
Abstract
The methylotrophic yeast Pichia pastoris is widely used for the production of recombinant glycoproteins. With the aim to generate biologically active 15N-labeled glycohormones for conformational studies focused on the unravelling of the NMR structures in solution, the P. pastoris strains GS115 and X-33 were explored for the expression of human chorionic gonadotropin (phCG) and human follicle-stimulating hormone (phFSH). In agreement with recent investigations on the N-glycosylation of phCG, produced in P. pastoris GS115, using ammonia/glycerol-methanol as nitrogen/carbon sources, the N-glycosylation pattern of phCG, synthesized using NH4Cl/glucose–glycerol–methanol, comprised neutral and charged, phosphorylated high-mannose-type N-glycans (Man8–15GlcNAc2). However, the changed culturing protocol led to much higher amounts of glycoprotein material, which is of importance for an economical realistic approach of the aimed NMR research. In the context of these studies, attention was also paid to the site specific N-glycosylation in phCG produced in P. pastoris GS115. In contrast to the rather simple N-glycosylation pattern of phCG expressed in the GS115 strain, phCG and phFSH expressed in the X-33 strain revealed, besides neutral high-mannose-type N-glycans, also high concentrations of neutral hypermannose-type N-glycans (Manup-to-30GlcNAc2). The latter finding made the X-33 strain not very suitable for generating 15N-labeled material. Therefore, 15N-phCG was expressed in the GS115 strain using the new optimized protocol. The 15N-enrichment was evaluated by 15N-HSQC NMR spectroscopy and GLC-EI/MS. Circular dichroism studies indicated that 15N-phCG/GS115 had the same folding as urinary hCG. Furthermore, 15N-phCG/GS115 was found to be similar to the unlabeled protein in every respect as judged by radioimmunoassay, radioreceptor assays, and in vitro bioassays.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
One of the popular host-cell systems for the expression of recombinant proteins derived from eukaryotic genomes [1] is the methylotrophic yeast Pichia pastoris. This species, which is able to perform post-translational modifications such as glycosylation, is easier to manipulate genetically and to culture than mammalian cells. The P. pastoris expression system uses only pure reagents, such as methanol, glucose or glycerol, biotin, trace salts, and water. The media are free of toxins, and in addition bacterial contaminations are prevented by the use of methanol, which suits perfectly with pharmaceutical requirements. Furthermore, P. pastoris cell cultures can be grown at high cell densities and protein purification is usually simple because of the low level of native yeast proteins secreted [2].
Biologically active hCG and hFSH have successfully been expressed in P. pastoris using the GS115 and X-33 strains, yielding phCG and phFSH, respectively [3, 4] (unpublished data for the X-33 strain). The wild-type P. pastoris X-33 strain has been created from the GS115 strain by PCR of the wild type HIS4 gene, and transforming GS115 to His+ by homologous recombination. The strains X-33 and GS115 differ only by one basepair in the HIS4 gene. Recently, we have investigated in high detail the glycosylation pattern of phCG expressed in P. pastoris strain GS115, demonstrating the occurrence of (phosphorylated) high-mannose-type structures [5].
The glycosylation studies on phCG, as mentioned above, were carried out in the context of a larger project focused on the unraveling of the NMR structures in solution of hCG and hFSH. So far, this research has led to the unraveling of the NMR structure in solution of the α-subunit of native, urinary hCG [6–9], a structure that shows some differences with the α-subunit in the crystal structure of hCG [10, 11]. Of special interest is that the solution structure of the α-subunit exhibits an increased structural disorder [8] compared to the α-subunit in the crystal structure of the α,β-dimer.
In order to generate biologically active 15N-labeled glycohormones in P. pastoris for NMR studies, in the present paper an optimization of the earlier reported fermentation protocol [4] is presented. Furthermore, attention is paid to the influence of the GS115 and X-33 strains on the N-glycosylation of P. pastoris-expressed hCG and hFSH. Finally, the most optimal protocol and strain have been used to generate biologically active 15N-labeled phCG.
2 Materials and methods
2.1 General
[125I]NaI and [1,2,6,7,16,17-3H]-testosterone were obtained from Perkin Elmer Life Sciences (Boston, MA) and 15NH4Cl was purchased from Isotec (St Louis, MO). Pichia expression vectors, yeast extract, peptone, and yeast nitrogen base without amino acids, required for growing P. pastoris cells, were obtained from Invitrogen Corp (Carlsbad, CA).
All the DNA manipulations were according to standard techniques [12]. For hCG expressions using the P. pastoris strain GS115 and the pPIC9k expression vector, full details have been reported [3, 4]. For expressions using the P. pastoris strain X-33, the pGAPZalpha vector was used to prepare phFSH/X-33 and phCG/X-33 (unpublished data).
2.2 Growth media
The preculturing of P. pastoris cells was carried out in YPD (1% yeast extract, 2% peptone, and 2% dextrose), containing 100 μg/ml geneticin. The 1-liter medium for the culturing in the fermenter vessel contained 42.9 g KH2PO4, 1 g CaSO4·2H2O, 14.3 g K2SO4, and 11.7 g MgSO4·7H2O. The nitrogen and carbon sources needed were prepared separately. The medium used is an adaptation of the FM22 medium [13, 14], wherein (NH4)2SO4 has been substituted by NH4Cl. To this end, 55 g NH4Cl were dissolved in 550 ml H2O, and the solution was sterilized by filtration through a 0.22 μm filter. One hundred milliliter of this solution were pumped into the fermenter vessel every 24 h except on day 2, when 50 ml were pumped. A 200 ml solution of 50% (w/v) glucose, containing 1 ml of PTM1 trace salts, replaced the previously used glycerol [4] during the batch phase, and a 50% glycerol solution (2 g glycerol in total) was used during the fed-batch phase of 20 min. PTM1 consisted of 24 mM CuSO4, 0.53 mM NaI, 19.87 mM MnSO4, 0.83 mM Na2MoO4, 0.32 mM boric acid, 2.10 mM CoCl2, 0.15 mM ZnCl2, 0.23 mM FeSO4, and 0.82 mM biotin.
2.3 Production of phCG in P. pastoris GS115
The production of phCG/GS115 was performed using a BioFlo 3000 fermenter (New Brunswick Scientific, Edison, NJ), equipped with a 3-liter bioreactor constantly maintained at 28°C.
The P. pastoris cells were revived by inoculation from a frozen glycerol stock of a single colony into 5 ml YPD, containing 100 μg/ml geneticin (preculturing). After 2 days of incubation at 30°C, 1 ml of the culture was transferred into 100 ml of the same medium, and incubated for 3 days at 30°C. Then, the culture was spun down at 5,000 rpm, and the cells were resuspended in 10 ml of the supernatant.
After autoclaving the fermentation medium (1 l) in the fermenter vessel, 100 ml of the NH4Cl solution, 50 ml of the glucose solution (see above), and 6 ml of trace salts were added in the vessel before inoculation of the cells. The pH was automatically maintained at 5.0 by addition of 5.0 M KOH. The agitation was set to 600 rpm, and O2 was automatically mixed with air to maintain the dissolved oxygen (DO) level at the set point of 30%. The console automatically adjusted the gas flow, varying between one and two vessel volume. The cell density was estimated every 24 h by measuring the wet cell weight and the absorbance at 600 nm. Foaming was prevented by the addition of 100 μl Antifoam 289. The culture used the glucose present in the vessel in about 3.5 h, and then the glucose feeding was gradually adjusted according to the DO reading at 6 ml/h. The cells started to utilize O2 while they were actively growing, the agitation was set to 750 rpm, and towards the end of the batch phase they were utilizing 90% of pure O2. In total, 100 g of glucose was used during this batch growth phase, which lasted for about 20 h. Then, when all the glucose was consumed, an increase in both DO and pH was observed. The glycerol fed-batch phase was initiated by feeding the culture with the glycerol solution and lasted 20 min. The cells were starved for 30 min before starting the induction phase. Then, the culture was fed with a 50% (v/v) methanol solution (containing 6 ml/l of trace salts) at a rate of 6 ml/h. Once the culture was adapted to grow on methanol, the rate was increased and the feeding solution was replaced by a 90% (v/v) methanol solution (containing 6 ml/l of trace salts). DO spikes were regularly monitored to ensure the viability of the culture; the induction phase lasted 96 h.
At the end of the fermentation, the pH of the medium was adjusted to 7.4, EDTA and NaN3 were added to a concentration of 10 and 1 mM, respectively, and the cells were separated from the 3-liter of supernatant by centrifugation at 5,000 rpm with a Beckman centrifuge for 20 min at 20°C.
phCG/GS115 was purified using an earlier described protocol using sequential Phenyl Sepharose (Amersham Biosciences, Piscataway, NJ) and SP-Sepharose Fast Flow (Amersham Biosciences) column chromatography [4], yielding about 25 mg/l full biological active glycohormone.
2.4 Production of phCG and phFSH in P. pastoris X-33
The production of phCG/X-33 and phFSH/X-33 was carried out in a 10-liter fermentor vessel using ammonia/glycerol–methanol as nitrogen/carbon sources (unpublished data of R.R. Dighe), yielding about 17 and 22 mg/l biological active glycohormone, respectively.
2.5 Release and isolation of N-glycans of phCG and phFSH with PNGase F or Endo H
Glycoprotein samples (phCG or phFSH) were reduced and S-carboxymethylated according to standard procedures [5, 15], except that the reducing glycoprotein solution was boiled for 3 min instead of being incubated for 2 h at 37°C.
Reduced and carboxymethylated glycoprotein or glycopeptide (derived from phCG) samples were dissolved (2 mg/ml) in 20 mM NaH2PO4/Na2HPO4, pH 7.2, containing 10 mM EDTA, and digested with peptide-N 4-(N-acetyl-β-glucosaminyl)asparagine amidase F (PNGase F, EC 3.5.1.52; Roche Applied Science, IN) (0.5 U/mg) for 24 h at 37°C. Alternatively, samples were dissolved (2 mg/ml) in 20 mM NaH2PO4/Na2HPO4, pH 5.5, containing 10 mM EDTA, and digested with endo-β-N-acetylglucosaminidase H (Endo H, EC 3.2.1.96; Roche Applied Science, IN) (1 mU/100 μg).
For the N-glycan profiling of phCG/GS115, the PNGase F digest was separated on a Superdex G75 column (60 × 2.6 cm; Pharmacia, Uppsala, Sweden) using 50 mM NH4HCO3, pH 7.0, as eluent at a flow rate of 1 ml/min, monitored at 214 nm (Uvicord, LKB). In the case of phCG/X-33 and phFSH/X-33, the PNGase F digest was fractionated on a Bio-Gel P-10 column (75 × 2 cm, BioRad) using 50 mM NH4HCO3, pH 7.0, as eluent at a flow rate of 13 ml/h, monitored by refractive index detection (Bischoff 8100 RI detector). The N-deglycosylated glycoproteins and the carbohydrates were isolated and lyophilized four times.
N-glycans released from glycopeptides were directly labeled with 2-aminobenzamide (2AB) without prior separation from the peptide moiety.
2.6 Preparation, isolation, and fractionation of phCG/GS115 glycopeptides
Reduced and carboxymethylated phCG/GS115 (15 mg), dissolved in 7.5 ml 20 mM NaH2PO4/Na2HPO4, pH 8.0, was digested with 100 μg trypsin (EC 3.4.21.4; Roche Applied Science, IN) for 2 h at 37°C. The digest was stored for 20 min at −80°C to deactivate the enzyme, then lyophilized. The tryptic digest was dissolved in buffer A (20 mM Tris/HCl, pH 7.5, containing 0.3 M NaCl, 1 mM CaCl2, 1 mM MnCl2, and 0.02% NaN3) and applied to a 2-ml ConA-Sepharose (Sigma-Aldrich, St Louis, MO) column, equilibrated in the same buffer. After washing with buffer A, the column was eluted with 100 mM methyl α-d-mannopyranoside in buffer A, and the glycopeptide-containing fraction obtained was lyophilized.
HPLC fractionations were carried out on a Waters 600 HPLC system. The glycopeptide fraction was separated on a Vydac 214TP510 C4 column (4.6 × 250 mm; Grace Vydac, Hesperia, CA). Elutions were performed with a gradient of acetonitrile in 0.1 M triethylamine phosphate, pH 6.5 (built up from buffers B and C), at a flow rate of 1.5 ml/min, monitored at 214 nm. Buffer B consisted of 5% acetonitrile in 0.1 M triethylamine phosphate, pH 6.5; buffer C of 60% acetonitrile in 0.1 M triethylamine phosphate, pH 6.5. For gradient details, see relevant figure captions. Isolated glycopeptides were desalted on a HiTrap column (Pharmacia), then lyophilized three times. One of the Vydac fractions was further fractionated on a Lichrosorb-NH2 10 μm column (25 × 0.46 cm, Alltech, Breda, The Netherlands), equipped with a LiChrospher Amino 5 μm guard column (7.5 × 4.6 mm). Elutions were performed with a gradient of 30 mM K2HPO4/KH2PO4, pH 6.8, in acetonitrile, at a flow rate of 1.5 ml/min, monitored at 214 nm. Relevant fractions were concentrated under a N2 stream, and lyophilized.
2.7 Characterization of 15N-phCG/GS115
2.7.1 SDS-PAGE
The purity of 15N-phCG/GS115 was checked by SDS-PAGE under reducing conditions. The gel was composed of a 4% stacking gel at pH 6.8 and a 12% running gel at pH 8.8. The bands were visualized with Coomassie Blue.
2.7.2 Radioimmunoassay (RIA)
The hCG activity of the purified 15N-phCG/GS115 material was determined by RIA using various polyclonal and monoclonal antibody-based RIAs as described earlier [3, 16]. Predetermined quantities of urinary hCG or purified 15N-phCG/GS115 material were diluted in RIA buffer (0.5 M sodium phosphate buffer, pH 7.4, containing 150 mM NaCl and 50 mM EDTA), then incubated with appropriate dilutions of either hCG polyclonal antiserum or monoclonal antibodies against hCG and [125I]hCG (100,000 cpm/tube) overnight at room temperature. The antigen-antibody complexes were precipitated by adding an appropriate dilution of normal rabbit/ mouse serum, goat anti-rabbit/ anti-mouse IgG and 2.5% polyethylene glycol. The tubes were centrifugated at 5,000×g, the supernatant was discarded, and the radioactivity in the pellet was counted in a gamma counter.
2.7.3 Radioreceptor assays (RRA)
The ability of 15N-phCG/GS115 to bind to luteinizing hormone (LH) receptors was assayed by RRA, using a crude rat testicular homogenate as a source of LH receptors [3, 17]. The receptor preparation was incubated with varying concentrations of 15N-phCG/GS115 or urinary hCG in the presence of [125I]hCG (200,000 cpm/tube) as the tracer. The non-specific binding was determined by evaluating the binding of [125I]hCG to the receptor in the presence of a large excess of unlabeled hCG (400 ng/ml). The samples were incubated at room temperature for 2–3 h, followed by addition of 1 ml RRA buffer consisting of 0.05 M phosphate buffer, pH 7.4, containing 5 mM MgCl2 and 1 mg/ml BSA. The samples were centrifugated for 15 min at 4,000×g at 4°C. The supernatant was discarded and the pellet was counted using a Perkin Elmer autogamma counter.
2.7.4 Mouse Leydig cell in vitro bioassay
The ability of 15N-phCG/GS115 to stimulate testosterone production in mouse Leydig cells, obtained from adult male Swiss mice (2–3 months old) was assayed as described [17]. Testes were decapsulated, suspended in Dulbecco’s minimum essential medium (DMEM), pH 7.4, containing 0.05 M Hepes and 0.1% bovine serum albumin, and stirred in an ice bath on a magnetic stirrer for 15 min. The cell suspension per testis was filtered through a nylon cloth, oxygenated, and incubated for 1 h at 34°C (shaking water bath). After centrifugation for 10 min at 750×g, the cells were washed twice with DMEM, and then resuspended in 5 ml medium. Aliquots of the cell suspension (0.4 ml/tube) were incubated with different dilutions of 15N-phCG/GS115 or urinary hCG for 4 h at 34°C (shaking water bath) following oxygenation. Testosterone, secreted upon stimulation with 15N-phCG/GS115 or urinary hCG, was determined by a testosterone specific RIA, as described earlier [17], using specific testosterone antiserum (at 1:50,000 dilution) and [1,2,6,7,16,17-3H]-testosterone (6,000 cpm/tube) in a total volume of 400 μl. Binding reactions were carried out for 1 h at room temperature followed by 1 h at 4°C. At the end of the incubation period, free testosterone was removed by adding an equal volume of dextran-coated charcoal (1 g charcoal and 0.1 g dextran/ml). Subsequently, the tubes were centrifugated at 5,000×g for 15 min at 4°C. 700 μl of each supernatant were thoroughly mixed with 1 ml scintillation cocktail (2.5 g 2,5-diphenyloxazole, 200 mg p-bis[2-(5-phenyloxazolyl)]-benzene, 10.5 ml methanol in 500 ml toluene). The solutions were incubated at room temperature in the dark for at least 4 h, and radioactivity was counted in a Pharmacia Liquid Scintillation counter.
2.8 Analysis methodologies
2.8.1 Monosaccharide analysis
Desalted glycoprotein samples were subjected to methanolysis, followed by re-N-acetylation and trimethylsilylation, and the generated mixtures of trimethylsilylated methyl glycosides were analyzed by GLC-EI/MS, as described [5, 18].
2.8.2 Methylation analysis
The hypermannose-type chain fraction derived from phFSH/X-33 (1 mg) was permethylated using methyl iodide and solid sodium hydroxide in dimethyl sulfoxide. After hydrolysis, the mixture of partially methylated monosaccharides was converted into a mixture of partially methylated alditol acetates, which was analyzed by GLC-EI/MS, as described [19, 20].
2.8.3 2AB-labeling of N-glycans and HPLC profiling
Enzymatically released glycoprotein N-glycans were derivatized with 2-aminobenzamide, as described [5, 21]. Neutral N-glycan profiling on normal phase TSKgel Amide-80 and charged N-glycan profiling on weak anion-exchange Vydac 301 VHP5410 were carried out as reported previously [5].
2.8.4 Matrix-assisted laser desorption ionization time-of-flight mass spectrometry
MALDI-TOF mass spectra were recorded on a Voyager-DE PRO mass spectrometer (Applied Biosystems, Nieuwerkerk aan de IJssel, The Netherlands) with implemented delayed extraction technique, equipped with a N2 laser (337 nm, 3 ns pulse width). Spectra (positive-ion mode) were recorded in the linear mode at an accelerating voltage of 25 kV using an extraction delay of 600 ns. Peptide samples (0.5 μl), when necessary desalted with Omix tips C18 (Varian, Lake Forest, CA), were mixed in a 1:1 ratio on the target with α-cyano-4-hydroxycinnaminic acid (10 mg/ml) dissolved in acetonitrile—0.3% aqueous trifluoroacetic acid (50:50, v/v). 15N-phCG/GS115 (2 mg/ml) was dissolved in 0.2% trifluoroacetic acid and boiled for 3 min. Then, 0.5-μl samples were mixed in a 1:1 ratio on the target with 3,5-dimethoxy-4-hydroxycinnaminic acid (sinapinic acid, 10 mg/ml) dissolved in acetonitrile—0.3% aqueous trifluoroacetic acid (50:50, v/v).
2.8.5 Amino acid analysis
15N-phCG/GS115 (400 μg) was hydrolyzed in 6 M HCl at 110°C for 22 h. After trimethylsilylation, the derivatized amino acids were analyzed by GLC-EI/MS on a Fisons Instruments GC 8060/MD 800 system (Interscience), equipped with an AT-1 column (30 m × 0.25 mm, Alltech, Breda, The Netherlands). The temperature was maintained for 2 min at 80°C, and then increased at 4°C/min until 180°C, which was kept for 5 min.
2.8.6 Circular dichroism (CD)
Samples were prepared by dissolving 0.5 mg 15N-phCG/GS115 in 300 μl 50 mM sodium phosphate buffer, pH 5.6. CD measurements were carried out between 260 and 190 nm at room temperature on a Jasco J-810 spectropolarimeter, using a 1-mm path length cell, 1-nm bandwidth, 1-s response time, and a scan speed of 50 nm/min.
2.8.7 NMR spectroscopy
NMR spectra were recorded on a 900-MHz NMR spectrometer (Bruker Avance DRX). Lyophilized 15N-phCG/GS115 (15 mg) was dissolved in 500 μl 90% (v/v) H2O, containing 10 mM EDTA and 10% 2H2O (99.9%; Cambridge Isotope Laboratories Inc., MA). The 15N-HSQC spectrum was recorded at a probe temperature of 57°C and pH 4.4, using Echo/Antiecho-TPPI gradient selection with decoupling during acquisition [22–24]. The experiments were obtained with 128 × 1024 points, 128 scans using a spectral width of 2737 Hz in the 15N dimension and 9920 Hz in the 1H direction. NMR data sets were processed using in house developed software with zero filling, π/2 shifted sine bell window function for t1, π/2 shifted squared sine bell window function for t2, and automatic spline baseline correction in both dimensions.
3 Results and discussion
3.1 Optimization of the production of recombinant phCG in P. pastoris GS115
In general, P. pastoris can provide high expression levels of foreign proteins [3, 13], being strongly dependent on the methanol-regulated promoter of the alcohol oxidase 1 gene (AOX1) [25, 26]. In order to produce 15N-labeled glycohormones in an economical way, an optimization of earlier protocols [4, 14, 27] was investigated. Due to the high costs of 15N-labeled ammonia, the use of ammonia was not explored. Although both 15NH4Cl and (15NH4)2SO4 are equally priced, the use of (NH4)2SO4 results in a precipitation of K2SO4, which can hamper cell growth. As an exchange of medium would be required during the fermentation to remove K2SO4 [27], 15NH4Cl was selected instead. Looking for a reduction of the costs of a potential 13C labeling, introductory experiments showed that partial replacement of glycerol [14, 28] by glucose turned out to be an excellent option. Therefore, in the present work the nitrogen/carbon sources NH4Cl/glucose-glycerol-methanol were explored to replace the earlier used [4] ammonia/glycerol-methanol. The finally developed production process, yielding the highest cell densities and yields (about 25 mg/l phCG/GS115) so far, has been detailed in “Materials and methods.”
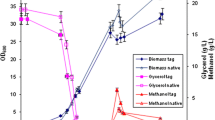
For protein expression in P. pastoris, it is crucial to supply an optimal amount of NH4Cl. However, continuous diluted NH4Cl feeding was not explored. Previous protocols [14] recommended to feed the culture every 24 h with 10 g/l of NH4Cl. Following this strategy, however, we found that on the second day of the fermentation, 5 g/l were enough to ensure acidification of the medium by the cells because there was no change in the DO. The short glycerol fed-batch phase of 30 min was long enough to derepress the AOX1 promoter [14]. At the end of the batch phase (48 h), just before methanol induction, the wet cell weight (wcw) reached 109 g/l (Fig. 1), and 48 h after the beginning of the methanol induction, the wcw reached 233 g/l, then decreased to 198 g/l because of the dilution effect. It should be noted that this is the first time that such high cell densities are reported for recombinant glycoprotein hormones in P. pastoris. This is probably due to the use of NH4Cl instead of ammonia [4] or (NH4)2SO4 for which in both cases the highest wcw never exceeded 62 g/l (OD 150) (unpublished data). During the adaptation phase of the methanol induction, 50% aqueous methanol was supplied at a flow rate of 6 ml/h for 8 h. The choice of diluted methanol was to avoid a toxic methanol accumulation, which would provoke cell death. Methanol feeding was progressively increased in order to maintain a good O2 drop, and after 30 h, a solution of 90% aqueous methanol was used as carbon source.

Growth curve of Pichia pastoris cells during the fermentation with NH4Cl/glucose–glycerol–methanol as nitrogen/carbon sources
Summarising, a highly efficient protocol using the nitrogen/carbon sources NH4Cl/glucose-glycerol-methanol for the production of phCG in P. pastoris GS115 could be established.
3.2 Comparison of the N-glycan profiles of phCG/GS115, phCG/X-33, and phFSH/X-33
Recently, we have investigated the glycosylation pattern of phCG/GS115 expressed in P. pastoris strain GS115 using ammonia/glycerol-methanol as nitrogen/carbon sources [5]. The N-glycans consisted of neutral (80%) and charged, phosphate-containing (20%) high-mannose-type structures. Mannosyl O-glycans were not detected. Neutral oligosaccharides were composed of Man8GlcNAc2 (11%), Man9GlcNAc2 (47%), Man10GlcNAc2 (28%), Man11GlcNAc2 (10%), Man12GlcNAc2 (4%), and traces of Man13–15GlcNAc2. Mono- and di-phosphate-containing oligosaccharides were present, whereby the mono-phosphate compounds ranged from Man9 PGlcNAc2 to Man13 PGlcNAc2. For the detailed isomer composition of the various Man8–11GlcNAc2 ensembles, see [5].
In order to search for differences in N-glycosylation when glycoprotein hormones are expressed in P. pastoris strain GS115 or X-33, of importance for future NMR studies, N-glycan screenings were carried out on the phCG/GS115 (new conditions), phCG/X-33, and phFSH/X-33 batches. Monosaccharide analysis performed on phCG/GS115 revealed a carbohydrate content of 30% (by mass), and the presence of Man and GlcNAc in the molar ratio of 9:2, comparable with phCG/GS115 when using ammonia/glycerol-methanol as nitrogen/carbon sources [5]. However, monosaccharide analysis of phCG/X-33 and phFSH/X-33 revealed a carbohydrate content of 37 and 39% (by mass), respectively, and Man and GlcNAc were present in the molar ratio of 33:2 and 36:2, respectively. In view of the relatively high amounts of mannose found, the possible presence of free mannose in the samples was excluded via an additional purification by size-exclusion chromatography (Hitrap). As no differences were found between the first and the second monosaccharide analysis of each probe, these data indicate the presence of hypermannosylation in both phCG/X-33 and phFSH/X-33.
After reduction and carboxymethylation, the three samples were digested with PNGase F, and the completeness of the N-deglycosylation was checked by SDS-PAGE.
In the case of phCG/GS115, the pool of released N-glycans was isolated via size-exclusion chromatography, then 2AB-labeled, and analyzed by HPLC. The molar ratio of Man8GlcNAc2:Man9GlcNAc2:Man10GlcNAc2:Man11GlcNAc2 was shown to be 19:38:27:16. Glycans larger than Man11GlcNAc2 were present in negligible amounts. Evidently, the neutral N-glycosylation patterns of the phCG/GS115 batches prepared using the nitrogen/carbon sources NH4Cl/glucose–glycerol–methanol (this study) and ammonia/glycerol–methanol [5] contain the same components, although in slightly different molar ratios. The same holds for the charged N-glycans. Note that the Man9GlcNAc2 isomer mainly represents [5]:

In the case of phCG/X-33 and phFSH/X-33, the pools of N-glycans were fractionated by size-exclusion chromatography, yielding three fractions, denoted P10.1, P10.2, and P10.3 (Fig. 2). Fraction P10.1 contained the protein moiety. Monosaccharide analysis demonstrated the occurrence of carbohydrate in fractions P10.2 (Man:GlcNAc = 33:2 for phCG/X-33, and 36:2 for phFSH/X-33) and P10.3 (Man:GlcNAc = 10:2 for both samples); fraction P10.2 contained small amounts of protein.

Bio-Gel P-10 fractionation pattern of PNGase F-treated phFSH/X-33. Elutions were performed at a flow rate of 13 ml/h and monitored by refractive index detection. A similar profile was obtained with phCG/X-33. Peak 1, protein; peak 2, hypermannose-type N-glycans; peak 3, high-mannose-type N-glycans
HPLC profiling of the 2AB-labeled P10.3 fractions derived from phCG/X-33 and phFSH/X-33, showed in both cases a series of neutral oligomannose-type N-glycans, ranging from Man9GlcNAc2 to Man11GlcNAc2 (Fig. 3a and b); molar ratios of Man9GlcNAc2:Man10GlcNAc2:Man11GlcNAc2 are 43:39:17 for phCG/X-33, and 40:42:18 for phFSH/X-33. Comparing Fig. 3a and b, it is postulated that phCG/X-33 and phFSH/X-33 present slight differences in their Man10GlcNAc2 and Man11GlcNAc2 isomeric populations. In contrast to phCG/GS115 [5], phCG/X-33 and phFSH/X-33 did not contain phosphorylated N-glycans.

HPLC profile of 2AB-labeled neutral oligosaccharides derived from glycoprotein samples expressed in strain X-33 on a normal-phase TSKgel Amide-80 column. Elutions were carried out with a gradient of ammonium formate, pH 4.4, in acetonitrile, at a flow rate of 0.8 ml/min [5]. a N-glycans of phCG/X-33; b N-glycans of phFSH/X-33. M = Man, GN = GlcNAc. Under the applied conditions, the acidic compounds elute in the void volume, not shown in the chromatograms
Fraction P10.2 of phFSH/X-33 was subjected to methylation analysis, and it was found that the P10.2 glycans are built up from terminal Man, 2-substituted Man, and 2,6-disubstituted Man in the molar ratio 1.2:2.3:1.0; very small amounts of 6- and 3,6-di-substituted Man were detected. It should be noted that, as expected, the hypermannose-type chains did not contain (α1–3)-linked Man residues in the outer part [29].
Based on the finding of hypermannosylation when using the X-33 strain, the GS115 strain, yielding a more simple N-glycosylation pattern, was selected for generating 15N-labeled phCG for NMR studies.
This is the first glycosylation study on glycoproteins expressed in the P. pastoris wild type X-33 strain, a strain that has become commercially available recently. The found hypermannosylation confirms the recent findings of [30] for the glycosylated chicken cystatin expressed in the X-33 strain, whereby hypermannose-type N-glycans up to 50 Man residues were indicated. Probably, the difference of one basepair in the HIS4 gene of the X-33 strain causes a frame shift, and therefore induces translation modifications. P. pastoris strains are generally preferred to Saccharomyces cerevisiae strains, because they were known for their suitability to express recombinant glycoproteins with only high-mannose-type, and not hypermannose-type N-glycans. With the introduction of the P. pastoris wild type X-33 strain, one should really realize the differences in glycosylation machinery among P. pastoris strains.
3.3 Site-specific N-glycosylation of phCG/GS115
phCG/GS115 (N-glycosylation sites at αAsn52, αAsn78, βAsn13, and βAsn30) was reduced, carboxymethylated, and digested with trypsin. The resulting mixture of peptides and glycopeptides was subjected to ConA-Sepharose chromatography, and the glycopeptide-containing fraction, eluted with 100 mM methyl α-d-mannopyranoside, was subjected to HPLC analysis, yielding 3 subfractions denoted 1, 2, and 3 (Fig. 4). Each subfraction was digested with either PNGase F or Endo H, and analyzed by positive-ion mode MALDI-TOF-MS. Under these conditions, being specific for peptides, released carbohydrates are not detected as they do not crystallize with the matrix and/or fail to protonate. Due to the limited amount of material available, attention was paid only to the neutral N-glycans, representing 80% of the total N-glycan pool. Fraction 1, digested with PNGase F, presented [M+H]+ signals at m/z 1373.2 and 2070.9, corresponding to the peptides β9–20 and α76–92, respectively [31, 32]. Amino acid analysis performed on fraction 1 revealed the presence of about 50% β9–20 and 50% α76–92. Subfractionation of fraction 1 on Lichrosorb-NH2 yielded 2 fractions, denoted 1-1 and 1-2. MALDI-TOF-MS analysis of these fractions, digested with PNGase F, showed that fraction 1-1 corresponded to β9–20 (Fig. 5a) and fraction 1-2 to α76–92 (Fig. 5d) [31, 32]. Fraction 2 was resistant to PNGase F, but, digested with Endo H, it gave rise to a single [M+H]+ peak at m/z 1561.3, corresponding to peptide α52–63 with one GlcNAc residue attached at αAsn52 (Fig. 5b). It should be noted that PNGase F does not release N-glycans from Asn residues that occur as N- or C-terminus in a peptide [33]. MALDI-TOF-MS analysis of fraction 3, digested with PNGase F, demonstrated the presence of a single [M+H]+ peak at m/z 2667.2, corresponding to peptide β21–43 (Fig. 5c) [31, 32]. Although the HPLC profile suggests a poor separation between fractions 2 and 3 (Fig. 4), MALDI-TOF-MS analysis of these fractions showed pure α52–63 and β21–43, respectively. Finally, the released 2AB-derivatized N-glycans of the fractions 1-1, 1-2, 2, and 3 were subjected to HPLC analysis on TSKgel amide-80 (normal phase) and Vydac 301 VHP5410 (weak-anion exchange), and the results are presented in Table 1.

HPLC elution profile at 214 nm of the phCG/GS115-derived tryptic glycopeptide fraction on a reversed-phase Vydac C4 column. Elutions were carried out with a gradient of 0–80% (v/v) solvent C [60% acetonitrile in triethylamine phosphate] in solvent B [5% acetonitrile in triethylamine phosphate] at 1.5 ml/min

MALDI-TOF mass spectra of deglycosylated tryptic glycopeptides derived from phCG/GS115, recorded in the positive-ion mode: a glycopeptide β9–20 (PNGase F); b glycopeptide α52–63 (endo H); c glycopeptide α21–43 (PNGase F); d glycopeptide α76–92 (PNGase F)
As is evident from Table 1, the N-glycosylation profiles at αAsn52 and αAsn78 are highly similar with dominating Man9GlcNAc2 and Man10GlcNAc2 structures. The N-glycosylation profile of βAsn13 presents similar amounts of Man8–11GlcNAc2, whereas for βAsn30 the Man9GlcNAc2 structure strongly dominates. Interestingly, compared with the other glycosylation sites, βAsn30 contains a very low amount of Man11GlcNAc2. It seems that the N-glycans in the α-subunit are more processed than those in the β-subunit. Within the β-subunit the N-glycans at βAsn13 are more processed than those at βAsn30. It should be noted that the N-glycan at αAsn52 in urinary hCG, located at the subunit interface [10, 11], is involved in receptor binding and in steroidogenic activity [34–36]. As phCG/GS115 is biologically active, the high-mannose-type N-glycans at αAsn52 seem to replace efficiently the sialylated glycan at αAsn52 in urinary hCG.
The difference in processing between βAsn13 and βAsn30 could be explained in terms of space availability. Crystallographic studies have shown that the N-glycans at βAsn13 and βAsn30 are located less than 7 Å from each other on the outward faces of the two adjacent β-strands [11]. Due to this proximity, it is postulated that there is a competition between the mannosyltransferase-directed growing of the more bulky high-mannose-type chains, as compared with the complex-type structures in urinary hCG, at both sites, whereby the faster growing bulky glycan at βAsn13 might prevent any further processing at βAsn30.
3.4 Production and characterization of 15N-phCG/GS115
Performing the culturing protocol using 15NH4Cl/glucose-glycerol-methanol as nitrogen/carbon sources for the preparation of phCG in P. pastoris strain GS115, it was possible to isolate from 3 l culture supernatant about 25 mg of 15N-phCG/GS115.
The purified protein showed two bands on SDS-PAGE with apparent molecular masses of 25 kDa and 35 kDa, corresponding to N-glycosylated α- and β-15N-phCG/GS115, respectively (Fig. 6, lane 2). Theoretical masses of α- and β-15N-phCG/GS115 (15.9 kDa and 21.3 kDa, respectively) were calculated considering that a Man9GlcNAc2 high-mannose-type structure was present at each N-glycosylation site. Apparent molecular masses are larger than theoretical masses, presumably because of partial shielding of the protein moiety, hindering the binding of SDS to the protein. The 15N incorporation in the labeled phCG/GS115 was estimated by GLC-EI/MS of a trimethylsilylated protein hydrolysate. Six amino acids (valine, leucine, glycine, serine, threonine, and proline) could be clearly identified in the TIC chromatogram by comparing their mass spectra (data not shown) with reference data [37]. Calculation of the ratio of 15N-enriched / non-enriched fragments using the mass spectra of the mentioned amino acids demonstrated that phCG/GS115 was 15N-labeled for about 70%. The non-completed labeling of the hCG is due to the fact that the preculturing was performed with 14NH4Cl.

12% SDS-PAGE gel of purified 15N-phCG/GS115 stained with Coomassie Blue. Lane 1 indicates the protein molecular markers (phosphorylase B, 94 kDa; bovine serum albumin, 67 kDa; ovalbumin, 43 kDa; carbonic anhydrase, 30 kDa; trypsin inhibitor, 20.1 kDa; α-lactalbumin, 14.4 kDa). Lane 2 shows 15N-phCG/GS115 (α-, β-subunits)
As reported previously for glycoprotein hormones [38–40], characterization of intact hCG is quite difficult by MALDI-TOF-MS. Very broad peaks are obtained due to glycan heterogeneity and also here to the 15N-incorporation of 70%. A typical mass spectrum of 15N-phCG/GS115 contained peaks corresponding to the α- and β-subunits at 15.9 and 21.3 kDa, respectively (Fig. 7). Unfortunately, the 15N-enrichment of phCG/GS115 could not be detected by this technique.

MALDI-TOF mass spectrum of purified 15N-phCG/GS115. The matrix consisted of 3,5-dimethoxy-4-hydroxycinnaminic acid (sinapinic acid, 10 mg/ml) dissolved in acetonitrile-0.3% aqueous trifluoroacetic acid (50:50, v/v). Before analysis, the sample was dissolved in 0.2% aqueous trifluoroacetic acid, and boiled for 3 min
The secondary structure of 15N-phCG/GS115 was studied by circular dichroism (CD), and compared with urinary hCG. Previous studies carried out on urinary hCG have suggested that covalent binding of carbohydrate results in an increase of the optical activity at 207 nm [41]. As is evident from Fig. 8, the CD spectra of both compounds, recorded under the same conditions, are superimposable, indicating that the structure of 15N-phCG/GS115 is similar to that of urinary hCG. It can be inferred that 15N-phCG/GS115 possesses the proper assembly of subunits. The high-mannose-type N-glycans of 15N-phCG/GS115 [5], being partially phosphorylated, seem to have no influence on the secondary structure of the protein.

CD spectra of urinary hCG (thin line) and 15N-phCG/GS115 (thick line), recorded at room temperature. Samples (0.5 mg) were dissolved in 300 μl 50 mM sodium phosphate buffer, pH 5.6
The reconstituted glycohormone showed similar biological activities as urinary hCG in terms of receptor binding, cAMP stimulation and steroidogenesis. The overall conformation of 15N-phCG/GS115 was judged by radioimmunoassay using polyclonal antiserum against hCG [17]. All the RIA curves were analyzed using Graphpad prism software (version 3.03). As shown in Fig. 9a, the RIA displacement curve obtained with 15N-phCG/GS115 is parallel to that observed with an authentic urinary hCG preparation, suggesting that the overall conformation in 15N-hCG/GS115 is comparable to that of the natural hormone. To demonstrate that 15N-phCG/GS115 is biologically active, its ability to bind to the LH receptor and elicit a biological response was next determined. As shown in Fig. 9b, 15N-phCG/GS115 was able to inhibit completely the binding of 125I-hCG to the receptor. Furthermore, the biological activity was demonstrated by incubating the mouse Leydig cells with equivalent receptor activities of hCG, 15N-phCG/GS115 and phCG/GS115 for 4 h, and testosterone secreted into the medium was determined. Typical dose response curves are shown in Fig. 9c, where 15N-phCG/GS115, phCG/GS115, and urinary hCG showed almost identical activities. 15N-phCG/GS115 has therefore full biological activity as judged by RIA, RRA, and in vitro bioassays.

Characterization of the hCG activity of purified 15N-phCG/GS115. a Radioimmunoassay with B52/12 MAb. b Receptor binding activity of 15N-phCG/GS115. Incubation with a rat LH receptor preparation along with [125I]hCG; radioactivity bound to the receptor was determined. c Biological activity of 15N-phCG/GS115. Incubation of urinary hCG, phCG/GS115 and 15N-phCG/GS115 with mouse Leydig cells; testosterone secreted in the medium was measured by RIA
The 15N-HSQC NMR spectrum of 15N-phCG/GS115 (Fig. 10) presents a broad chemical shift dispersion of backbone amide protons from 9.7 to 7.2 ppm, being characteristic of structured proteins. 15N-phCG/GS115 (with 30 Pro residues and 2 N-termini) contains 233 amino acid residues that could potentially be observed in a 15N-HSQC spectrum. In fact, approximately 220 cross-peaks are observed in the spectrum of Fig. 10, which indicates that the large hCG (molecular mass 37.2 kDa, including the oligosaccharide chains and the 15N incorporation) is not aggregating at the NMR conditions, and that it has relatively uniform hydrodynamic properties. The cross-peaks in the 15N-HSQC spectrum show a wide distribution in peak intensity. Although this could be due to sample heterogeneity of 15N-phCG/GS115 due to different glycosylation patterns, or partially due to anisotropic tumbling, it appears more likely that this reflects differences in dynamics. Approximately 40 NH cross-peaks, that are more intense than most other signals, are all located in a narrow region of the spectrum (F2 = 8.5–7.8 ppm/F1 = 125–130 ppm), and thus show limited chemical shift dispersion. These residues are likely located in an unstructured highly mobile part of 15N-phCG/GS115.

1H-15N HSQC spectrum of 15N-phCG/GS115, recorded at a probe temperature of 57°C and pH 4.4
The crystallographic structure of dimeric hCG [11] showed a disordered region with no electron density for the carboxylic terminal extension of β-hCG (βAsp111- βGln145). An intense cross-peak can be observed at 8.43/112.5 ppm, the random coil frequencies for Gly, which can then be assigned to βGly136. Therefore, we ascribe most of the intense signals to this part of the protein, which then demonstrates that the disorder in the X-ray structure is due to high mobility of hCG.
3.5 Final remarks
Until now, the production of labeled proteins for NMR purposes has been carried out mainly in bacterial systems, such as E. coli. Previously, also α- and β-hCG have been expressed in these bacteria [42, 43], and 6–10 mg/l of unfolded proteins could be produced. Refolding of each subunit followed by dimer assembly is then necessary to obtain an hCG heterodimer. However, non-glycosylated systems are obtained, usually not fully biologically active [42], which are not useful in case of studying native glycoproteins. Also eukaryotic systems were investigated for hCG expression, e.g. insect cells (α-hCG [44], β-hCG [45], and hCG [46]) as well as Dictyostelium discoideum (hCG [47]). hCG has also been expressed in Chinese hamster ovary (CHO) cells, and its glycosylation pattern was established [48]. This recombinant hCG is used for in vitro fertilization treatments [49, 50]. CHO cells [51, 52] were also used to produce uniformly 13C- and 15N-labeled hCG, but NMR structures have not been solved. P. pastoris was selected because this yeast can perform post-translational modifications, is easier to grow than CHO cells, and requires media of a simpler formulation. The present investigation has shown that this choice can generate very efficiently 15N-labeled hCG to be used in NMR studies.
Abbreviations
- 2AB:
-
2-Aminobenzamide
- CD:
-
Circular dichroism
- CHO:
-
Chinese hamster ovary
- DMEM:
-
Dulbecco’s minimum essential medium
- DO:
-
Dissolved oxygen
- GLC-EI/MS:
-
Gas-liquid chromatography–electron impact mass spectrometry
- hCG:
-
Human chorionic gonadotropin
- hFSH:
-
Human follicle stimulating hormone
- HPLC:
-
High-performance liquid chromatography
- HSQC:
-
Heteronuclear single quantum coherence
- LH:
-
Luteinizing hormone
- MAbs:
-
Monoclonal antibodies
- MALDI-TOF-MS:
-
Matrix-assisted laser desorption ionization time-of-flight mass spectrometry
- NMR:
-
Nuclear magnetic resonance
- PCR:
-
Polymerase chain reaction
- phCG:
-
Human chorionic gonadotropin expressed in Pichia pastoris
- phFSH:
-
Human follicle stimulating hormone expressed in Pichia pastoris
- PNGase F:
-
Peptide-N 4-(N-acetyl-β-glucosaminyl) asparagine amidase F
- RIA:
-
Radioimmunoassay
- RRA:
-
Radioreceptor assays
- TIC:
-
Total ion current
- TPPI:
-
Time proportional phase incrementation
- wcw:
-
Weight cell weight
- YPD:
-
Yeast peptone dextrose
References
Eckart, M.R., Bussineau, C.M.: Quality and authenticity of heterologous proteins synthesized in yeast. Curr. Opin. Biotechnol. 7, 525–530 (1996)
Cereghino, J.L., Cregg, J.M.: Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol. Rev. 24, 45–66 (2000)
Sen Gupta, C., Dighe, R.R.: Hyperexpression of biologically active human chorionic gonadotropin using the methylotropic yeast, Pichia pastoris. J. Mol. Endocrinol. 22, 273–283 (1999)
Gadkari, R., Deshpande, R., Dighe, R.R.: Hyperexpression and purification of biologically active human luteinizing hormone and human chorionic gonadotropin using the methylotrophic yeast, Pichia pastoris. Protein Expr. Purif. 32, 175–184 (2003)
Blanchard, V., Gadkari, R.A., Gerwig, G.J., Dighe, R.R., Leeflang, B.R., Kamerling, J.P.: Characterization of the N-linked oligosaccharides from human chorionic gonadotropin expressed in the methylotrophic yeast Pichia pastoris. Glycoconj. J. 24, 33–47 (2007)
De Beer, T., van Zuylen, C.W., Leeflang, B.R., Hård, K., Boelens, R., Kaptein, R., Kamerling, J.P., Vliegenthart, J.F.G.: NMR studies of the free α-subunit of human chorionic gonadotropin. Structural influences of N-glycosylation and the β-subunit on the conformation of the α-subunit. Eur. J. Biochem. 241, 229–242 (1996)
Van Zuylen, C.W., de Beer, T., Leeflang, B.R., Boelens, R., Kaptein, R., Kamerling, J.P., Vliegenthart, J.F.G.: Mobilities of the inner three core residues and the Man(α1–6) branch of the glycan at Asn78 of the α-subunit of human chorionic gonadotropin are restricted by the protein. Biochemistry 37, 1933–1940 (1998)
Erbel, P.J., Karimi-Nejad, Y., de Beer, T., Boelens, R., Kamerling, J.P., Vliegenthart, J.F.G.: Solution structure of the α-subunit of human chorionic gonadotropin. Eur. J. Biochem. 260, 490–498 (1999)
Erbel, P.J., Karimi-Nejad, Y., van Kuik, J.A., Boelens, R., Kamerling, J.P., Vliegenthart, J.F.G.: Effects of the N-linked glycans on the 3D structure of the free α-subunit of human chorionic gonadotropin. Biochemistry 39, 6012–6021 (2000)
Wu, H., Lustbader, J.W., Liu, Y., Canfield, R.E., Hendrickson, W.A.: Structure of human chorionic gonadotropin at 2.6 Å resolution from MAD analysis of the selenomethionyl protein. Structure 2, 545–558 (1994)
Lapthorn, A.J., Harris, D.C., Littlejohn, A., Lustbader, J.W., Canfield, R.E., Machin, K.J., Morgan, F.J., Isaacs, N.W.: Crystal structure of human chorionic gonadotropin. Nature 369, 455–461 (1994)
Sambrook, J., Fritsch, E.F., Maniatis, T.: Molecular Cloning: A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, New York (1989)
Laroche, Y., Storme, V., De Meutter, J., Messens, J., Lauwereys, M.: High-level secretion and very efficient isotopic labeling of tick anticoagulant peptide (TAP) expressed in the methylotrophic yeast, Pichia pastoris. Bio/Technology 12, 1119–1124 (1994)
Van den Burg, H.A., de Wit, P.J.G.M., Vervoort, J.: Efficient 13C/15N double labeling of the avirulence protein AVR4 in a methanol-utilizing strain (Mut+) of Pichia pastoris. J. Biomol. NMR 20, 251–261 (2001)
Lustbader, J.W., Birken, S., Pollak, S., Levinson, L., Berstine, E., Hsiung, N., Cornfield, R.: Characterization of the expression products of recombinant human choriogonadotropin and subunits. J. Biol. Chem. 262, 14204–14212 (1987)
Dighe, R.R., Murthy, G.S., Moudgal, N.R.: Two simple and rapid methods to detect monoclonal antibodies with identical epitope specificities in a large population of monoclonal antibodies. J. Immunol. Methods 131, 229–236 (1990)
Dighe, R.R., Moudgal, N.R.: Use of α- and β-subunit specific antibodies in studying interaction of hCG with Leydig cell receptors. Arch. Biochem. Biophys. 225, 490–499 (1983)
Kamerling, J.P., Vliegenthart, J.F.G.: Carbohydrates. In: Lawson, A.M. (ed.) Clinical Biochemistry—Principles, Methods, Applications, Vol. 1, Mass Spectrometry, pp. 175–263. Walter de Gruyter, Berlin, Germany (1989)
Ciucanu, I., and Kerek, F.: A simple and rapid method for the permethylation of carbohydrates. Carbohydr. Res. 131, 209–219 (1984)
Faber, E.J., van den Haak, M.J., Kamerling, J.P., Vliegenthart, J.F.G.: Structure of the exopolysaccharide produced by Streptococcus thermophilus S3. Carbohydr. Res. 331, 173–182 (2001)
Bigge, J.C., Patel, T.P., Bruce, J.A., Goulding, P.N., Charles, S.M., Parekh, R.B.: Nonselective and efficient fluorescent labeling of glycans using 2-aminobenzamide and anthranilic acid. Anal. Biochem. 230, 229–238 (1995)
Palmer, A.G., III, Cavanagh, J., Wright, P.E., Rance, M.: Sensitivity improvement in proton-detected two-dimensional heteronuclear correlation NMR spectroscopy. J. Magn. Reson. 93, 151–170 (1991)
Kay, L.E., Keifer, P., Saarinen, T.: Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J. Am. Chem. Soc. 114, 10663–10665 (1992)
Schleucher, J., Schwendinger, M., Sattler, M., Schmidt, P., Schedletzky, O., Glaser, S.J., Sørensen, O.W., Griesinger, C.: A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients. J. Biomol. NMR 4, 301–306 (1994)
Ellis, S.B., Brust, P.F., Koutz, P.J., Waters, A.F., Harpold, M.M., Gingeras, T.R.: Isolation of alcohol oxidase and two other methanol regulatable genes from the yeast Pichia pastoris. Mol. Cell. Biol. 5, 1111–1121 (1985)
Tschopp, J.F., Brust, P.F., Cregg, J.M., Stillman, C.A., Gingeras, T.R.: Expression of the lacZ gene from two methanol-regulated promoters in Pichia pastoris. Nucleic Acids Res. 9, 3859–3876 (1987)
Wood, M.J., Komives, E.A.: Production of isotopically labeled protein in Pichia pastoris by fermentation. J. Biomol. NMR 13, 149–159 (1999)
Rodriguez, E., Krishna, N.R.: An economical method for 15N/13C isotopic labeling of proteins expressed in Pichia pastoris. J. Biochem. 130, 19–22 (2001)
Bretthauer, R.K., Castellino, F.J.: Glycosylation of Pichia pastoris-derived proteins. Biotechnol. Appl. Biochem. 30, 191–192 (1999)
Tzeng, S.S., Jiang, S.T.: Glycosylation modification improved the characteristics of recombinant chicken cystatin and its application on mackerel surimi. J. Agric. Food Chem. 52, 3612–3616 (2004)
Weisshaar, G., Hiyama, J., Renwick, A.G.C.: Site-specific N-glycosylation of human chorionic gonadotropin, Structural analysis of glycopeptides by one- and two-dimensional 1H NMR spectroscopy. Glycobiology 1, 393–404 (1991)
Liu, C., Bowers, L.D.: Mass spectrometric characterization of nicked fragments of the β-subunit of human chorionic gonadotropin. Clin. Chem. 43, 1172–1181 (1997)
Maley, F., Trimble, R.B., Tarentino, A.L., Plummer, T.H. Jr.: Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal. Biochem. 180, 195–204 (1989)
Matzuk, M.M., Keene, J.L., Boime, I.: Site-specificity of the chorionic gonadotropin N-linked oligosaccharides in signal transduction. J. Biol. Chem. 264, 2409–2414 (1989)
Sairam, M.R., Jiang, L.G.: Comparison of the biological and immunological properties of glycosylation deficient human chorionic gonadotropin variants produced by site directed mutagenesis and chemical deglycosylation. Mol. Cell. Endocrinol. 85, 227–235 (1992)
Erbel, P.J., Haseley, S.R., Kamerling, J.P., Vliegenthart, J.F.G.: Studies on the relevance of the glycan at Asn-52 of the α-subunit of human chorionic gonadotropin in the αβ dimer. Biochem. J. 364, 485–495 (2002)
Leimer, K.R., Rice, R.H., Gehrke, C.W.: Complete mass spectra of the per-trimethylsilylated amino acids. J. Chromatogr. A 141, 355–375 (1977)
Laidler, P., Cowan, D.A., Hider, R.C., Keane, A., Kicman, A.T.: Tryptic mapping of human chorionic gonadotropin by matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom. 9, 1021–1026 (1995)
Jacoby, E.S., Kicman, A.T., Laidler, P., Iles, R.K.: Determination of the glycoforms of human chorionic gonadotropin β-core fragment by matrix-assisted laser desorption/ionization mass spectrometry. Clin. Chem. 46, 1796–1803 (2000)
Gervais, A., Hammel, Y.A., Pelloux, S., Lepage, P., Baer, G., Carte, N., Sorokine, O., Strub, J.M., Koerner, R., Leize, E., Van Dorsselaer, A.: Glycosylation of human recombinant gonadotrophins: characterization and batch-to-batch consistency. Glycobiology 13, 179–189 (2003)
Holladay, L.A., Puett, D.: Gonadotropin and subunit conformation. Arch. Biochem. Biophys. 171, 708–720 (1975)
Ren, P., Sairam, M.R., Yarney, T.A.: Bacterial expression of human chorionic gonadotropin α-subunit: studies on refolding, dimer assembly and interaction with two different β-subunits. Mol. Cell. Endocrinol. 113, 39–51 (1995)
Huth, J.R., Norton, S.E., Lockridge, O., Shikone, T., Hsueh, A.J.W., Ruddon, R.W.: Bacterial expression and in vitro folding of the β-subunit of human chorionic gonadotropin (hCGβ) and functional assembly of recombinant hCGβ with hCGα. Endocrinology 135, 911–918 (1994)
Nakhai, B., Sridhar, P., Talwar, G.P., Hasnain, S.E.: Construction, purification and characterization of a recombinant baculovirus containing the gene for α-subunit of human chorionic gonadotropin. Indian J. Biochem. Biophys. 28, 237–242 (1991)
Nakhai, B., Sridhar, P., Pal, R., Talwar, G.P., Hasnain, S.E.: Over-expression and characterization of recombinant β-subunit of the human chorionic gonadotropin hormone synthesized in insect cells infected with a genetically engineered baculovirus. Indian J. Biochem. Biophys. 29, 315–321 (1992)
Belzhelarskaya, S.N., Sutugina, L.P., Filenko, A.M., Buchatskii, L.P., Rubtsov, P.M.: Expression of human chorionic gonadotropin cDNA in insect cell culture. Mol. Biol. 30, 306–309 (1996)
Linskens, M.K.H., Grootenhuis, P.D.J., Blaauw, M., Huisman de Winkel, B., van Ravenstein, A., van Haastert, P.J.M., Heikoop, J.C.: Random mutagenesis and screening of complex glycoproteins: expression of human gonadotropins in Dictyostelium discoideum. FASEB J. 13, 639–645 (1999)
Kamerling, J.P., Hård, K., Vliegenthart, J.F.G. Structural analysis of carbohydrate chains of native and recombinant-DNA glycoproteins. In: Crommelin, D.J.A., Schellekens, H. (eds.) Developments in Biotherapy, vol. 1, From Clone to Clinic, pp. 295–304. Kluwer, Dordrecht, The Netherlands (1990)
The European Recombinant Human Chorionic Gonadotrophin Study Group: Induction of final follicular maturation and early luteinization in women undergoing ovulation induction for assisted reproduction treatment-recombinant hCG versus urinary hCG. Hum. Reprod. 15, 1446–1451 (2000)
Chang, P., Kenley, S., Burns, T., Denton, G., Currie, K., DeVane, G, O’Dea, L.: Recombinant human chorionic gonadotropin (rhCG) in assisted reproductive technology: results of a clinical trial comparing two doses of rhCG (Ovidrel) to urinary hCG (Profasi) for induction of final follicular maturation in in vitro fertilization-embryo transfer. Fertil. Steril. 76, 67–74 (2001)
Lustbader, J.W., Birken, S., Pollak, S., Pound, A., Chait, B.T., Mirza, U.A., Ramnarain, S., Canfield, R.E., Miles Brown, J.: Expression of human chorionic gonadotropin uniformly labeled with NMR isotopes in Chinese hamster ovary cells: an advance towards rapid determination of glycoprotein structures. J. Biomol. NMR 7, 295–304 (1996)
Weller, C.T., Lustbader, J., Seshadri, K., Brown, J.M., Chadwick, C.A., Kolthoff, C.E., Ramnarain, S., Pollak, S., Canfield, R., Homans, S.W.: Structural and conformational analysis of glycan moieties in situ on isotopically 13C, 15N-enriched recombinant human chorionic gonadotropin. Biochemistry 35, 8815–8823 (1996)
Acknowledgments
The authors acknowledge Prof. Dr. A. Verkleij for the use of the fermenter. This research has been financially supported by the Council for Chemical Sciences of the Netherlands Organization for Scientific Research (NWO-CW), the Department of Biotechnology/Indian Council of Medical research (New Dehli), and CSIR (fellowship to Satapura Roy). NWO-CW is acknowledged for using the 900 MHz NMR facility.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Véronique Blanchard and Rupali A. Gadkari contributed equally.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Blanchard, V., Gadkari, R.A., George, A.V.E. et al. High-level expression of biologically active glycoprotein hormones in Pichia pastoris strains—selection of strain GS115, and not X-33, for the production of biologically active N-glycosylated 15N-labeled phCG. Glycoconj J 25, 245–257 (2008). https://doi.org/10.1007/s10719-007-9082-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-007-9082-8