
Abstract
Various oligosaccharides containing galactose(s) and one glucosamine (or N-acetylglucosamine) residues with β1–4, α1–6 and β1–6 glycosidic bond were synthesized; Galβ1–4GlcNH2, Galα1–6GlcNH2, Galα1–6GlcNAc, Galβ1–6GlcNH2, Galβ1–4Galβ1–4GlcNH2 and Galβ1–4Galβ1–4GlcNAc. Galα1–6GlcNH2 (MelNH2) and glucosamine (GlcNH2) had a suppressive effect on the proliferation of K562 cells, but none of the other saccharides tested containing GlcNAc showed this effect. On the other hand, the proliferation of the human normal umbilical cord fibroblast was suppressed by none of the saccharides other than GlcNH2. Adding Galα1–6GlcNH2 or glucosamine to the culture of K562 cell, the cell number decreased strikingly after 72 h. Staining the remaining cells with Cellstain Hoechst 33258, chromatin aggregation was found in many cells, indicating the occurrence of cell death. Furthermore, all of the cells were stained with Galα1–6GlcNH-FITC (MelNH-FITC). Neither the control cells nor the cells incubated with glucosamine were stained. On the other hand, when GlcNH-FITC was also added to cell cultures, some of them incubated with Galα1–6GlcNH2 were stained. The difference in the stainability of the K562 cells by Galα1–6GlcNH-FITC and GlcNH-FITC suggests that the intake of Galα1–6GlcNH2 and the cell death induced by this saccharide is not same as those of glucosamine. The isolation of the Galα1–6GlcNH2 binding protein was performed by affinity chromatography (melibiose-agarose) and LC-MS/MS, and we identified the human heterogeneous ribonucleoprotein (hnRNP) A1 (34.3 kDa) isoform protein (30.8 kDa). The hnRNP A1 protein was also detected from the eluate(s) of the MelNH-agarose column by the immunological method (anti-hnRNP-A1 and HRP-labeled anti-mouse IgG (γ) antibodies).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
It has been reported that glucosamine (GlcNH2) is an amino monosaccharide occurring in connective and cartilage tissues of humans and other animals [1–3]. It is well known that this amino sugar is a precursor substance of glycosaminoglycans [4], and that it has an effect on nitric oxide synthesis [5] and a reparation effect on arthrodial cartilage such as in case of osteoarthritis [6–8]. Furthermore, it has been suggested that glucosamine benefited some patients with knee osteoarthritis and that it aided wound healing [9, 10]. Quastel and Cantero observed that GlcNH2 injection into mice with tumor sarcoma 37 inhibited the tumor growth, and suggested that administration of GlcNH2 might divert adenosine triphosphate (ATP) activity to affect the rates of the metabolic paths involved in tumor proliferation [11]. Molnar and Bekesi reported that addition of GlcNH2 or mannosamine (ManNH2) in Ehrlich ascites carcinoma and Sarcoma 180 ascites tumor cells in vitro provoked striking cytoplasmic and nuclear changes, including vacuolization of the cytoplasm and various degrees of disintegration [12]. Moreover, they found that continuous vein infusion with glucosamine resulted in a high rate of tumor regression in rats with i.m. Walker 256 carcinosarcoma [13]. Ichikawa et al. reported that incubation of mastocytoma P-815 cells with 5 mM glucosamine resulted in a marked inhibition of growth and significant reduction of cellular uptake and oxidation of glucose and cellular level ATP, and that glucosamine also reduced the uridine nucleotides and accumulated UDP-GlcNAc [14].
It has also been reported that a melibiose-binding protein composed of −58, 32, 26 kDa polypeptides was isolated from human spleen, and the protein was mainly anti-αGal antibody and it preferred Galα1–6 to Galα1–3 [15]. Galectin-1 (14 kDa), which is a generously β-galactose binding protein, was isolated from bovine heart (BHL-14) and reexamined regarding the binding specificity against saccharides, and it was observed that terminal α-linked galactose, rather than β-galactose in N-acetyllactosamine was the ligand for the galectin-1 [16]. Moreover, it is supposed that galectin-1 (β-galactose binding protein), which was expressed by stromal cells in human thymus and lymph nodes, induced apoptosis of activated human T-cells and human T-leukemia cell lines, and that galectin-1, which induced apoptosis, required expression of CD45 [17]. McReynolds et al. showed that human immunodeficiency virus (HIV-1) recombinant gp 120 (rgp 120) recognized non-natural glycosphingolipids, cellobiosyl and melibiosyl ceramides but not those lacking a lipid component [18].
We have taken notice of the various functions of glucosamine, such as its anti-inflammatory and other properties in humans and other animals, and have been furthering the development of novel functional oligosaccharides. Thus, we synthesized several oligosaccharides having a glucosamine residue at the reducing terminal, using several α- and β-galactosidases. Moreover, we examined their effects on some human cancer and normal cells, and we attempted to identify Galα1–6GlcNH2 binding protein from K562 cells.
2 Materials and methods
2.1 Saccharides
Lactosamine (LacNH2, Galβ1–4GlcNH2), allolactosamine (AlloLacNH2, Galβ1–6GlcNH2), MelNAc (Galα1–6GlcNAc), MelNH2 (Galα1–6GlcNH2), galactosyl-N-acetyllactosamine (GalLacNAc, Galβ1–4Galβ1–4GlcNAc) and galactosyllactosamine (GalLacNH2, Galβ1–4Galβ1–4GlcNH2) were synthesized by reverse reactions of α- and β-galactosidases of bacteria, Aspergillus niger and Aspergillus oryzae, respectively (PT No. 2004–352672 and 2004–352673). N-acetyllactosamine (LacNAc, Galβ1–4GlcNAc), N-acetyl-allolactosamine (AlloLacNAc, Galβ1–6GlcNH2) and chito oligosaccharides (bi-, tri-, tetra- and penta-: GlcNH2[β1–4GlcNH2]1~4) were supplied by Yaizu Suisankagaku Ind. Co. (Yaizu, Japan). Each oligosaccharide was dissolved in the 0.05 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.0, buffer containing 0.15 M NaCl, and filtrated (pore size 0.45 μm, Advantec Co., Tokyo) to sterilize. Fluorescein isothiocyanate-labeled Galα1–6GlcNH2 and GlcNH2 (MelNH-FITC and GlcNH-FITC) were synthesized by us. Briefly, reaction mixtures contained equal moles of each saccharide and FITC in 0.025 M phosphate buffer, pH 9.0, and were incubated for 30 min at room temperature and purified by thin layer chromatography (TLC) using chloroform/methanol (70/30) developmental solution. After TLC development, the synthesized MelNH- and GlcNH-FITC were isolated and recovered. Cellstain Hoechst 33258 (H33258) was purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Antibodies, mouse monoclonal anti-hnRNP-A1 antibody and horse radish peroxidase (HRP)-labeled goat anti-mouse IgG(γ), and polyvinylidene fluoride (PVDF) membrane for immunological detection were purchased from KPL (Kirkegaard and Perry Laboratories, Gaithersburg, MD, USA), Sigma and Millipore (Bedford, MA, USA), respectively. Other chemicals of analytical grade were obtained from Wako Pure Chemical Industries, Ltd. (Osaka, Japan) and Sigma.
2.2 Cell culture
Cells (human leukemia cell, K562 cells; human normal umbilical cord fibroblast (HUC-F2) cells) were purchased from the Riken Cell Bank (Tsukuba, Japan). Cell culture was carried out in Ham F-12 medium [Sigma; containing 10% fetal bovine serum (FBS)] for K562 cells and in αMEM (alpha minimum essential medium, containing 10% FBS, Sigma) for HUC-F2 cells in a 5% CO2 humidified air environment for 3–4 days at 37°C using Petri dishes (ϕ70 × 15 mm). Cells were collected by centrifugation at 1,000 rpm for 10 min, carefully aspirated and resuspended with fresh medium. In the case of HUC-F2 cell culture, confluent cells were treated with 0.25% trypsin-0.2% ethylenediaminetetraacetic acid (EDTA) in medium (Invitrogen, Co., Carlsbad, CA. USA), carefully shaken for 5–10 min at 37°C, centrifuged at 1,000 rpm for 10 min and washed with αMEM medium (10% FBS). Cells were suspended again in fresh medium and the number of cells was adjusted to 1.0 ∼ 1.7 × 105 cells/ml.
2.3 Suppression experiment of cell proliferation by saccharides
The suppression experiments of the cell proliferation were carried out using cells (K562 and HUC-F2) and saccharides as inhibitors in triplet. The cell culture medium (0.36 ml) of K562 was added to a 24-well plate, and incubated with 5 ∼ 10 mM saccharides using 50 and 100 mM as their stock solutions for 24 ∼ 72 h in the CO2 incubator. As a blank control, an aliquot of 50 mM HEPES buffered saline (pH 7.0), in place of saccharide, was added to the cell culture. After incubation, the cell number was counted using a cell counting chamber, except for the injured and shrunk cells, under the microscope at ×100 ∼ 200 magnifications, without cell staining by dye. After incubation of HUC-F2 with or without saccharides, the cell culture was aspirated carefully and 0.2 ml of 0.25% trypsin-0.2% EDTA was added to each well. Careful shaking was performed as described above, and fresh medium (0.2 ml) was added and the cell number was counted in the same manner as described above.
2.4 Cell staining with Cellstain Hoechst 33258, MelNH-FITC and GlcNH-FITC
In order to determine cell death, we observed nuclear chromatin aggregates of cultured cells with or without saccharides (GlcNH2 or MelNH2) using Cellstain Hoechst 33258 fluorescence dye, H33258, (2 mM solution in 50 mM Tris-HCl, pH 7.4, buffered saline). After staining cells (15 μl of suspension) with dye (1 μl) about 5 min at room temperature, aliquots (5 ∼ 10 μl) of cell suspension were dropped on slide glass and covered with cover glass without washing with fresh medium. MelNH-FITC and GlcNH-FITC fluorescent dyes (2 mM solution in 50 mM Tris-HCl, pH 7.4, buffered saline) were used for investigation of stainability of the cells which were cultured with oligosaccharide, using the same procedure as described above. The fluorescent cells were observed by fluorescent microscope at 346 nm for H33258 and 492 nm for MelNH- and GlcNH-FITC, respectively. Each cell sample dyed with H33258 was also observed optically.
2.5 Extraction of melibiose binding protein
K562 cells (about 5 × 109) cultured without any saccharide were collected by centrifugation at 1,000 rpm for 10 min at room temperature, washed thoroughly with 10 mM Tris-HCl buffer, pH 7.4, containing 0.15 M NaCl and exposed to 2 ml of 0.1% Triton X-100 and 100 mM melibiose in the same buffer. After gentle shaking by magnetic stirrer, the cell lysate was sonicated at 1.5 kW for 2 min at 4°C and centrifuged at 12,000 rpm for 30 min at 4°C. The supernatant was collected and the precipitate was washed with the same buffer. The supernatants were mixed and dialyzed two times against 500 ml of the same Tris-HCl buffer without melibiose.
2.6 Affinity chromatography
The dialyzed sample was applied to affinity column chromatography at 4°C, and the melibiose-binding material(s) was isolated, using a modification of the method of Sharma et al. [15]. Briefly, the cell extract was applied to the immobilized melibiose (Seikagaku Co. Tokyo) column, 1.0 × 5 cm, equilibrated with 10 mM Tris-HCl buffer, pH 7.4, containing 0.1% Triton X-100, 1 mM CaCl2 and 1 mM MgCl2, and the column was washed with about 100 ml of the same buffer. The melibiose-binding material(s) was eluted with 0.4 M melibiose in the same buffer, and each fraction (1.4 ml) was collected. The elutant was used for the following experiments.
Furthermore, another affinity chromatography (MelNH-agarose), in which the column was made by us using HiTrap™ NHS-activated HP column (1 mll; GE Healthcare, Uppsala, Sweden), was done as described above except for elution by 0.1 M MelNH2. Each sample, crude extract, unbound fraction and bound fraction (concentrated to about ten-fold), was tested for detection of the hnRNP A1 by immunological method.
2.7 Sodium dodecyl sulfate polyacrylamide gel electrophoresis
An aliquot (15 μl) of each fraction eluted from the affinity (melibiose-) column was treated with the loading buffer (Invitrogen) without the SH-reagent, 2-mercaptoethanol (2 ME), for 3 min at 90°C and applied to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; NuPAGE, 10% gel, Invitrogen) using NuPAGE MOPS buffer in accordance with the manufacturer’s instructions. After electrophoresis, the slab gel was stained with silver staining (Daiichi Pure Chemicals Co. Ltd. Tokyo). The fractions, which contained melibiose binding material(s) were pooled and concentrated by Amicon Ultra-4 (Millipore, Co. Bedford, MA, USA) at 3,500 rpm for 40 ∼ 60 min at 4°C. A part (15 μl) of the concentrated sample was loaded again on NuPAGE under reducing and non-reducing conditions for 3 min at 90°C and the gel was stained with silver staining.
2.8 LC-MS/MS analysis of the melibiose binding protein
In order to identify the MelNH2 binding protein, the sample (30 μl) described above and acrylamide (20 μmol) were mixed and treated for 3 min at 95°C. The mixture was applied on a stacking gel and the cysteine residues were alkylated with acrylamide during SDS-PAGE [19]. After electrophoresis and silver staining, the corresponding band on the gel was cut out and digested with trypsin. The triptic peptides were subjected to LC-MS/MS analysis using API QSTAR Pulsar (I) hybrid mass spectrometer (Applied Biosystem, Foster City, CA, USA) with a nano-liquid chromatography (DiNa, KYA TECH Corporation, Tokyo, Japan). The nano-LC was carried out using Magic C18 column (0.2 mm ID × 50 mm) and peptides were eluted with 0.1% formic acid and 0.1% formic acid 90% CH3CN.
Protein on SDS-PAGE was identified with LC-MS/MS using Mascot search engine (Matrix Science, UK) and NCBI database.
Aliquot (10 μl) of each sample (crude extract, unbound and bound fractions), which was obtained from MelNH-agarose affinity chromatography was blotted on PVDF membrane. Membrane strips were treated or not treated with 0.3% H2O2-methanol prior to immunostaining using mouse anti-hnRNP A1 monoclonal antibody and HRP-labeled goat anti-mouse IgG (γ) antibody.
3 Results
3.1 Suppressive effect of novel oligosaccharides on cancer cell proliferation
Among the oligosaccharides and monosaccharides tested, Galα1–6GlcNH2 (MelNH2) had the strongest suppressive effect on the proliferation of K562 cells (Fig. 1A). GlcNH2 was also an effective suppressor, and other oligosaccharides containing glucosamine (LacNH2, AlloLacNH2 and GalLacNH2), but not N-acetylglucosamine (GlcNAc, LacNAc, AlloLacNAc and MelNAc), controlled on the proliferation of K562 cells slightly. Moreover, although AlloLacNH2 and MelNH2 both have the 1–6 glycosidic bond between galactose and GlcNH2 residues, the former showed only a weak inhibitory effect on K562 cell proliferation. As shown in Fig. 1B, chito-oligosaccharides (chito-biose, chito-triose, chito-tetraose and chito-pentaose) were tested for their influence on the cell proliferation, and chito-biose (GlcNH2β1–4GlcNH2) seemed to be slightly inhibitive against K562 cell proliferation. However, other chito oligosaccharides (chito-triose ~ chito-pentaose) seemed to have no effect on the cell proliferation.

Effects of various saccharides on human leukemia K562 cell proliferation (A) Each saccharide was added to the cell culture at 5 mM concentration. (B) Chito-oligosaccharide (di, tri, tetra and penta: CH-2, CH-3, CH-4 and CH-5, 5 mM concentration) effect on cell proliferation after 72 h. Data are shown as the mean ± SD. (n = 3 for all saccharide samples). **p < 0.01 compared with the control
Neutral saccharides (such as glucose, galactose, lactose and melibiose) tested showed no influence on proliferation of K562 cells (part of the data not shown).
On the other hand, when human normal umbilical cord fibroblasts, HUC-F2 cells, were incubated with a series of oligosaccharides, the cell proliferation was influenced only by GlcNH2 (Fig. 2).

Effects of saccharides on the proliferation of human normal HUC-F2 cells. Data are shown as the mean ± SD (n = 3 for all saccharide samples). **p < 0.01 compared with the control
Mel and MelNH2 are structurally very similar, but their effects on the K562 cells are quite different. The question arises as to whether these saccharides share in part the same metabolic pathway, such as through an interaction with specific melibiose-binding protein(s). To clarify this, the effect of preincubation with Mel against anti-proliferation action of MelNH2 was examined using K562 cells (Fig. 3). Mel moderately inhibited MelNH2 action; after 72 h incubation the cell number increased about four-fold (compare left and middle columns in Fig. 3). However, simultaneous treatment of Mel and MelNH2 did not increase the cell number (data not shown), suggesting that they share the same binding molecule, at least in part.

Inhibition against MelNH2 suppressive effect on K562 cell by Mel The cell was cultured with 12.5 mM Mel for 1 ∼ 2 h prior to addition of 5 mM MelNH2, and after 72 h incubation the remaining cell was counted. Data are shown as the mean ± SD. (n = 3 for all saccharide samples). *p < 0.05 compared the MelNH2 with the Mel ± MelNH2
3.2 Cell staining with Cellstain Hoechst 33258, MelNH-FITC and GlcNH-FITC
In order to clarify the cell stainability with H33258, MelNH-FITC and GlcNH-FITC fluorescent dyes, the K562 cells were observed under the fluorescent microscope after dyeing. As shown in Fig. 4, 1a, when the cells were cultured for 24 h without any saccharide and stained with H33258, most cells were of a good shape and dark blue. Moreover, no cells showed any nuclear aggregation. With MelNH-FITC, however, most of the cells did not show staining and they were black under the fluorescent microscope (Fig. 4, 1c). Incubation of cells with 5 mM GlcNH2 or MelNH2 for 24 h showed that a few cells were bright with H33258 staining. But most cells did not show staining with MelNH-FITC (data not shown): thus the situation was almost the same as the control incubation for 24 h. The incubation for 72 h with no specially added saccharide gave the result that several cells were bright (H33258) and their nuclear chromatin was detected to be aggregated (Fig. 4, 2a), but almost all were not stained with MelNH-FITC (Fig. 4, 2c). Moreover, the incubation of cells with 5 mM GlcNH2 for 72 h gave the result that about half of them were brightly stained with H33258 and their chromatin was aggregated, but only several cells were greenish in the case of MelNH-FITC staining, as shown in Fig. 4, 3a and 3c. On the other hand, when cells were cultured with 5 mM MelNH2 for 72 h, all cells showed aggregated nuclear chromatin (Fig. 4, 4a), and they were also stained with MelNH-FITC (Fig. 4, 4c).

K562 cell staining by Hoechst 33258 and MelNH-FITC Each cell sample was stained with Hoechst 33258 and MelNH-FITC after 24 and 72 h cell incubation with or without saccharides (GlcNH2 and MelNH2), and observed under the fluorescent microscopy (a and c). Each cell sample (stained with H33258) was also observed optically (b) at ×200 magnification. Bar, 100 μm
When cell staining with GlcNH-FITC was carried out after incubation with 5 mM GlcNH2 and MelNH2 for 72 h, among the cells incubated with MelNH2 more than half were not stained (Fig. 5, 2c).

Stainability of K562 cells with GlcNH-FITC after adding of GlcNH2 and MelNH2 After 72 h incubation of cells with GlcNH2 and MelNH2, cell samples were stained by GlcNH-FITC and were observed optically (1a ∼ 1c) and under fluorescent (2a ∼ 2c) microscopy at ×200 magnification. Bar, 100 μm
Furthermore, when any oligosaccharide (5 mM) having a GlcNAc residue was added to the cell culture, chromatin aggregation was observed to be only of the same grade as that of the control (data not shown).
3.3 SDS-PAGE of the fraction eluted from the melibiose–agarose column
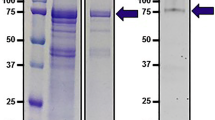
Each fraction which was eluted from the Mel-conjugated column was loaded on SDS-PAGE under non-reducing condition and the slab gel was stained by silver (data not shown). The fractions, Nos. 6 ∼ 9, were collected, concentrated to about 0.3 ml and applied again on SDS-PAGE. As shown in Fig. 6, one main (76.0 kDa) and one minor (48.0 kDa) band were detected under non-reducing conditions, and one major (30.8 kDa) band was detected under reducing conditions. Thus the Mel binding protein(s) was effectively purified by one step affinity chromatography.

SDS-PAGE of the Mel-binding protein obtained from the affinity chromatography Lane NR: Sample was not treated by reducing reagent. Lane R: The same sample was treated by two ME reducing reagent. Standard proteins were myosin (188 kDa), phosphorylase B (97 kDa), glutamic dehydrogenase (51 kDa), carbonic anhydrase (33 kDa), myoglobin blue (21 kDa) and myoglobin red (19 kDa)
3.4 Identification of the MelNH2 binding protein by LC-MS/MS analysis and immunological method
Sequences of several peptides which were obtained from the 30.8 kDa protein band on the SDS-PAGE were analyzed and determined by trypsin digestion and LC-MS/MS analysis, and each peptide consisted of 12 ∼ 19 amino acids (Fig. 7). From the result of sequences of peptides hnRNP A1 (34.3 kDa) isoform protein (30.8 kDa) was identified as a candidate with sequence coverage of 25% as shown in Fig. 7.

Human hnRNP A1 amino acid sequence The amino acid sequence of human hnRNP A1 was determined by Buvoli et al. [28], and was about 34.3 kDa. Amino acids sequences circled with squares are identified as agreeing with our analysis data
As shown in Fig. 8, when the elutants from the MelNH-agarose column were immunostained by anti-hnRNP A1 monoclonal and HRP-anti-mouse IgG (γ) antibodies on PVDF membrane, the hnRNP A1 was only detected in the fraction eluted with 0.1 M MelNH2 after sample blotting and treating the membrane with 0.3% H2O2–methanol (Fig. 8). However, the dark brown spots were also detected on all untreated spots.

Detection of hnRNP A1 protein on the PVDF membrane by immunological method Crude Ext.; K562 cell extract fraction, UBf; unbound fraction to the MelNH-agarose column, Bf; bound fraction to the same column, TR; Each sample was treated with 0.3% H2O2-methanol at room temperature for 30 min in order to inactivate endogenous peroxidase activities, NT; not treated with H2O2-methanol
4 Discussion
We showed that novel synthesized oligosaccharides, especially MelNH2 (Galα1–6GlcNH2), had the strongest inhibitory activity on the cancer cell proliferation of human leukemia cells (K562), but not on normal cells (HUC-F2; Figs. 1 and 2). Moreover, GlcNH2 monosaccharide was also an inhibitor of the cancer and normal cells. Quastel and Cantero have reported that daily injection of GlcNH2, intraperitoneally, into mice with tumors showed suppressive effect on the growth of the tumors, and they expected that administration of GlcNH2 might divert ATP activity to affect the rates of the metabolic paths involved in tumor proliferation [11]. They also showed that the radioactive intermediates found following the administration of glucosamine-14C were GlcNAc-6P, GlcNAc-1P, GlcNH2–6P, UDP-GlcNAc, UDP-GalNAc and sialic acid, and they expected that glucosamine derivatives had a cytotoxic effect on the tumors, which should be most sensitive to glucosamine. Ichikawa et al. reported that incubation of mastocytoma P-815 cells with 5 mM glucosamine resulted in a marked inhibition of the cell growth [14]. They further reported that the cellular uptake and oxidation of glucose and the cellular level of ATP were significantly reduced by addition of glucosamine, and that a diminishing of uridine nucleotides and an accumulation of UDP-GlcNAc were observed. In our study, addition of GlcNH2 to the K562 cell culture decreased the cell numbers of the K562. Thus we suppose that GlcNH2 should also divert ATP activity, and that its derivatives should damage cancer cells. The precise mechanism of the reduction of the cancer cells, however, remains unclear.
On the other hand, the degree of suppressive activity of MelNH2 against the K562 cell proliferation was higher than that of GlcNH2, although the same concentrations of these saccharides (5 mM each) were used for this examination. Furthermore, the MelNH2 addition to the K562 cell culture decreased the cell numbers to a greater degree than those of other oligosaccharides which had GlcNH2 residue (LacNH2, AlloLacNH2 and GalLacNH2), as shown in Fig. 1A. These results suggest that it is unreasonable to think that MelNH2 might be hydrolyzed to two monosaccharides (Gal and GlcNH2) and that the GlcNH2 residue functions as an inhibitor against cancer cell proliferation. Supposing this assumption is correct, the influence of GlcNH2 on the K562 cells must be greater than that of MelNH2. Thus we expect that the administration effects of MelNH2 and GlcNH2 on the K562 cells indicate the differences between their metabolic pathways in cancer cells.
Furthermore, chito-disaccharide might have a slight inhibitory effect on K562 cells, but other chito-oligosaccharides (tri-, tetra- and penta-) showed no suppressive affect on the cell proliferation, indicating that chito-oligosaccharides were neither taken up into the cell nor metabolized (Fig. 1B). It seems quite likely that chito-disaccharide or other chito-oligosaccharides could be neither hydrolyzed in the cell culture systems nor absorbed on the cell surface.
The results of the preincubation examination of K562 cells with Mel prior to the incubation with MelNH2 showed a moderate decrease in the anti-proliferation activity of MelNH2. This suggests that the same receptor(s) against MelNH2 and Mel is present on the cell (Fig. 3), and that the former has a higher affinity than the latter.
After incubation of the K562 cell with MelNH2 or GlcNH2, the cell was stained with H33258, MelNH-FITC and GlcNH-FITC, as shown in Figs. 4, 1a ~ 4c and 5. The degree of stainability of the K562 cell changed with progress of time and a drastic change seemed to occur about 48 h incubation after. This suggests that the influence of MelNH2 or GlcNH2 upon the K562 leukemia cells increased slowly. After incubation for 72 h with MelNH2, every cell was stained with these two fluorescence dyes (H33258 and MelNH-FITC) and their nuclear chromatin aggregations were identified as light blue ones (Fig. 4, 4a and 4c). However, although GlcNH2 showed a suppressive effect against the K562 cell proliferation, only half of the cells were stained with H33258 after 72 h incubation and the cells were not sufficiently stained by MelNH-FITC (Fig. 4, 3a and 3c). Furthermore, as described in these figures, the brightly stained cells which showed nuclear chromatin aggregation with H33258 were determined to have shown cell death induced by these saccharides and most cells died after MelNH2 addition (Fig. 4, 4a and 4c). On the other hand, staining of the cells with GlcNH-FITC showed a difference from that with MelNH-FITC (Figs. 4, 4 and 5), suggesting that there might be some uptake pathways of these saccharides. Thus these results suggest that the cell death pathway of K562 cells induced by MelNH2 differs from that induced by GlcNH2 and that at least some cell death pathways induced by saccharides might be present. We hope that the MelNH2 and Mel specific receptor protein(s) exists in/on the K562 cell membrane and that the cell takes up and transport these saccharides by this molecule(s).
We determined by LC-MS/MS analysis and immunological method that the Mel/MelNH2 binding protein is the hnRNP A1 isoform, and that this protein had an apparent molecular mass of 30.8 kDa on SDS-PAGE (Figs. 6, 7 and 8). Human hnRNP A1 is a single-stranded nucleic acid-binding protein, which has many functions in various aspects of mRNA maturation and in telomere length regulation [20, 21]. On the other hand, Murzin et al. have observed a novel folding motif in four different proteins, which bind oligonucleotides or oligosaccharides: in nuclease, anticodon binding domain of asp-tRNA synthetase and B-subunits of heat-labile enterotoxin and verotoxin-1 (in some enzymes and toxins) [22]. They called the OB-fold in which a five-stranded β-sheet was coiled to form a closed β-barrel. Moreover, the hnRNP protein is predicted to have a very closely related motif and to bind to oligosaccharide (e.g. lactose) as well as oligonucleotide [22, 23]. It was shown that the hnRNP was shuttling between the nucleus and the cytoplasm [24]. Celis et al. and LeStourgeon et al. reported that there was a marked difference in the concentration of hnRNP A1 between resting or dividing cells and rapidly dividing cells [25, 26], and Xu et al. demonstrated that levels of hnRNP A1 were elevated in some cancers [27]. We observed that MelNH2 and GlcNH2 did not significantly influence the K562 cells, which had been cultivating for about 6 months and that the division speed decreased (data not shown), and that the human normal cell (HUC-F2) was controlled by only GlcNH2 but not MelNH2. We have carried out an experiment on the influence of some saccharides on the human B cell derived leukemia cell line (BALL cells), and found that the BALL cells was also controlled with MelNH2, GlcNH2 and alloLacNH2 (data not shown). We think that novel oligosaccharides containing amino-sugar (e. g. glucosamine) show a certain effect on the leukemia cell lines in vitro. Thus we suppose that a molelule having specific affinity against MelNH2 appears in the K562 cells and that it is elevated on/in the cancer cell membrane, and that MelNH2 taken into cells disturbs the abilities of the hnRNP to mature mRNA regulating telomere length or to perform other significant function(s) for the cancer cell proliferation and survival. Thus, although it is not clear how MelNH2 is taken into the K562 cells, we hope that the MelNH2 binding protein (hnRNP A1 isoform) plays important roles in transporting MelNH2 into the cell nucleus via the cytoplasm. However, the molecule which was expected to be the receptor on/in the cell membrane was not detected in this study. This will be a subject of our research in the near future.
Abbreviations
- GlcNAc:
-
N-acetylglucosamine
- GlcNH2 :
-
glucosamine
- Mel:
-
melibiose
- MelNAc:
-
Galα1–6GlcNAc
- MelNH2 :
-
Galα1–6GlcNH2
- LacNAc:
-
N-acetyllactosamine
- LacNH2 :
-
lactosamine
- alloLacNAc:
-
N-acetyl-allolactosamine
- alloLacNH2 :
-
allolactosamine
- GalLacNAc:
-
galactosyl-N-acetyllactosamine
- GalLacNH2 :
-
galactosyllactosamine
- heterogeneous nuclear ribonucleoprotein A1:
-
hnRNP A1
References
Stockwell, R.A., Scott, J.E.: Distribution of acid glycosaminoglycans in human articular cartilage. Nature 215, 1376–1378 (1967). doi:10.1038/2151376a0
Partrige, S.M., Davis, H.F., Adair, G.S.: The chemistry of connective tissues. 6. The constitution of the chondroitin sulphate-protein complex in cartilage. Biochem. J. 79, 15–26 (1961)
Baxter, B., Muir, H.: The nature of the protein moieties of cartilage proteoglycans of pig and ox. Biochem. J. 149, 657–668 (1975)
Malemud, C.J., Goldberg, V.M., Moskowitz, R.W., Getzy, L.L., Papay, R.S., Norby, D.P.: Biosynthesis of proteoglycan in vitro by cartilage from human osteochondrophytic spurs. Biochem. J. 206, 329–341 (1982)
Meininger, C.J., Kelly, K.A., Li, H., Haynes, T.E., Wu, G.: Glucosamine inhibits inducible nitric oxide synthesis. Biochem. Biophys. Res. Commun. 279, 234–239 (2000). doi:10.1006/bbrc.2000.3912
Zerkak, D., Dougados, M.: The use of glucosamine therapy in osteoarthritis. Curr. Rheumatol. Rep. 6, 41–45 (2004). doi:10.1007/s11926-004-0082-4
Fenton, J.I., Chlebek-Brown, K.A., Peters, T.L., Caron, J.P., Orth, M.W.: Glucosamine HCl reduces equine articular cartilage degradation in explant culture. Osteoarthritis Cartilage 8, 258–265 (2000). doi:10.1053/joca.1999.0299
Barclay, T.S., Tsourounis, C., McCart, G.M.: Glucosamine. Ann. Pharmacother. 32, 574–579 (1998). doi:10.1345/aph.17235
Houpt, J., McMillan, R., Wein, C., Paget-Dellio, S.D.: Effect on glucosamine hydrochloride in the treatment of pain of osteoarthritis of the knee. J. Rheumatol. 26, 2423–2430 (1999)
McCarty, M.F.: Glucosamine for wound healing. Med. Hypotheses 47, 273–275 (1996). doi:10.1016/S0306-9877(96)90066-3
Quastel, J.H., Cantero, A.: Inhibition of tumor growth by D-glucosamine. Nature 171, 252–254 (1953). doi:10.1038/171252a0
Molnar, Z., Bekesi, J.G.: Effects of D-glucosamine, D-mannosamine, and 2-deoxy-D-glucose on the ultrastructure of ascites tumor cells in vitro. Cancer Res. 32, 380–389 (1972a)
Molnar, Z., Bekesi, J.G.: Cytotoxic effects of D-glucosamine on the ultrastructures of normal and neoplastic tissues in vivo. Cancer Res. 32, 756–765 (1972b)
Ichikawa, A., Takagi, M., Tomita, K.: Effect of D-glucosamine on growth and several functions of cultured mastocytoma P-815 cells. J. Pharmacobiodyn. 3, 577–588 (1980)
Sharma, A., Ahmed, H., Allen, H.J.: Isolation of a melibiose-binding protein from human spleen. Glycoconj. J. 12, 17–21 (1995). doi:10.1007/BF00731864
Appukuttan, P.S.: Terminal a-linked galactose rather than N-acetyllactosamine is ligand for bovine heart galectin-1 in N-linked oligosaccharides of glycoproteins. J. Mol. Recognit. 15, 180–187 (2002). doi:10.1002/jmr.573
Perillo, N.L., Pace, K.E., Seilhamer, J.J., Baum, L.G.: Apoptosis of T cells mediated by galectin-1. Nature 378, 736–739 (1995). doi:10.1038/378736a0
McReynolds, K.D., Bhat, A., Conboy, J.C., Saavedra, S.S., Gervay-Hague, J.: Non-natural glycosphingolipids and structurally simpler analogues bind HIV-1 recombinant Gp120. Bioorg. Med. Chem. 10, 625–637 (2002). doi:10.1016/S0968-0896(01)00325-X
Mineki, R., Taka, H., Fujimura, T., Kikkawa, M., Shindo, N., Murayama, K.: In situ alkylation with acrylamide for identification of cysteinyl residues in proteins during one- and two-dimensional sodium dodecyl sulphate-polyacrylamide gel electrophoresis. Proteomics 2, 1672–1681 (2002). doi:10.1002/1615-9861(200212)2:12<1672::AID-PROT1672>3.0.CO;2-#
Carpenter, B., Mackay, C., Alnabulsi, A., MacKay, M., Telfer, C., Melvin, W.T., et al.: The roles of heterogeneous nuclear ribonucleoproteins in tumour development and progression. Biochim. Biophys. Acta. 1765, 85–100 (2006)
Zhang, Q.S., Manche, L., Xu, R.M., Krainer, A.R.: hnRNP A1 associates with telomere ends and stimulates telomerase activity. RNA 12, 1116–1128 (2006). doi:10.1261/rna.58806
Murzin, A.G.: OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J. 12, 861–867 (1993)
Ding, J., Hayashi, K.M., Zhang, Y., Manche, L., Krainer, A.R., Rui-Ming, X.: Crystal structure of the two-RRM domain of hnRNP A1 (UP1) complexed with single-stranded telomeric DNA. Genes Dev. 13, 1102–1115 (1999). doi:10.1101/gad.13.9.1102
Pinol-Rome, S., Dreyfuss, G.: Shuttling of pre-mRNA binding proteins between nucleus and cytoplasm. Nature 355, 730–732 (1992). doi:10.1038/355730a0
Celis, J.E., Bravo, R., Arenstorf, H.P., Lestourgeon, W.M.: Identification of proliferation-sensitive human proteins amongst components of the 40 S hnRNP particles. Identity of hnRNP core proteins in the HeLa protein catalogue. FEBS Lett. 194, 101–109 (1986). doi:10.1016/0014-5793(86)80059-X
Lestourgeon, W.M.B.A., Christensen, M.E., Walker, B.W., Poupore, S.M., Daniels, L.P.: The packaging proteins of core hnRNP particles and the maintenance of proliferative cell stages. Cold Spring Harb. Symp. Quant. Biol. 42, 885–898 (1978)
Xu, X., Joh, H.D., Pin, S., Schiller, N.I., Prange, C., Burger, P.C., et al.: Expression of multiple larger-sized transcripts for several genes in oligodendrogliomas: potential markers for glioma subtype. Cancer Lett. 171, 67–77 (2001). doi:10.1016/S0304-3835(01)00573-0
Buvoli, M., Biamonti, G., Tsoulfas, P., Bassi, M.T., Ghetti, A., Riva, S., et al.: cDNA cloning of human hnRNP protein A1 reveals the existence of multiple mRNA isoforms. Nucleic Acids Res. 16, 3751–3770 (1988). doi:10.1093/nar/16.9.3751
Acknowledgements
This research was supported by grants from the Institute of Sports, Health and Medical Science, Juntendo University. We thank Drs. D. Ohmori, K. Ikeda, B. Allen R. Mineki and H. Taka for their excellent technology and suggestions.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hosomi, O., Misawa, Y., Takeya, A. et al. Novel oligosaccharide has suppressive activity against human leukemia cell proliferation. Glycoconj J 26, 189–198 (2009). https://doi.org/10.1007/s10719-008-9175-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-008-9175-z