from the same precursors in all cells and tissues – triosephosphates – but the pathways that sustain triosephosphate concentrations differ and may be multiple in cells and tissues.
In experimental models of obesity there is evidence of decreased Glo-1 activity in visceral adipose tissue [16]. Human Glo-1 protein is a dimer of 42 kDa. It undergoes post-translational modifications: C139 may form a mixed disulfide with GSH, inhibiting Glo-1 activity in vitro . Glo-1 may be S-nitrosylated on C139 and is a substrate for calcium, calmodulin-dependent protein kinase II, with phosphorylation at T107. There is acetylation at K148 [65] and likely de-acetylation by cytosolic sirtuin-2 [66]. Glo-1 down regulation in obesity may be driven through hypoxia signalling by hypoxia-inducible factor-1α (HIF1α), which down-regulates Glo-1 expression [67]. As adipose tissue expands, interstitial oxygen tension decreases. HIF1α protein is highly enriched in expanding adipocytes as it drives increased adipose tissue vascularization. Adipocyte-specific deletion of HIF1α decreased HFD–induced adipose tissue inflammation and insulin resistance [68]. Increased MG formation by glyceroneogenesis and decreased Glo-1 expression through HIF1α signalling therefore provides the conditions for dicarbonyl stress in obesity.
The consequences of dicarbonyl stress in white adipose tissue (WAT) are unknown but contribute to insulin resistance. Obesity is associated with decreased activity of the insulin sensitising effects of fibroblast growth factor-21 (FGF21) due to down regulation of the FGF21 receptor cofactor β-Klotho [69]. Decreased FGF21 activity also impairs uncoupling protein-1 expression in brown adipose tissue, energy utilisation for thermogenesis [70] and facilitating fat deposition and weight gain. MG-driven protein glycation decreased expression of β-Klotho [71] and thereby likely contributes to insulin resistance. Decreased β-Klotho is also permissible for induction of pro-inflammatory mediators, interleukin-8, monocyte chemotactic protein-1, intracellular adhesion molecule-1 and receptor for AGEs, RAGE [71]. Overexpression of Glo-1 prevented insulin resistance and inflammation in HFD-fed mice, suggesting a functional role of dicarbonyl stress in obesity. This will be tested clinically by evaluation of Glo-1 inducer therapeutics.
In dicarbonyl stress there is increased protein glycation by MG. MG modification is directed to arginine residues often at functional sites of proteins and leads to functional change or inactivation. Protein targets of MG modification are called the dicarbonyl proteome [72]. The extent of protein glycation by MG is usually 1–2 % but low level increases can have profound physiological effect [73]. Examples are: MG modification of mitochondrial proteins, which increases formation of reactive oxygen species and oxidative damage [74]; modification of extracellular matrix proteins produces endothelial cell detachment with exposure of the sub-endothelium, platelet activation and thrombosis [72]; and modification of apolipoprotein B100 of low density lipoprotein (LDL) producing a atherogenic transformation to small, dense LDL [75]. Moreover, MG modification of proteins stimulates their proteolysis, decreasing protein half-life and thereby concentrations of the unmodified protein unless there is compensatory increased transcription - as found for apolipoprotein A1 of high density lipoprotein [76]. In relation to this, increasing endogenous MG by Glo-1 silencing in aortic endothelial cells changed expression of >400 genes [77]. Dicarbonyl stress, therefore, has proteome, dicarbonyl proteome and transcriptome signatures.
Increased MG formation from glyceroneogenesis on adipose tissue and liver and decreased Glo-1 activity in obesity likely drives dicarbonyl stress in WAT increasing the dicarbonyl proteome and related dysfunction. The functional significance of this is indicated by protection from insulin resistance, inflammation and weight gain in Glo-1 transgenic mice in experimental model of over-eating induced obesity. The clinical significance will likely emerge from on-going clinical evaluation of inducers of Glo-1 expression in overweight and obese subjects – for example, Clinicaltrials.gov; NCT02095873.
Summary
MG metabolism and the glyoxalase system are disturbed in obesity leading to dicarbonyl stress. Functional genomics studies with Glo-1 in the overfeeding model of HFD-fed mice suggest dicarbonyl stress is a risk factor for health impairment and complications of obesity. Likely drivers of dicarbonyl stress in obesity are: increased formation of MG from increased glycerogenesis in triglyceride synthesis and decreased Glo-1 expression and activity through hypoxia and inflammatory signalling. A recent clinical intervention study with a Glo-1 inducer produced a profound improvement of insulin resistance, improved glycemic control and arterial function and decreased vascular inflammation, suggesting that Glo-1 inducer therapeutics may have a future key role in alleviating complications of obesity. Increased albumin TER in obesity associated with increased fat mass, hypertension and inflammation suggest glycated albumin is not a reliable measure of glycemic control and formation of AGEs in obesity.
Abbreviations
- A1C:
-
Glycated hemoglobin
- AGEs:
-
Advanced glycation endproducts
- CHD:
-
Coronary heart disease
- CML:
-
Nε-Carboxymethyl-lysine
- DHAP:
-
Dihydroxyacetonephosphate
- FFA:
-
Free fatty acid
- FGF21:
-
Fibroblast growth factor-21
- GA3P:
-
Glyceraldehyde-3-phosphate
- Glo-1:
-
Glyoxalase 1
- HFD:
-
High fat diet
- HIF1α:
-
Hypoxia-inducible factor-1α
- LC-MS/MS:
-
Liquid chromatography-tandem mass spectrometry
- MG:
-
Methylglyoxal
- MGdG:
-
3-(2′-deoxyribosyl)-6,7-dihydro-6,7-dihydroxy-6/7-methylimidazo-[2,3-b]purine-9(8)one
- MG-H1:
-
Nδ-(5-hydro-5-methyl-4-imidazolon-2-yl)-ornithine
- NAFLD:
-
Non-alcoholic fatty liver disease
- NASH:
-
Hepatic steatosis
- RAGE:
-
Receptor for advanced glycation endproducts
- T2DM:
-
Type 2 diabetes
- TG:
-
Triglyceride
- TER:
-
Transcapillary escape rate
- WAT:
-
White adipose tissue
References
WHO: World Health Statistics. (2012) In., pp. 1–176. WHO Press, Geneva, Switzerland
Ng, M., Fleming, T., Robinson, M., Thomson, B., Graetz, N., Margono, C., Mullany, E.C., Biryukov, S., Abbafati, C., Abera, S.F., Abraham, J.P., Abu-Rmeileh, N.M.E., Achoki, T., AlBuhairan, F.S., Alemu, Z.A., Alfonso, R., Ali, M.K., Ali, R., Guzman, N.A., Ammar, W., Anwari, P., Banerjee, A., Barquera, S., Basu, S., Bennett, D.A., Bhutta, Z., Blore, J., Cabral, N., Nonato, I.C., Chang, J.-C., Chowdhury, R., Courville, K.J., Criqui, M.H., Cundiff, D.K., Dabhadkar, K.C., Dandona, L., Davis, A., Dayama, A., Dharmaratne, S.D., Ding, E.L., Durrani, A.M., Esteghamati, A., Farzadfar, F., Fay, D.F.J., Feigin, V.L., Flaxman, A., Forouzanfar, M.H., Goto, A., Green, M.A., Gupta, R., Hafezi-Nejad, N., Hankey, G.J., Harewood, H.C., Havmoeller, R., Hay, S., Hernandez, L., Husseini, A., Idrisov, B.T., Ikeda, N., Islami, F., Jahangir, E., Jassal, S.K., Jee, S.H., Jeffreys, M., Jonas, J.B., Kabagambe, E.K., Khalifa, S.E.A.H., Kengne, A.P., Khader, Y.S., Khang, Y.-H., Kim, D., Kimokoti, R.W., Kinge, J.M., Kokubo, Y., Kosen, S., Kwan, G., Lai, T., Leinsalu, M., Li, Y., Liang, X., Liu, S., Logroscino, G., Lotufo, P.A., Lu, Y., Ma, J., Mainoo, N.K., Mensah, G.A., Merriman, T.R., Mokdad, A.H., Moschandreas, J., Naghavi, M., Naheed, A., Nand, D., Narayan, K.M.V., Nelson, E.L., Neuhouser, M.L., Nisar, M.I., Ohkubo, T., Oti, S.O., Pedroza, A., Prabhakaran, D., Roy, N., Sampson, U., Seo, H., Sepanlou, S.G., Shibuya, K., Shiri, R., Shiue, I., Singh, G.M., Singh, J.A., Skirbekk, V., Stapelberg, N.J.C., Sturua, L., Sykes, B.L., Tobias, M., Tran, B.X., Trasande, L., Toyoshima, H., van de Vijver, S., Vasankari, T.J., Veerman, J.L., Velasquez-Melendez, G., Vlassov, V.V., Vollset, S.E., Vos, T., Wang, C., Wang, X., Weiderpass, E., Werdecker, A., Wright, J.L., Yang, Y.C., Yatsuya, H., Yoon, J., Yoon, S.-J., Zhao, Y., Zhou, M., Zhu, S., Lopez, A.D., Murray, C.J.L., Gakidou, E.: Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384(9945), 766–781 (2014)
Lu Y., Hajifathalian K., Ezzati M., Woodward M., Rimm E.B., Danaei G., Global Burden Metab Risk Factors, C.: Metabolic mediators of the effects of body-mass index, overweight, and obesity on coronary heart disease and stroke: a pooled analysis of 97 prospective cohorts with 1.8 million participants. Lancet. 383(9921), 970–983 (2014)
Karlas T., Wiegand J., Berg T.: Gastrointestinal complications of obesity: non-alcoholic fatty liver disease (NAFLD) and its sequelae. Best Pract. Res. Clin. Endocrinol. Metab. 27(2), 195–208 (2013)
Rabbani N., Thornalley P.J.: Glyoxalase in diabetes, obesity and related disorders. Semin. Cell Dev. Biol. 22(3), 309–317 (2011)
Rabbani N., Xue M., Thornalley P.J.: Activity, regulation, copy number and function in the glyoxalase system. Biochem. Soc. Trans. 42(2), 419–424 (2014)
Maessen D.E.M., Stehouwer C.D.A., Schalkwijk C.G.: The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin. Sci. 128(12), 839–861 (2015)
Rabbani N., Thornalley P.J.: Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophys. Res. Commun. 458(2), 221–226 (2015)
Rankinen T., Zuberi A., Chagnon Y.C., Weisnagel S.J., Argyropoulos G., Walts B., Perusse L., Bouchard C.: The human obesity Gene map: the 2005 update. Obesity. 14(4), 529–644 (2006)
Thornalley P.J., Battah S., Ahmed N., Karachalias N., Agalou S., Babaei-Jadidi R., Dawnay A.: Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 375(3), 581–592 (2003)
Thornalley P.J., Waris S., Fleming T., Santarius T., Larkin S.J., Winklhofer-Roob B.M., Stratton M.R., Rabbani N.: Imidazopurinones are markers of physiological genomic damage linked to DNA instability and glyoxalase 1-associated tumour multidrug resistance. Nucleic Acids Res. 38(16), 5432–5442 (2010)
Wilson A.F., Elston R.C., Tran L.D., Siervogel R.M.: Use of the robust sib-pair method to screen for single-locus, multiple-locus, and pleiotropic effects: application to traits related to hypertension. Am. J. Human Genet. 48(5), 862–872 (1991)
Wuschke S., Dahm S., Schmidt C., Joost H.G., Al Hasani H.: A meta-analysis of quantitative trait loci associated with body weight and adiposity in mice. Int. J. Obes. 31(5), 829–841 (2006)
Sanchez J.C., Converse V., Nolan A., Schmid G., Wang S., Heller M., Sennitt M.V., Hochstrasser D.F., Cawthorne M.A.: Effect of rosiglitazone on the differential expression of diabetes-associated proteins in pancreatic islets of C57BI/6 lep/lep mice. Mol. Cell. Proteomics. 1(7), 509–516 (2002)
Wortmann, M., Hakimi, M., Fleming, T., Peters, A., Nawroth, P., Bockler, D., Dihlmann, S.: The role of glyoxalase-1 (Glo-1) in mouse metabolism and atherosclerosis. Biochem. Soc. Trans., www.biochemistry.org/Portals/0/Conferences/abstracts/SA158/SA158P005.pdf (2013)
Maessen, D., Brouwers, O., Miyata, T., Stehouwer, C., Schalkwijk, C.: Glyoxalase-1 overexpression reduces body weight and adipokine expression, and improves insulin sensitivity in high-fat diet-induced obese mice. Diabetologia 57, Supplement 1, 713 (2014)
Maldonado, E.M., Rabbani, N., Thornalley, P.J., Wang, H., Miller, M., Dillon, J.F., Moore, J.B.: Examination of methylglyoxal levels in an in vitro model of steatosis and serum from patients with non-alcoholic fatty liver disease. Proc. Nutrit. Soc. 74(OCE1), E1 (2015)
Li S.Y., Liu Y., Sigmon V.K., McCort A., Ren J.: High-fat diet enhances visceral advanced glycation end products, nuclear O-Glc-Nac modification, p38 mitogen-activated protein kinase activation and apoptosis. Diabetes Obes. Metab. 7(4), 448–454 (2005)
Zender L., Xue W., Zuber J., Semighini C.P., Krasnitz A., Ma B., Zender P., Kubicka S., Luk J.M., Schirmacher P., Richard McCombie W., Wigler M., Hicks J., Hannon G.J., Powers S., Lowe S.W.: An Oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell. 135(5), 852–864 (2008)
Pourmotabbed T., Creighton D.J.: Substrate specificity of bovine liver formaldehyde dehydrogenase. J. Biol. Chem. 261(30), 14240–14244 (1986)
McLellan A.C., Thornalley P.J.: Synthesis and chromatography of 1,2-diamino-4,5-dimethoxybenzene, 6,7-dimethoxy-2-methylquinoxaline and 6,7-dimethoxy-2,3-dimethylquinoxaline for use in a liquid chromatographic fluorimetric assay of methylglyoxal. Anal. Chim. Acta. 263(1–2), 137–142 (1992)
Rabbani N., Thornalley P.J.: Measurement of methylglyoxal by stable isotopic dilution analysis LC-MS/MS with corroborative prediction in physiological samples. Nat. Protoc. 9(8), 1969–1979 (2014)
Rabbani N., Thornalley P.J.: Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids. 42(4), 1133–1142 (2012)
McLellan A.C., Thornalley P.J., Benn J., Sonksen P.H.: The glyoxalase system in clinical diabetes mellitus and correlation with diabetic complications. Clin. Sci. 87(1), 21–29 (1994)
Rabbani, N., Thornalley, P.J.: Dicarbonyls (glyoxal, methylglyoxal, and 3-Deoxyglucosone). In: Uremic Toxins. pp. pp. 177–192. John Wiley & Sons, Inc., (2012)
Shinohara M., Thornalley P.J., Giardino I., Beisswenger P.J., Thorpe S.R., Onorato J., Brownlee M.: Overexpression of glyoxalase I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycaemia-induced increases in macromolecular endocytosis. J. Clin. Invest. 101(5), 1142–1147 (1998)
Dhar A., Dhar I., Jiang B., Desai K.M., Wu L.: Chronic methylglyoxal infusion by Minipump causes pancreatic beta-cell dysfunction and induces type 2 diabetes in Sprague-Dawley rats. Diabetes. 60(3), 899–908 (2011)
Nigro C., Raciti G., Leone A., Fleming T., Longo M., Prevenzano I., Fiory F., Mirra P., D’Esposito V., Ulianich L., Nawroth P., Formisano P., Beguinot F., Miele C.: Methylglyoxal impairs endothelial insulin sensitivity both in vitro and in vivo. Diabetologia. 57(7), 1485–1494 (2014)
Riboulet-Chavey A., Pierron A., Durand I., Murdaca J., Giudicelli J., Van Obberghen E.: Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes. 55(5), 1289–1299 (2006)
Fiory, F., Lombardi, A., Miele, C., Giudicelli, J., Beguinot, F., Van Obberghen, E.: Methylglyoxal impairs insulin signalling and insulin action on glucose-induced insulin secretion in the pancreatic beta cell line INS-1E. Diabetologia 54(11), 2941–2952.
Matafome P., Santos-Silva D., Crisostomo J., Rodrigues T., Rodrigues L., Sena C.M., Pereira P., Seica R.: Methylglyoxal causes structural and functional alterations in adipose tissue independently of obesity. Arch. Physiol. Biochem. 118(2), 58–68 (2012)
Berlanga J., Cibrian D., Guillen I., Freyre F., Alba J.S., Merino N., Aldama A., Quintela A.M., Triana M.E., Ajameich H., Lopez P., Ahmed N., Thornalley P.J.: Methylglyoxal administration induces diabetes-like microvascular changes and perturbs the healing process of skin wounds. Clin. Sci. 109(1), 83–95 (2005)
McLellan A.C., Phillips S.A., Thornalley P.J.: Fluorimetric assay of D-lactate. Anal. Biochem. 206(1), 12–16 (1992)
Bierhaus A., Fleming T., Stoyanov S., Leffler A., Babes A., Neacsu C., Sauer S.K., Eberhardt M., Schnolzer M., Lasischka F., Neuhuber W.L., Kichko T.I., Konrade I., Elvert R., Mier W., Pirags V., Lukic I.K., Morcos M., Dehmer T., Rabbani N., Thornalley P.J., Edelstein D., Nau C., Forbes J., Humpert P.M., Schwaninger M., Ziegler D., Stern D.M., Cooper M.E., Haberkorn U., Brownlee M., Reeh P.W., Nawroth P.P.: Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 18(6), 926–933 (2012)
Beisswenger P.J., Howell S.K., Touchette A., Lal S., Szwergold B.S.: Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes. 48(1), 198–202 (1999)
Scheijen, J.L.J.M., Hanssen, N.M.J., van de Waarenburg, M.P.H., Jonkers, D.M.A.E., Stehouwer, C.D.A., Schalkwijk, C.G.: L(+) and D(−) lactate are increased in plasma and urine samples of type 2 diabetes as measured by a simultaneous quantification of L(+) and D(−) lactate by reversed-phase liquid chromatography tandem mass spectrometry. Exp. Diabetes Res. 2012, 234812 (2012)
Oh M.S., Uribarri J., Alveranga D., Lazar I., Bazilinski N., Carroll H.J.: Metabolic utilisation and renal handling of D-lactate in man. Metab. Clin. Exp. 34, 621–625 (1985)
Rabbani N., Shaheen F., Anwar A., Masania J., Thornalley P.J.: Assay of methylglyoxal-derived protein and nucleotide AGEs. Biochem. Soc. Trans. 42(2), 511–517 (2014)
Maessen D.E., Brouwers O., Gaens K.H., Wouters K., Cleutjens J.P., Janssen B.J., Miyata T., Stehouwer C.D., Schalkwijk C.G.: Delayed intervention with Pyridoxamine improves metabolic function and prevents adipose tissue inflammation and insulin resistance in high-fat diet–induced obese mice. Diabetes. 65(4), 956–966 (2016)
Thornalley P.J., Rabbani N.: Assay of methylglyoxal and glyoxal and control of peroxidase interference Biochem. Soc. Transit. 42(2), 504–510 (2014)
Fysekidis M., Cosson E., Banu I., Duteil R., Cyrille C., Valensi P.: Increased glycemic variability and decrease of the postprandial glucose contribution to HbA1c in obese subjects across the glycemic continuum from normal glycemia to first time diagnosed diabetes. Metabolism. 63(12), 1553–1561 (2014)
Wang F., Ma X., Hao Y., Yang R., Ni J., Xiao Y., Tang J., Bao Y., Jia W.: Serum glycated albumin is inversely influenced by fat mass and visceral adipose tissue in Chinese with normal glucose tolerance. PLoS One. 7(11), e51098 (2012)
Miyashita Y., Nishimura R., Morimoto A., Matsudaira T., Sano H., Tajima N.: Glycated albumin is low in obese, type 2 diabetic patients. Diabetes Res. Clin. Pract. 78(1), 51–55 (2007)
Peters T.: All about albumin. Academic Press, New York (1996)
Margarson M.P., Soni N.: Serum albumin: touchstone or totem? Anaesthesia. 53(8), 789–803 (1998)
Maggs D.G., Jacob R., Rife F., Lange R., Leone P., During M.J., Tamborlane W.V., Sherwin R.S.: Interstitial fluid concentrations of glycerol, glucose, and amino acids in human quadricep muscle and adipose tissue. Evidence for significant lipolysis in skeletal muscle. J. Clin. Invest. 96(1), 370–377 (1995)
Parving H.-H., Gyntelberg F.: Transcapillary escape rate of albumin and plasma volume in essential hypertension. Circ. Res. 32(5), 643–652 (1973)
Dell’Omo G., Penno G., Pucci L., Mariani M., Del Prato S., Pedrinelli R.: Abnormal capillary permeability and endothelial dysfunction in hypertension with comorbid metabolic syndrome. Atherosclerosis. 172(2), 383–389 (2004)
Bhave G., Neilson E.G.: Body fluid dynamics: back to the future. J. Amer. Soc. Nephrol. 22(12), 2166–2181 (2011)
Tessari P., Cosma A., Vettore M., Millioni R., Puricelli L., Cogo P., Cecchet D., Carnielli V., Kiwanuka E.: Fibrinogen kinetics and protein turnover in obese non-diabetic males: effects of insulin. Diabetes Metab. Res. Rev. 26(1), 50–58 (2010)
Tessari P., Kiwanuka E., Millioni R., Vettore M., Puricelli L., Zanetti M., Gucciardi A., Tosolini M., Cogo P., Carnielli V., Tiengo A., Barazzoni R.: Albumin and fibrinogen synthesis and insulin effect in type 2 diabetic patients with Normoalbuminuria. Diabetes Care. 29(2), 323–328 (2006)
Murtiashaw M.H., Baynes J.W., Thorpe S.R.: Albumin catabolism in diabetic rats. Arch. Biochem. Biophys. 225(1), 256–262 (1983)
Johnson R.N., Easdale R.W., Tatnell M., Baker J.R.: Significance of variation in turnover of glycated albumin on indexes of diabetic control. Clin. Chim. Acta. 198(3), 229–238 (1991)
Tessari P., Kiwanuka E., Barazzoni R., Vettore M., Zanetti M.: Diabetic nephropathy is associated with increased albumin and fibrinogen production in patients with type 2 diabetes. Diabetologia. 49(8), 1955–1961 (2006)
Semba R.D., Arab L., Sun K., Nicklett E.J., Ferrucci L.: Fat mass is inversely associated with serum Carboxymethyl-lysine, an advanced glycation end product, in adults. J. Nutr. 141(9), 1726–1730 (2011)
Accacha S., Rosenfeld W., Jacobson A., Michel L., Schnurr F.J., Shelov S., Ten S., Boucher-Berry C., Carey D.E., Speiser P.W., Lowell B., Conroy R., Klein M., Fennoy I., Rapaport R., Rosenbaum M.: Plasma advanced glycation end products (AGEs), receptors for AGEs and their correlation with inflammatory markers in middle school-age children. Horm. Res. Paediatr. 80(5), 318–327 (2013)
Sebekova K., Somoza V., Jarcuskova M., Heidland A., Podracka L.: Plasma advanced glycation end products are decreased in obese children compared with lean controls. Int. J. Pediatr. Obes. 4(2), 112–118 (2009)
Gaens K.H.J., Ferreira I., van de Waarenburg M.P.H., van Greevenbroek M.M., van der Kallen C.J.H., Dekker J.M., Nijpels G., Rensen S.S., Stehouwer C.D.A., Schalkwijk C.G.: Protein-bound plasma N-epsilon-(Carboxymethyl) lysine is inversely associated with central obesity and inflammation and significantly explain a part of the central obesity-related increase in inflammation the Hoorn and CODAM studies. Arterioscler. Thromb. Vasc. Biol. 35(12), 2707–2713 (2015)
Gaens K.H.J., Goossens G.H., Niessen P.M., van Greevenbroek M.M., van der Kallen C.J.H., Niessen H.W., Rensen S.S., Buurman W.A., Greve J.W.M., Blaak E.E., van Zandvoort M.A., Bierhaus A., Stehouwer C.D.A., Schalkwijk C.G.: Nε-(Carboxymethyl)lysine-receptor for advanced glycation end product Axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arterioscler. Thromb. Vasc. Biol. 34(6), 1199–1208 (2014)
Nicholson J.P., Wolmarans M.R., Park G.R.: The role of albumin in critical illness. Brit. J. Anaesthesia. 85(4), 599–610 (2000)
Xue M., Weickert M.O., Qureshi S., Ngianga-Bakwin K., Anwar A., Waldron M., Shafie A., Messenger D., Fowler M., Jenkins G., Rabbani N., Thornalley P.J.: Improved glycemic control and vascular function in overweight and obese subjects by glyoxalase 1 inducer formulation. Diabetes. (2016). doi:10.2337/db16-0153
Degen J., Vogel M., Richter D., Hellwig M., Henle T.: Metabolic transit of dietary methylglyoxal. J. Agric. Food Chem. 61(43), 10253–10260 (2013)
Franckhauser S., Muñoz S., Elias I., Ferre T., Bosch F.: Adipose overexpression of phosphoenolpyruvate Carboxykinase leads to high susceptibility to diet-induced insulin resistance and obesity. Diabetes. 55(2), 273–280 (2006)
Nye C., Kim J., Kalhan S.C., Hanson R.W.: Reassessing triglyceride synthesis in adipose tissue. Trends Endocrinol. Metab. 19(10), 356–361 (2008)
Lundby A., Lage K., Weinert B.T., Bekker-Jensen D.B., Secher A., Skovgaard T., Kelstrup C.D., Dmytriyev A., Choudhary C., Lundby C., Olsen J.V.: Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2(2), 419–431 (2012)
Rauh D., Fischer F., Gertz M., Lakshminarasimhan M., Bergbrede T., Aladini F., Kambach C., Becker C.F.W., Zerweck J., Schutkowski M., Steegborn C.: An acetylome peptide microarray reveals specificities and deacetylation substrates for all human sirtuin isoforms. Nat. Commun. 4, 2327 (2013)
Zhang H., Li H., Xi H.S., Li S.: HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood. 119(11), 2595–2607 (2012)
Lee Y.S., Kim J.-w., Osborne O., Oh D.Y., Sasik R., Schenk S., Chen A., Chung H., Murphy A., Watkins S.M., Quehenberger O., Johnson R.S., Olefsky J.M.: Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell. 157(6), 1339–1352 (2014)
Gallego-Escuredo J.M., Gomez-Ambrosi J., Catalan V., Domingo P., Giralt M., Fruhbeck G., Villarroya F.: Opposite alterations in FGF21 and FGF19 levels and disturbed expression of the receptor machinery for endocrine FGFs in obese patients. Int. J. Obes. 39(1), 121–129 (2015)
Furusawa Y., Uruno A., Yagishita Y., Higashi C., Yamamoto M.: Nrf2 induces fibroblast growth factor 21 in diabetic mice. Genes to Cells. 19(12), 864–878 (2014)
Zhao Y., Banerjee S., Dey N., LeJeune W.S., Sarkar P.S., Brobey R., Rosenblatt K.P., Tilton R.G., Choudhary S.: Klotho depletion contributes to increased inflammation in kidney of the db/db mouse model of diabetes via RelA (serine)536 phosphorylation. Diabetes. 60(7), 1907–1916 (2011)
Rabbani N., Thornalley P.J.: Dicarbonyl proteome and genome damage in metabolic and vascular disease. Biochem. Soc. Trans. 42(2), 425–432 (2014)
Thornalley P.J., Rabbani N.: Protein damage in diabetes and uremia - identifying hotspots of proteome damage where minimal modification is amplified to marked pathophysiological effect. Free Radic. Res. 45(1), 89–100 (2010)
Morcos M., Du X., Pfisterer F., Hutter H., Sayed A.A.R., Thornalley P., Ahmed N., Baynes J., Thorpe S., Kukudov G., Schlotterer A., Bozorgmehr F., El Baki R.A., Stern D., Moehrlen F., Ibrahim Y., Oikonomou D., Hamann A., Becker C., Zeier M., Schwenger V., Miftari N., Humpert P., Hammes H.P., Buechler M., Bierhaus A., Brownlee M., Nawroth P.P.: Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell. 7(2), 260–269 (2008)
Rabbani N., Godfrey L., Xue M., Shaheen F., Geoffrion M., Milne R., Thornalley P.J.: Conversion of low density lipoprotein to the pro-atherogenic form by methylglyoxal with increased arterial proteoglycan binding and aortal retention. Diabetes. 60(7), 1973–1980 (2011)
Godfrey L., Yamada-Fowler N., Smith J.A., Thornalley P.J., Rabbani N.: Arginine-directed glycation and decreased HDL plasma concentration and functionality. Nutr Diabetes. 4, e134 (2014)
Mäkinen, V.-P., Civelek, M., Meng, Q., Zhang, B., Zhu, J., Levian, C., Huan, T., Segrè, A.V., Ghosh, S., Vivar, J., Nikpay, M., Stewart, A.F.R., Nelson, C.P., Willenborg, C., Erdmann, J., Blakenberg, S., O'Donnell, C.J., März, W., Laaksonen, R., Epstein, S.E., Kathiresan, S., Shah, S.H., Hazen, S.L., Reilly, M.P., Lusis, A.J., Samani, N.J., Schunkert, H., Quertermous, T., McPherson, R., Yang, X., Assimes, T.L., the Coronary, A.D.G.-W.R., Meta-Analysis, C.: Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS Genet. 10(7), e1004502 (2014)
Acknowledgments
This work was supported by the European Union’s Seventh Framework Program FP7 2007-2013 under grant agreement no. 244995 (BIOCLAIMS Project).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Masania, J., Malczewska-Malec, M., Razny, U. et al. Dicarbonyl stress in clinical obesity. Glycoconj J 33, 581–589 (2016). https://doi.org/10.1007/s10719-016-9692-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-016-9692-0