
Abstract
The evidence supporting the recent hypothesis of a homoploid hybrid origin for the butterfly species Heliconius heurippa is evaluated. Data from selective breeding experiments, mate-choice studies, and a wide variety of DNA markers are reviewed, and an alternative hypothesis for the origin of the species and its close relatives is proposed. A scenario of occasional red wing-pattern mutations in peripheral populations of Heliconius cydno with subsequent adaptive convergence towards sympatric mimicry rings involving H. melpomene and H. erato is offered as an alternative to the HHS hypothesis. Recent twists of this tale are addressed in a postscript.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The hybrid individuals cannot be distinguished from other individuals of H. cydno, indicating that multiple generations of backcrossing must have occurred. (Mavárez et al. 2006: Fig. S4)
If a similar scenario were involved in the origin of H. heurippa, then it would be very difficult to detect a significant signal of the hybrid founding event—the genome would primarily be derived from H. cydno despite the crucial role of introgression of the patterning genes in the formation of the novel lineage. (Jiggins et al. 2008)
One likely possibility is that timareta is another form that has stabilized after hybridization between Amazonian rayed melpomene and an unidentified, maybe extinct form of cydno. (Mallet 2009)
Some genuinely testable theories, when found to be false, are still upheld by their admirers—for example by introducing ad hoc some auxiliary assumption, or by reinterpreting the theory ad hoc in such a way that it escapes refutation. Such a procedure is always possible, but it rescues the theory from refutation only at the price of destroying, or at least lowering, its scientific status. (Popper 1968:37)
Heliconius butterflies (Lepidoptera: Nymphalidae) have long been celebrated for their bold aposematic coloration, Müllerian mimicry and striking intraspecific polymorphism (Bates 1862; Eltringham 1916; Turner 1976; Brown 1979). The diversity of wing color patterns exhibited by the co-mimetic pair Heliconius erato (L.) and Heliconius melpomene (L.) is unparalleled by any other insects, with at least ten highly distinct phenotypes shared between geographical races of the two species (Sheppard et al. 1985). The genetic architecture underlying this pattern diversity is likewise quite distinct from the norm. In “typical” butterfly wing patterns, the expression of pattern elements is more or less developmentally integrated over the entire wing surface (Monteiro et al. 1994; Beldade and Brakefield 2002) such that allelic variation in shapes, sizes and colors of the wing pattern may be expressed as single allelic “supergene” differences (Clarke and Sheppard 1960). By contrast, in Heliconius, different wing pattern elements are controlled by genes on separate chromosomes (Sheppard et al. 1985; Jiggins et al. 2005; Kapan et al. 2006) and assort independently in experimental crosses, as well as in natural hybrid zones (Sheppard 1963; Turner 1971; Mallet 1993). This diversity of pattern elements under independent genetic control has been referred to as a “toolbox” for the generation of phenotypic diversity (Gilbert 2003).
Müllerian mimicry is an “honest” signal between unpalatable prey and potential predator, and is selectively beneficial to the taxa that employ it because a smaller number of individuals per species must be attacked before a predator learns the aposematic signal and ceases pursuit of insects exhibiting it (Müller 1879). Mimetic aposematism is maintained by positive frequency-dependent selection: the most abundant pattern in a given habitat confers the greatest protection to individuals that display it, and exhibiting a novel phenotype is strongly disadvantageous (Mallet and Barton 1989). The diversity of mimetic forms within and among Heliconius species in spite of selection to conform to a single aposematic pattern has thus been viewed as an evolutionary paradox (Ford 1953; Turner 1977; Joron and Mallet 1998), explained variously as a product of vicariance in Pleistocene refugia (Brown et al. 1974), selective divergence across ecotones (Endler 1982; Mallet 1993), or some combination thereof (Turner 1982, 1983).
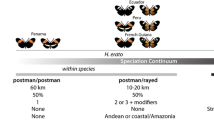
Hybrid zones both challenge the axiomatic view of an orderly natural hierarchy imposed by taxonomy, and provide a natural laboratory for the study of speciation (Harrison 1990, 1993). For decades, there has been an interest in Heliconius hybridization, both to understand the diversity of phenotypes produced in hybrid zones, and to characterize the putative selective forces that constrain the width of hybrid zones between phenotypically divergent yet interfertile geographical races (Descimon and Mast de Maeght 1983; Sheppard et al. 1985; Mallet 1989; Mallet et al. 1990; Jiggins et al. 1997, 2001a; Linares 1997a; Blum 2008). More recently, the focus of hybrid zone research has expanded to explore putative interspecific hybridization events (Naisbit et al. 2001; Gilbert 2003; Salazar et al. 2005; Bull et al. 2006; Kronforst et al. 2006). Although to date there is only a single wild-caught specimen that has been conclusively determined to be of interspecific hybrid origin (Dasmahapatra et al. 2007), Mallet et al. (2007) have argued, based on the occurrence of phenotypically atypical specimens from natural history museums that they infer to be natural interspecific hybrids, that the species boundary in Heliconius is a porous continuum, ranging from relatively free hybridization between close relatives, and exhibiting a logarithmic decrease in hybridization events as genetic divergence increases.
Perhaps the most dramatic development in recent times regarding theories of diversification of Heliconius butterflies was the widely-publicized paper by Mavárez et al. (2006; presaged by Salazar et al. 2005), proposing that the origin of the species Heliconius heurippa Hewitson stemmed from interspecific hybridization between geographical races of Heliconius cydno Doubleday and Heliconius melpomene. While self-perpetuating polyploid and/or parthenogenetic lineages may occasionally form as a result of hybridization between diploid sexual species (reviewed in Mallet 2007), these are typically instantly reproductively isolated from the parental lineage(s) by differences in chromosome number that prevent successful meiosis in backcrosses. The production of a reproductively-isolated, sexually reproducing, diploid species from two sexually reproducing parental species (known as homoploid hybrid speciation, HHS) is considered to be improbable, because chromosomal isolating mechanisms are not in place, and the genetic compatibility that allows hybridization between the parental species should not, in theory, promote the establishment of a reproductively isolated, genetically distinct yet sympatric hybrid offspring (Coyne and Orr 2004).
Most of the recently documented putative cases of HHS in animals (e.g., Nolte et al. 2005; Schwarz et al. 2005; Gompert et al. 2006; Kuusela et al. 2007) rely upon the hybrids’ occupation of novel habitats where they have neither the opportunity to compete against nor to interbreed with their parental species. By contrast, in the H. heurippa case, the putative hybrid offspring species is sympatric with one of its parents, and is hypothesized (according to the Mavárez et al. 2006 model; see also Duenez-Guzman et al. 2009) to have competitively excluded the other. As discussed above, the origin of a novel, phenotypically distinctive hybrid species such as H. heurippa arising from parent species belonging to different Müllerian mimetic complexes ought to be further impeded by the hurdle of overcoming the strong positive frequency-dependent selection by predators that maintains homogeneity within and differences between the parental phenotypes (Mallet and Barton 1989). Although several reviews discussing the H. heurippa case have already been published (Mallet 2007, 2009; Jiggins 2008; Jiggins et al. 2008; Mavárez and Linares 2008), most advance hybrid speciation of H. heurippa as received wisdom or hedge the original hypothesis with modified concepts such as “hybrid trait speciation” or “ecological speciation,” and none attempts to critically reevaluate the data purported to support the case. The object of this paper is to review the experimental and genetic evidence from the H. cydno—H. melpomene clade supporting the emerging Heliconius hybrid speciation paradigm, and to explore alternative hypotheses for the origin of H. heurippa.
Systematic background
Historically, members of the geographically variable species H. cydno and H. melpomene have been viewed as intrinsically partitioned by ecological differences into separate habitats, larval host plants and mimicry complexes (e.g., Smiley 1978; Mallet and Gilbert 1995; Srygley and Ellington 1999). The two species are broadly sympatric in their ranges from southern Mexico to Venezuela and western Ecuador, and each is involved in Müllerian mimicry with one or more members of the H. erato-H. sara “pupal mating” clade (Brown 1981; see Fig. 1). H. cydno is a comimic of black and white or black and yellow forms of H. sapho (Drury) and H. eleuchia Hewitson, while H. melpomene participates in the famous geographically diverse red, yellow and black association with H. erato (Eltringham 1916). There is one notable exception to this pattern: in the upper Cauca Valley, Colombia, H. melpomene, H. sapho and H. eleuchia are absent, and H. erato chestertonii Hewitson lacks a red forewing band and mimics H. cydno weymeri Staudinger form “gustavi,”Footnote 1 both species exhibiting a satiny bluish black ground coloration with a yellow transverse hindwing stripe.

Phylogenetic relationships among species of the genus Heliconius, as inferred by Beltrán et al. (2007), with presence or absence of red wing pattern elements optimized (assuming accelerated transformation) on internal nodes (red = red; yellow = no red; striped = equivocal). Basal red dots on the ventral surface of the hindwing are not considered “pattern elements.” If the gain and loss of red pattern elements has occurred parsimoniously, it was present in the ancestor of the genus, lost at least six times, but unequivocally regained only once (in H. elevatus Nöldner). Note that this cladogram represents H. melpomene as a single terminal and thus does not reflect its inferred paraphyly discussed in the text
Although H. cydno and H. melpomene have long been considered to be closely-related (e.g., Stichel and Riffarth 1905), Brower (1996a) presented the first empirically-supported cladogram examining relationships among geographical races of H. cydno, H. melpomene, and closely-related “species” H. heurippa, H. pachinus Salvin and H. timareta Hewitson (none of which, based upon the mtDNA sequences, appeared to be any more differentiated from H. cydno than the intraspecific variation among races within H. cydno, H. melpomene or H. erato). Brower (1996b) described H. tristero, a fourth H. cydno satellite, from the upper Putumayo valley in southeastern Colombia, geographically between the ranges of H. heurippa and H. timareta. The phylogenetic relationships implied by mtDNA COI-COII sequences in these papers suggest that H. melpomene is paraphyletic with respect to H. cydno and its satellites, with H. cydno and H. melpomene from Central America and Amazonia more closely related to one another than to H. melpomene from French Guiana. The paraphyletic H. melpomene pattern has generally been supported in subsequent work (Bull et al. 2006; Beltrán et al. 2007, Quek et al. 2010), although the sister-relationship between the H. cydno clade and a particular H. melpomene clade is not stable.
Heliconius heurippa was described in 1854 by W. C. Hewitson, based on material from eastern Colombia. The species, along with the eastern Ecuadorian H. timareta, was viewed by subsequent authors as a member of the “melpomene-group” (Riffarth 1901; Stichel 1906; Neustetter 1929; Emsley 1965; but see Brown 1979), based upon the presence of a red forewing band, although as early as 1901, Riffarth considered it to “stand between” H. melpomene and H. cydno. H. heurippa is sympatric with H. melpomene melpomeneFootnote 2 in the foothills above Villavicencio in Meta Prov., Colombia (I have collected both species at the same locality). As far as is known, H. heurippa is allopatric with H. cydno, the races in closest proximity being H. cydno wanningeri Neukirchen across the Cordillera Oriental of the Andes to the west (which is too high for easy dispersal), and H. cydno cordula Neustetter on the eastern slope of the Cordillera Oriental to the north in northern Colombia and western Venezuela (Salazar et al. 2005 suggest that this race may abut with H. heurippa near Yopal). Based on historical collection records, there is no evidence of natural hybridization in the wild between H. heurippa and H. cydno or H. melpomene (Mallet et al. 2007).
The toolbox
As noted above, Gilbert (2003) described a qualitative model for the evolution of wing pattern diversity in Heliconius as the “toolbox,” the “tools” being “those pattern genes that behave essentially the same” in H. melpomene and H. cydno. Gilbert’s scheme hypothesizes that after some initial diversification via mutation, that “hybridization and introgression would have begun to accelerate in importance and quickly would have replaced mutation as the proximate generator of novel pattern genotypes in Heliconius.” There is thus an unexplained reduction of the rate of wing-pattern altering mutations after the initial “ur-phenotypes” are established (the ur-cydno pattern is bluish black with white or yellow forewing and hindwing bands, and the ur-melpomene pattern is brownish black with at least some red pattern elements). White pattern elements observed in H. melpomene races, such as the hindwing marginal band of H. melpomene cythera Hewitson are hypothesized to have originated by introgression from sympatric cydno races (e.g., H. cydno alithea Hewitson), while red bands or rays in H. cydno-cognate taxa such as H. heurippa and H. timareta are thought to have originated via introgression from various H. melpomene forms.
While intuitively pleasing, Gilbert’s tool-swapping scheme is supported neither by phylogenetic evidence nor by comparative genomic data. Mapping the distribution of red wing pattern elements shows that presence of red pigment is plesiomorphic in Heliconius (Fig. 1) (the presence of red basal spots on the ventral surface of the hindwing may be a plesiomorphic feature of the entire subtribe). Thus, Heliconius species that do not exhibit red bands and/or rays are likely to have lost them. This is particularly relevant in the H. cydno-H. melpomene group, since it implies that the red wing pattern elements (or at least the genes that encode them) of putative “hybrid” taxa such as H. timareta and H. heurippa could be ancestral. Note also that while hybridization could explain novel patterns in those species, it cannot explain them in the equally phenotypically diverse H. erato or the sapho clade, members of which have no sympatric relatives with which they can exchange alleles (Gilbert 2003). In these taxa, all novel phenotypes, such as the white hindwing band in H. erato cyrbia Godart (the mimic of H. melpomene cythera) must have arisen by mutations.
From a genomic perspective, several recent papers have begun to reveal that the underlying genetic architecture of wing patterns in Heliconius is conserved between H. erato and H. melpomene (Joron et al. 2006; Papa et al. 2008; Baxter et al. 2010; Counterman et al. 2010), indicating that homologous genes have independently produced convergent phenotypes in different clades (an instance of homoiology, sensu Plate 1928; cf. Hennig 1966). If the alleles encoding, for example, red forewing patches (or absence thereof) have evolved independently in H. erato and H. melpomene, then there is no reason to assume that wing-pattern elements should not also arise in an unparsimonious manner within species, a hypothesis advanced by Brower (1996a). Thus, although hybridization may be a source of novel genetic variation, it is not unreasonable to invoke the occasional parallel switching on or off of shared genes responsible for similar-looking wing patterns as another potential source of novel variation within Heliconius species.
Evidence for hybrid speciation
Three types of evidence have been used in support of the hypothesis of hybrid origin of H. heurippa: the “recreation” of the heurippa phenotype by selective interbreeding of H. melpomene and H. cydno in the lab, studies of the species’ capacity to interbreed with its putative parent species, and patterns of genetic variation in sequences of mtDNA, several nuclear genes, and batteries of microsatellite loci. These will each be examined in turn.
“Re-creating” the H. heurippa phenotype in the laboratory
Mavárez et al. (2006) selected a true-breeding hybrid laboratory strain with a yellow and red banded forewing phenotype similar to that of H. heurippa, by crossing captive H. cydno cordula with H. melpomene melpomene, backcrossing F1 males to H. cydno, and selecting heurippa-like backcross individuals for subsequent matings. After an unstated number of generations, homozygous heurippa-like phenotypes were produced, which the authors claim breed true when crossed to H. heurippa (only one representative of this cross is illustrated).
Mavárez et al. indicate that there are three homologous genes shared between H. cydno and H. melpomene with allelic differences responsible for the major color pattern differences between H. cydno cordula and H. melpomene melpomene: B/b, presence/absence of a red forewing band; NN/NB, presence/absence of a yellow forewing band (heterozygotes intermediate); and Br/br, expression of the brown “forceps” on the ventral hindwing. The wild type genotype of H. cydno cordula is bbNNNNBrBr, while the wild type genotype of H. melpomene melpomene is BBNBNBbrbr, and the genotype of the heurippa-like hybrids is BBNNNNbrbr. Mavárez et al. apparently assume that the alleles making up this genotype are identical by descent with the alleles that produce the same phenotype in H. heurippa, but there are subtle differences between the phenotypes, such as the shape of the yellow band and the shape and the color of the red band, that cast some doubt upon that assumption. As has been corroborated by genome mapping efforts (e.g., Baxter et al. 2008), the genes responsible for various pattern elements are the same among divergent, mimetic Heliconius clades, albeit with different homoiologous alleles. There is thus no reason to assume that alleles resulting in a mimetic or otherwise convergent wing pattern element are IBD among all taxa that exhibit that phenotype, nor, more specifically, any reason why alleles for the expression of a red forewing patch and the masking of the brown hindwing “forceps” could not have arisen independently in H. heurippa. Since the loci responsible for the patterns are the same, selecting a hybrid strain of H. melpomene x H. cydno that looks like H. heurippa is no more evidence of the latter’s hybrid origin from those species than would a selected strain of H. erato that looks like H.heurippa be evidence that H. heurippa is descended from H. erato. Footnote 3
Intrinsic barriers to gene exchange
There is substantial evidence that Heliconius melpomene and H. cydno are capable of mating and producing offspring in the laboratory (Brown 1981; Mallet et al. 1998; Gilbert 2003), and there is likewise mounting circumstantial evidence that the two species occasionally hybridize in the wild (Salazar 1993; Mallet et al. 1998, 2007). However, Gilbert (2003) noted that “it is possible to keep cydno and melpomene in the same 13 ft × 21 ft greenhouse for years without the occurrence of interspecific courtship or mating.” Controlled mating experiments produce the same result: Naisbit et al. (2001) and Jiggins et al. (2001b) showed, respectively, in no-choice and tetrad mating experiments in the lab that H. melpomene rosina Boisduval and H. cydno chioneus Bates from Panama are completely behaviorally isolated from one another: males of one species will not court females of the other. Mavárez et al. (2006; their Table 2) reported similar results for attempted crosses between H. melpomene melpomene and H. cydno cordula from allopatric populations on the eastern slope of the Colombian Andes: in tetrad mate choice experiments, H. cydno and H. melpomene never mated with one another.
Mavárez et al. also performed reciprocal tetrad experiments to test H. heurippa’s capacity to mate with its two hypothetical parental species. They found that neither H. cydno nor H. melpomene males will mate with H. heurippa females, that H. heurippa males will not mate with H. melpomene females, but that male H. heurippa will mate with female H. cydno. In summary, H. cydno and H. melpomene are prezygotically isolated from one another, as are H. melpomene and H. heurippa. H. cydno and H. heurippa are not prezygotically isolated. Note that all of these experiments assess the mating predilections of males, while the females employed are usually sacrificial virgins with neither the opportunity, nor apparently the will, to discriminate. Salazar et al. (2005) indicate that “sexually mature females show a low mating probability with males that are not from their own species” and “H. heurippa females show strong assortative mating when tested against both H. melpomene and H. cydno.” Thus, in addition to male courtship, it is likely that in the wild, mate choice behavior of females of these species also contributes to premating isolation.
If these prezygotically isolated species are forced to mate or happen to mate out of desperation in the insectary, what is the fate of the resultant offspring? Offspring of the H. cydno x H. melpomene cross are viable and partially infertile, following Haldane’s rule: heterogametic females are sterile (Linares 1989; Nijhout et al. 1990). Salazar et al. (2005) performed crosses between both of these species and H. heurippa, finding complete hybrid viability and fertility in the H. cydno x H. heurippa crosses, and an asymmetrical pattern of offspring fertility in the H. melpomene x H. heurippa crosses. Male H. melpomene x female H. heurippa F1 females are sterile, while male H. heurippa x female H. melpomene F1 females are fertile (males from both crosses are viable and fertile). The patterns of pre- and postzygotic mating success described here are summarized in Fig. 2.

Schematic summary of pre-and postzygotic isolation among H. cydno cordula (top left), H. melpomene melpomene (top right), and H. heurippa (bottom). Red arrows indicate isolation, green arrows indicate compatibility. Barred arrows indicate asymmetrical patterns
Mavárez et al. (2006) stated, “… the phenotype of H. heurippa reproductively isolates it from both parental species.” This is simply not the case (see Mavárez et al. 2006, Table 2). While it is true that neither H. melpomene nor H. cydno males will mate with H. heurippa females, H. heurippa males are perfectly willing and able to mate with H. cydno females (although, as noted, “mature” H. cydno females might reject attempted matings by H. heurippa males). Therefore, it is likely that H. heurippa and H. cydno remain distinct from one another in nature due solely to their geographical disjunction from one another. These authors also claimed that their study provided, “the first example of a hybrid trait causing pre-mating isolation through assortative mating.” In fact, the trait that causes (partial) isolation between H. cydno and H. heurippa is the symplesiomorphic aversive response to red wing pattern elements by H. cydno males, a behavior that also deters them from mating with H. melpomene females (Jiggins et al. 2001b). H. heurippa males share with H. cydno the positive courtship stimulus of white/yellow wing pattern elements (also a symplesiomorphy), which explains why they will pursue H. cydno females but not H. melpomene females.
Patterns of genetic differentiation
Multiple “known” loci, as well as several batteries of microsatellite and AFLP markers, have been examined to assess the genetic structure of the melpomene-cydno group and the H. heurippa hybrid speciation hypothesis. The strategy employed in this paper was to obtain and reanalyze all relevant sequence data for each gene for which a H. heurippa has been sequenced, using the parsimony algorithm as implemented in TNT (Goloboff et al. 2003) (1,000 random addition replicates; gaps encoded as a fifth character; equal weights). Sequence data were transcribed from GenBank. Terminals are labeled with individual voucher codes as reported in the GenBank annotations or corresponding publications. Note that some sequences were published on more than one occasion and are represented in GenBank with more than one accession code. Aligned data matrices for each of the genes are available as Nexus files at http://www.mtsu.edu/~abrower/datasets. Alignments were performed by eye.
Data which were unavailable for reanalysis were evaluated based upon the authors’ descriptions of them in the original publications.
Mitochondrial DNA
As noted earlier, phylogenetic analysis of mtDNA COI-COII sequences has usually placed H. heurippa in a clade with H. cydno, nested in turn within a paraphyletic H. melpomene (Brower 1996a, b; Beltrán et al. 2007; Quek et al. 2010). There has evidently been some confusion about this pattern. Bull et al. (2006), in a rather contorted argument that contradicted their own results, claimed that “mutual monophyly (of H. cydno and H. melpomene) cannot be rejected,” and Quek et al. (2010), citing Brower (1996a), stated that “early mtDNA studies indicated a sister species relationship,” despite the cited paper’s clearly stating, “… intraspecific variation in H. melpomene is complicated by the apparent paraphyly of the species with respect to H. cydno and its close relatives ….”
To clarify the current understanding of relationships implied by this gene region, all published COI–COII sequences for the melpomene-cydno complex (371ingroup exemplars, from papers cited above, as well as Beltrán et al. 2002; Kronforst et al. 2006; Giraldo et al. 2008; Chamberlain et al. 2009) were extracted from Genbank, aligned (no length variation) and analyzed via parsimony (MP). A strict consensus tree is presented in Fig. 3.

Phylogenetic relationships of mtDNA COI–COII haplotypes. Strict consensus of 98 trees; length = 559 steps; CIx = 0.4846; RI = 0.9715. H. cydno and relatives form a clade embedded among various clades representing geographically separated groups of H. melpomene races. There is no implied introgression
The salient features of this tree are the same as those revealed by Brower (1996a): a basal melpomene clade from the Guianas, and then a polytomy comprising melpomene clades from southeastern Brazil, Amazonia, and the trans-Andean region and a clade of cydno cognates. Thus H. melpomene in the broad sense is still “paraphyletic” with respect to H. cydno and its satellites. Note that under the Phylogenetic Species Concept of Nixon and Wheeler (1990), two such entities are indeed considered to be “sister taxa”Footnote 4 (see Brower 1999). As Brower (1996a) found, there is little resolution within each of these clades, and no obvious correspondence between haplotypes and wing-patterns or finer scale geography. H. heurippa, H. timareta, and H. tristero individuals are interspersed within the H. cydno clade. Two “new” clades appear: a second clade of H. melpomene individuals from French Guiana and Trinidad (which will not be discussed here), and a cluster of “H. melpomene” haplotypes from Chirajara, Cundinamarca, Colombia and Santa Ana, Mérida, Venezuela, which is sister to the H. cydno clade. More of these anon.
Triose phosphate isomerase
Tpi is a polymorphic, sex-linked nuclear gene, a ~530 bp region of which was developed for examining Heliconius relationships by Beltrán et al. (2002). Additional members of the H.melpomene-H. cydno complex have been sequenced by Flanagan et al. (2004), Bull et al. (2006), Kronforst et al. (2006), Dasmahapatra et al. (2007), Giraldo et al. (2008), Kronforst (2008) and Salazar et al. (2008). 186 sequences were extracted from Genbank and analyzed via MP. Unlike the mtDNA, the Tpi gene region contains a length-variable segment spanning an intron. Alignment is not trivial, and approximately 2/3 of the phylogenetically informative sites occur in the noncoding region.
Analyses of these data, either including the gaps as a fifth character state or treating them as “missing,” reveal a weakly supported clade of alleles from various H. melpomene races, embedded within a paraphyletic cluster of alleles from H. cydno races, H. timareta and H. heurippa (Fig. 4). There is no apparent correlation of the gene tree topology with finer-scale phenotypic or geographical patterns. Single alleles are shared by multiple geographical races of H. melpomene and H. cydno, and among H. heurippa and H. timareta. Alternate alleles from some individuals are highly divergent from one another (although no alleles are shared between H. cydno and H. melpomene). There is no evidence for interspecific H. melpomene—H. cydno hybridization at this locus.

Phylogenetic relationships of Tpi alleles. Strict consensus of 174 trees; length = 612 steps; CIx = 0.6927; RI = 0.9685. H. melpomene alleles form a clade with H. cydno and relatives forming a paraphyletic basal grade. There is no implied introgression
Mannose-6-phosphate isomerase
Beltrán et al. (2002) also developed a region of the autosomal Mpi as a marker for examining relationships in Heliconius. The amplified region is composed of short exon sequences (9 and 13 codons) flanking a highly length-variable intron ranging from <100 to >400 bp in the current alignment. Additional sequences generated by Flanagan et al. (2004), Bull et al. (2006), Kronforst et al. (2006) and Kronforst (2008) were compiled into a matrix of 118 ingroup taxa plus five outgroups. The data exhibit long, highly similar insertions and deletions that are shared among a variety of taxa, including a 250 bp deletion shared among H. cydno chioneus and H. melpomene rosina, both from Panama, and H. melpomene melpomene from French Guiana. There is also a 65 bp insertion which occurs in several H. pachinus, H. cydno chioneus and H. cydno galanthus Bates, as well as one H. melpomene rosina.
Most of the authors working with Mpi have viewed the sharing of alleles among these species as evidence for current interspecific gene flow. How hybridization explains the distribution of the 250 bp deletion in specimens from Central America and French Guiana (more than 2,000 km apart) is not clear. Further, when one takes into account the phylogenetic distribution of alleles present in heterozygous individuals, the pattern suggests that the diversity of alleles is nearly panmictic, not only among members of the H. cydno-H. melpomene group, but also among outgroup H. hecale (Fabricius) and H. ethilla (Godart). In regards to the “hybrid speciation” question, the alleles from the single H. heurippa sampled for this gene fall into two distinct clades (Fig. 5), each of which contains alleles from both H. cydno and H. melpomene. Given that the patterns of change in the Mpi marker are incoherent with respect to species boundaries and biogeography and because the variation is evidently not driven by single nucleotide substitutions, it seems premature to ascribe much evidentiary value to phylogenetic interpretations of this gene region.

Phylogenetic relationships of Mpi alleles. Strict consensus of 236 trees; length = 777 steps; CIx = 0.7341; RI = 0.9442. While there are several clades containing only H. melpomene alleles, all clades containing H. cydno alleles also contain multiple H. melpomene alleles
Distal-less
Kronforst et al. (2006) and Mavárez et al. (2006) have examined two sets of sequences from the autosomal developmental gene Dll, each study interpreting the distribution of alleles to support the hypothesis of interspecific gene flow. Kronforst’s specimens were all Costa Rican, and Mavárez’s were all from the eastern slopes of the Andes in Colombia and Venezuela. Again, the bulk of informative variation in this gene region comes from a length-variable intron. Data from both these studies comprising 99 ingroup alleles were combined and analyzed via MP with gap characters from the intron included as a fifth character state. Mavárez et al. (2006) reported “no allele sharing between H. cydno and H. melpomene, whereas the H. heurippa genome appears as an admixture, sharing allelic variation with both putative parental species.” The tree based on the combined dataset (Fig. 6; see also Kronforst et al. 2007) complicates the interspecific hybridization scenario by revealing nearly identical alleles shared not only among sympatric H. melpomene and H. cydno, but also between Costa Rican and eastern Colombian H. melpomene, and between Costa Rican H. cydno galanthus and H. heurippa. Such a pattern suggests that ancestral polymorphism could also explain the maintenance of allelic diversity across the ranges of these taxa. It seems that further sampling of this locus from the geographical diversity of H. melpomene and H. cydno forms will be necessary before the extent and mode of gene flow throughout the clade may be clearly discerned.

Phylogenetic relationships of Distal-less alleles. Strict consensus of 86 trees; length = 638 steps; CIx = 0.6201; RI = 0.9715. H. cydno and H. melpomene are not distinct due to two H. melpomene rosina alleles in the H. cydno clade. H. heurippa alleles are widespread in both clades
Invected
The data for this gene have the same provenances as the Dll data, and were also invoked by their respective authors as evidence of the origin of H. heurippa via interspecific hybridization. Once again, most informative variation in the inv fragment comes from a length-variable intron. The two data sets were combined into a matrix of 67 ingroup alleles × 479 bp and analyzed via MP with gap characters from the intron included as a fifth character state. The combined tree (Fig. 7; see also Kronforst et al. 2007) exhibits near reciprocal monophyly of H. cydno-group alleles and H. melpomene alleles, but several H. heurippa alleles and one H. pachinus allele cluster among the H. melpomene alleles. This pattern represents the most convincing evidence of hybridization from any of the genes discussed so far. However, it is again evident that very closely-related alleles are distributed between Venezuela and Costa Rica, suggesting not only sympatric interspecific hybridization, but also extreme long-distance dispersal or some other mechanism that results in homologous (or convergent) intron variation among remote areas. Further, no H. heurippa allele is identical to any H. melpomene allele, and one of the H. heurippa alleles is sister taxon to all H. melpomene alleles save one, suggesting that if these alleles were acquired via hybridization, the hybridization events happened long ago and on several separate occasions. As in Dll, the entire picture of allelic diversity at this locus may not be represented by the limited geographical samples examined to date.

Phylogenetic relationships of Invected alleles. Strict consensus of 267 trees; length = 387 steps; CIx = 0.6655; RI = 0.9322. H. melpomene and H. cydno are not monophyletic with respect to the outgroup. H. heurippa alleles are distributed throughout the tree
White
Sequences of the ommochrome biosynthesis gene w for 37 ingroup alleles were obtained from Kronforst et al. (2006) and Salazar et al. (2008), representing the same taxa as Dll and inv. Again, the aligned region is short (434 bp) and contains a length-variable intron. Parsimony analysis resulted in a consensus tree (Fig. 8) with all species’ alleles polyphyletic with respect to one another, and no obvious geographical structure. If the distribution of alleles among species at this locus is due to hybridization, then H. cydno- H. melpomene group members are exchanging alleles not only among themselves, but also with the outgroup H. hecale.

Phylogenetic relationships of white alleles. Strict consensus of 8 trees; length = 351 steps; CIx = 0.7299; RI = 0.9343. Little resolution is evident among included species
Scalloped
Sequences of a region of this putative developmental gene representing 46 ingroup alleles were obtained from Kronforst et al. (2006) and Salazar et al. (2008), representing the same taxa as the preceding three genes. The aligned region is 555 bp and contains a length-variable intron. Parsimony analysis resulted in a consensus tree (Fig. 9) with all species’ alleles polyphyletic with respect to one another, and no obvious geographical structure.

Phylogenetic relationships of scalloped alleles. Strict consensus of 147 trees; length = 295 steps; CIx = 0.7677; RI = 0.9521. Little resolution is evident among included species
Wingless and elongation factor 1-alpha
Sequence data for these genes have been useful for a wide variety of higher-level studies of butterfly relationships (e.g., Brower 2000a; Wahlberg et al. 2009), but have proven to exhibit little variability within or among closely-related Heliconius species (Brower and Egan 1997; Beltrán et al. 2007). 33 wg sequences and 31 Ef-1 alpha sequences representing the H. cydno-H. melpomene group were obtained from Genbank, aligned and analyzed, but resulted in largely unresolved polytomies (data not shown).
Microsatellite and AFLP data
Microsatellites and AFLP data are the allozymes of the twenty-first century, and their use as a source of evidence to infer population structure has resurrected many of the dubious phenetic approaches that were superceded by cladistics in the late 1970s. Unlike sequences of known genes, these markers are “anonymous,” their putative homology is not testable, and they cannot be compared across studies in the manner performed for the loci discussed above. Their inscrutability is further exacerbated by the unfortunate tendency by researchers who employ them to not make available the raw data for critical reexamination. Thus, the would-be intersubjective corroborator is relegated to deciphering recondite graphical summaries and discursive interpretations as a means of evaluating these “data.” Several of the following studies did not examine H. heurippa, but the patterns found among other Heliconius species bear upon the interpretation of the H. heurippa story.
Mavárez et al. (2006) examined 12 microsatellite loci from five populations of H. cydno and H. melpomene and one population of H. heurippa using Structure 2.1 (Pritchard et al. 2000), a Bayesian model that simultaneously infers how many clusters of similar multilocus genotypes exist in the data and assigns individuals to those clusters. They reported that the “best model” specified three clusters, allowed for admixture and independent estimations of allele frequencies. They did not report whether or not the “best model” was significantly more likely than any other.Footnote 5 Nor do they mention the interesting phenomenon that the degree of inferred admixture among H. heurippa, H. melpomene and H. cydno appears to be highest between the populations of the latter two that are the most geographically remote (Pipeline Road, Panama) from the range of H. heurippa. In sum, the methods used to analyze these data were so inadequately described that the results are indeterminate. Their diagram (their Fig. 1) shows individuals of the three species organized into blocks of three different colors, suggesting that H. cydno, H. melpomene and H. heurippa are different from one another. But how different? Are these differences nested?
Mavárez et al. (2006, their supplementary Fig. 4) conducted a separate, also inadequately–described, microsatellite analysis of 36 H. melpomene melpomene, 44 H. cydno cordula and 9 putative cydno x melpomene hybrids from San Cristóbal, Venezuela. The microsatellite profiles of these “hybrid” specimens in Structure are apparently identical to those of H. cydno cordula (see epigram #1), suggesting under the hybrid speciation hypothesis that the only alleles from H. melpomene they have retained from their interbreeding are specifically those responsible for their “hybrid” wing patterns.
Jesús Mavárez (pers. comm., Nov. 2005) reported results of a to-date unpublished Factorial Analysis of Correspondence based on microsatellite variation at 12 loci for the two specimens of H. tristero known at that time,Footnote 6 compared to H. melpomene and H. cydno from Panama, Colombia and Venezuela. H. tristero clustered within H. cydno and not within H. melpomene, and no H. melpomene individual ever fell within the H. cydno cluster. He also indicated that his results show that H. pachinus and H. timareta are also closely related to H. cydno.
Kronforst et al. (2006, 2007) examined a large data set of AFLP loci to assess interspecific gene flow among Costa Rican H. pachinus, H. cydno galanthus and H. melpomene rosina. They found (2006) “multiple instances of mixed ancestry in all three species,” but concluded (2007) based on the same data that all their analyses “supported a close genetic relationship between H. pachinus and H. cydno and none suggested a genetic contribution from H. melpomene in the origin of H. pachinus.” They drew an important distinction between hybridization as an incidental process and hybridization as a source of evolutionarily significant genetic variation, ruling out the latter in H. pachinus, but invoked the inv and Dll data discussed above to suggest that H. heurippa may have a hybrid origin.
Giraldo et al. (2008) used microsatellite data in Structure to sort an unspecified number of individuals of H. melpomene malleti Lamas and H. florencia Giraldo et al.Footnote 7 into populations, and found five individuals with microsatellite profiles containing markers assigned to multiple species that they interpreted to be hybrids. As in Mavárez et al. (2006), there is no way to assess the robustness of this interpretation from the data as presented.
Chamberlain et al. (2009) examined microsatellites from samples of H. cydno alithea, H. cydno galanthus and H. pachinus to look for possible genetic differences between yellow and white polymorphic forms of the former. They didn’t find any, but interestingly, when they ran Structure on their data allowing admixture, as in the studies reviewed above, all of their H. cydno galanthus specimens appear as “hybrids” (sharing alleles from allopatric H. cydno alithea and H. pachinus!) with an equal or higher degree of admixture than most of the “hybrid” specimens discussed above (their Fig. 3b).
Most recently, Quek et al. (2010) have used 3,186 AFLP loci to examine geographical structure among populations of H. melpomene throughout its range. Their NJ phenogram for these data is largely congruent with trees inferred from mtDNA (e.g., Fig. 3), although bootstrap values indicate rather high instability of relationships among most races and geographical areas. Specimens from “Colombia” represent three different geographical areas—the Pacific slope, the Magdalena Valley and the eastern slope of the Andes, and it would have been a surprise if they did form a “clade.” Quek et al. also performed a Structure analysis of the same data, which as in the studies discussed above revealed “admixture” between various sampled individuals, including H. cydno from Panama and H. melpomene from southeastern Brazil.
So, to summarize these various Structure analyses, it seems that when one wants to find admixture to support one’s hypothesis, then one can do so. Or when one wants to ignore admixture to support one’s hypothesis, then one can do so. It is indeed an excellent type of evidence that is so compliant to the desired interpretations of the researcher.
In general, the available genetic evidence support a limited degree of shared polymorphism between H. melpomene and H. cydno and its various offshoots. Kronforst et al. (2006) summed up the pattern nicely: “Our results indicate that the three Heliconius species [Costa Rican H. cydno, H. pachinus and H. melpomene] studied here hybridize frequently enough to leave recognizable evidence of admixture and introgression but not enough to erode species boundaries.” However, studies limited to populations of different species from a single geographical region may reveal shared alleles, but these patterns cannot be interpreted as the necessary result of recent hybridization when the same or closely-related alleles are also shared among remotely allopatric populations, implying additional historical processes besides exchange of alleles among sympatric relatives. The patterns are also made difficult to interpret by the fact that most of the informative variation in the genes studied occurs in length-variable intron regions. It is clear that these length variants do not evolve one nucleotide at a time, but the “gap characters” are treated as though they did (as in the above reanalyses) or ignored altogether as missing data. It is extremely doubtful that either of these approaches adequately captures the character state transformations as they have occurred, but until more is known about the tempo and mode of intron evolution, there does not seem to be a more defensible alternative. Again, when the assumptions of the analytical approach are not met by the patterns of variation in the data, one is treading on thin epistemological ice to interpret the results as an accurate reflection of history.
Thirty years ago, Turner et al. (1979) found a high degree of shared polymorphism among Heliconius species using allozymes, and argued that the genes related to wing patterns must be decoupled or behave independently from the remainder of the genome. This pattern has been corroborated by recent genomic studies (Baxter et al. 2010), who found limited signatures of selection around specific loci associated with wing pattern elements. It therefore may not be possible to obtain a clear understanding of the evolution of mimetic phenotypes in these butterflies until we are able to examine gene genealogies for the genes that are responsible for the wing pattern elements themselves. I predict that the allele producing a red band on the forewing of H. heurippa will not be homologous (IBD) to that of sympatric H. melpomene melpomene, a pattern that would lay to rest the H. heurippa HHS hypothesis.
Hopeless monster: the origin and fate of an interspecific hybrid
But let us ignore all of the above and suppose, for the moment, that H. heurippa is the homoploid hybrid offspring of H. melpomene melpomene and H. cydno cordula. What events would need to take place for the origination and establishment of a genetically homogeneous H. heurippa population, sympatric with one of its parental species?
First, we would need an interspecific mating between the parental species—something that occurs rarely, and only when one of the species is rare, thereby limiting its mating options (Gilbert 2003). Since H. heurippa is currently sympatric with H. melpomene and not with H. cydno, it is parsimonious to suppose that H. cydno was the rare parental species. F1 individuals of this cross have a red forewing band with a thin dusting of yellow scales on its proximal margin (a pattern much more H. melpomene-like than H. cydno-like). One would suppose, therefore, that selection would favor the fine-tuning of the color pattern to be more H. melpomene-like in subsequent generations, since that is the common parent and a participant in the local Müllerian mimicry scheme with the even more common H. erato hydara Hewitson. However, the H. heurippa pattern is a worse mimic of H. melpomene than the F1 (so much so that it is often referred to as “non-mimetic”), so the normally strong positive frequency-dependent selection on the wing pattern would apparently either have been relaxed or have been overwhelmed by some alternative selective force counter to the positive frequency-dependent pull of Müllerian mimicry.
Next our F1 generation would need mates. Since female offspring of both reciprocal crosses of the parental species are sterile, F1 individuals cannot mate with one another to produce F2 offspring. Instead, the F1 hybrid males need to backcross to rare H. cydno females in order for their offspring to gain the cydno-like mtDNA that is observed in H. heurippa. The phenotypes of these F1-backcrosses exhibit 25% red forewing bands (Bb/NNNB) 25% red-and-yellow forewing bands (Bb/NNNN), and 50% yellow bands only (bb/NN-). Once again, selection must favor individuals with the “non-mimetic” H. heurippa pattern over those with patterns like the parentals. In order to become homozygous for the H. heurippa phenotype (BB/NNNN/brbr), two “H. melpomene” color pattern alleles need to become fixed, despite indications that backcrossing, at least initially, must be to H. cydno. A further complication to this scenario is the observation by Naisbit et al. (2001) that F1 hybrid males prefer to mate with F1 hybrid females over females of either of their parental strains. That would seem like a clear path to prezygotic isolation and speciation (cf. Melo et al. 2009) except that H. cydno x H. melpomene F1 females are, of course, sterile and such matings are completely fruitless.Footnote 8
Duenez-Guzman et al. (2009) found that when their model considered “color patterns learned as a whole,” “hybrids never achieve high frequencies … and both parental species coexist sympatrically”. Predators do indeed perceive the entire butterfly, as attested by the enormous selection coefficient against novel phenotypes found in a Peruvian H. melpomene hybrid zone (Mallet and Barton 1989), despite the fact that the butterflies on either side are both black with red and yellow markings. Jacamars (Galbulidae), which are likely to be the main predators responsible for maintaining Müllerian mimicry in Heliconius, have acute perception of small pattern differences (Langham 2004, 2006).
So, in addition to overcoming the problem of selection against novel phenotypes by predators, incipient “H. heurippa” hybrids would also need to mate with females of an allopatric geographical race of H. cydno with whom they would prefer not to, and who would prefer to not mate with them. This does not seem like a recipe for success.
H. heurippa, hybrid species? An alternative hypothesis
As reviewed above, all of the genetic and breeding evidence indicate that H. heurippa is more closely-related to H. cydno than it is to H. melpomene. If H. heurippa were the only H. cydno-cognate to exhibit red wing pattern elements, then the homoploid hybridization hypothesis with introgression of some H. melpomene wing pattern alleles into a mainly H. cydno background might provide a reasonable, if unlikely, explanation for its existence. But what are the chances of the same improbable phenomenon occurring independently multiple times in other forms with different wing patterns? Could there be another explanation for this general pattern?
It is becoming increasingly evident that an allopatric or parapatric string of taxa occurs between 800 and 1,200 m. along the Amazonian slope of the northern Andes with red wing pattern elements and “H. cydno genes” (see Mallet 2009 for a distribution map). These are briefly described from south northwards: H. timareta, with differentiated races in northeastern Peru and eastern Ecuador is often characterized, like H. heurippa, as “non-mimetic,” but at least some of its forms are very good mimics of nearby H. melpomene and H. erato races. Mallet (2009) mentions another unnamed H. cydno cognate from the Mayo valley in San Martín, Peru which, based upon its mimetic wing pattern, has long been conflated with the local M. melpomene amaryllis Felder & Felder. In the Putumayo valley of southeastern Colombia, H. tristero is a dead-ringer for H. melpomene mocoa Brower and H. erato dignus Stichel. Not far to the northeast in the Río Hacha valley occurs the recently-described H. florencia, a very good mimic of H. melpomene malleti and H. erato lativitta Butler. Giraldo et al. (2008) illustrate an unnamed H. cydno cognate with a yellow forewing band like that of H. cydno cordula (and no red pattern elements) from the Río Pata valley near the Cordillera de Los Pichados. H. heurippa itself occurs in the Meta valley above Villavicencio. Then there is a gap, where no H. cydno cognate is known, until the range of H. cydno cordula in the Cordillera de Mérida in northern Colombia and western Venezuela. But, remember the “H. melpomenes” with red forewing patches but H. cydno mtDNA from Chirajara (sympatric with H. heurippa) and Santa Ana, Venezuela (Salazar et al. 2008) and the “hybrids” from San Cristóbal, Venezuela (latter two sites about 15 km apart)? What if these specimens represent another H. cydno cognate that mimics H. melpomene melpomene and H. erato hydara and occurs along the eastern slope of the northern Colombian Andes? That would explain both the “hybrid” wing patterns and the genetic similarity of the San Cristóbal “hybrids” to H. cydno much more simply than the “multiple generations of backcrossing” invoked by Mavárez et al. (2006). In general, given that the red wing pattern elements of these various taxa occur on different parts of the wing and are controlled by different genes (Sheppard et al. 1985), it seems all the more unlikely that they can all be explained as the result of HHS, since each of those hybridization-speciation events would need to occur independently.
Here is an alternative scenario for the origin of all these cis-Amazonian H. cydno cognates. Many authors have argued that H. cydno (and H. melpomene) have been subordinate participants in their mimicry rings, evolving to converge on the wing patterns of more abundant members of the “pupal mating” group—H. sapho/H. eleuchia and H. erato, respectively. Since H. melpomene is paraphyletic with respect to H. cydno, and its basal lineages occur in southeastern Brazil and the Guianas, it is reasonable to hypothesize that H. melpomene had a broad distribution before the origin of H. cydno. H. cydno and H. melpomene diverged from one another west of the Andes (perhaps even in southern Mexico, where H. melpomene does not occur), and H. cydno’s wing pattern converged on that of H. sapho. H. cydno began to expand its range southward and eastward, evolving to mimic a variety of locally common aposematic Heliconius (or even ithomiines such as Elzunia humboldt (Latreille); cf. Linares 1997b), but its distribution was limited by the distribution of its black-and-white comimics, which are restricted to the western side of the Andes (Brown 1979).Footnote 9
H. cydno appears to be one of the more phenotypically labile Heliconius species, exhibiting not only geographical variation in wing patterns, but also sympatric wing pattern polymorphism in several different areas: H. cydno chioneus is polymorphic in western Ecuador (Kapan 2001); H. cydno weymeri has at least three morphs in the upper Cauca valley on Colombia (Linares 1997b); H. timareta is polymorphic in eastern Ecuador (Brown 1979). Let us imagine that peripheral populations of H. cydno spilled over onto the eastern slopes of the Andes during Pleistocene warm periods, where they came into contact with more abundant red-and-yellow members of the H. erato-H. melpomene mimicry ring. Under such circumstances, mutations that resulted in red wing color patterns would confer a selective advantage (at least against less acute predators than jacamars), and might provide the initial impetus to switch mimicry rings. Recall that since H. cydno is descended from H. melpomene, such mutations would not represent evolutionary novelties, but rather reversals to an ancestral phenotype, and could be accomplished by switching on or off alleles already present in the underlying genetic architecture of wing pattern.
But doesn’t this seem implausible and ad hoc, given the apparent rarity of wing pattern mutants? Gilbert (2003) suggested a spontaneous wing pattern mutation rate of 1:4,000, which is not very different from Mallet et al.’s (2007) estimate of the rate of hybridization between H. melpomene and H. cydno, 1:2,000. Since the red forewing patch allele B is dominant in H. heurippa and other “red” alleles are usually dominant in H. melpomene (Sheppard et al. 1985), we can hypothesize that novel red-producing mutations would also be dominant in H. cydno and therefore expressed in heterozygotes. If such alleles were beneficial in a new mimetic environment, they would rapidly increase in frequency in the population. And the individuals bearing them would not suffer a loss in fitness due to hybrid sterility because they would not need to hybridize with H. melpomene. Furthermore, if a number of the aberrant specimens interpreted by Mallet et al. (2007) to be interspecific hybrids actually were mutants, that would imply that the mutation rate could be higher, while the inferred rate of interspecific hybridization between H. melpomene and H. cydno is concomitantly lower.
The evolution of mimetic diversity despite frequency-dependent selection for a common aposematic pattern remains a paradox, and no hypothesis explaining the origin of novel forms is immune from criticism. However, H. heurippa, H. florencia, H. timareta and H. tristero have all been shown by available data to be mainly reproductively compatible with/related to/descended from H. cydno; those alleles shared between H. heurippa and H. melpomene appear to be also widely shared among other members of the H. cydno-H. melpomene clade; and the underlying genetic architecture to produce red wing pattern elements is plesiomorphic for the entire genus. The HHS hypothesis has evolved to accommodate the lack of evidence of H. heurippa’s hybrid genome. The current version, “hybrid trait speciation” (Jiggins et al. 2008), requires only the genes controlling adaptive traits to move across species boundaries, while neutral genetic variation is either assorted by species or shared among all members of the clade (turning the Turner et al. 1979 “contrasted modes” hypothesis on its head). It may come to pass that incontrovertible H. melpomene alleles responsible for mate discrimination and selective divergence are discovered in H. heurippa and its red-winged cousins, but until that happy day, the alternative scenario for the origin of these species presented here seems a somewhat less onerous alternative.
Postscript: 3′ kinesin, smoking gun, or magic bullet?
While this manuscript was in review, a recent, highly relevant article by Salazar et al. (2010) came to my attention. In that paper, the authors announced the discovery of a region at the 3′ end of the kinesin gene with 14 single nucleotide polymorphisms (SNPs) that are shared between H. melpomene and H. heurippa, which the authors argue represents conclusive evidence of hybridization between those taxa. They describe this pattern as a “magic trait” (Gavrilets, 2004): a feature that promotes speciation by both causing assortative mating and experiencing divergent natural selection in a polymorphic population. One reviewer of my manuscript suggested that I withdraw the paper in light of this “molecular evidence for adaptive introgression during hybrid speciation” (Salazar et al. 2010). I have manifestly not obliged the reviewer’s desire, but instead will briefly take stock of the new evidence here.
Salazar et al. (2010) is an example of several disturbing emergent trends in genomic-era publications. The paper alludes to analysis of an enormous amount of data: nearly 45 kb of sequence from 30 individuals, representing nearly 3,000 individual GenBank accessions. The sheer quantity of data entails a somewhat telegraphic description of analytical methods, that makes results difficult to evaluate critically. For example, ten specimens each of H. heurippa, H. m. melpomene and H. cydno cordula were processed to produce a total of sixty terminal entities (“alleles”) for each locus, but it is never explained how the genomic DNA of individual butterflies was segregated for amplification and sequencing of two separate alleles from each diploid individual. Nor are these “alleles” labeled so that the composite genotypes of individual butterflies may be reassembled.
It would be tangential to the aim of this paper to conduct a comprehensive critical reappraisal of the diversity of analyses performed in Salazar et al. (2010). Perhaps most salient to the data analyses presented above are several phylogenetic trees (their Fig. 4) representing inferred relationships among “alleles” for three gene segments linked to the putative site of the gene encoding the red forewing band of H. melpomene and other red-banded Heliconius (Baxter et al. 2010): sorting nexin, 5′ kinesin, and 3′ kinesin. The authors state that their rooted 3′ kinesin tree “shows H. heurippa forming a well-supported and derived clade within H. melpomene” (note that the other two gene trees presented were not rooted with an outgroup). Reanalysis of the 3′ kinesin data (generously provided by C. Salazar) shows that the strict consensus MP tree (Fig. 10) is not the same as that shown in Salazar et al.’s Fig. 4. Unfortunately, it is not possible to directly compare the topologies from these two analyses, because Salazar et al. did not label their terminals, but it is evident from Fig. 10 that rather than being a “derived clade within H. melpomene,” the H. heurippa “alleles” form a basal polytomy with two H. melpomene “alleles” and another clade containing the other 18 H. melpomene “alleles.” The branch support for the melpomene + heurippa clade is 9, a relatively high value, but it is important to bear in mind that the gene region analyzed (6,493 bp) was specifically circumscribed because it contained a number of fixed character states supporting a heurippa + melpomene relationship.

Phylogenetic relationships of 3′ kinesin alleles. Strict consensus of 3,488 trees; length = 1,852 steps; CIx = 0.4649; RI = 0.8723. Note that although the branch support (sensu Brower 2006) = 9 for the node leading to H. melpomene and H. heurippa alleles, that the H. heurippa alleles form a polytomy with and are not embedded within the H. melpomene alleles, contra Salazar et al. (2010)
As noted, the 3′ kinesin data set includes an outgroup sequence from the closely-related silvaniform H. numata. While one outgroup is certainly better than none (as in Salazar et al.’s sorting nexin and 5′ kinesin trees), it is evident from recent analyses of other Heliconius genes that inclusion or exclusion of relevant additional taxa can impact phylogenetic relationships among members of the ingroup (cf. Brower 1996a vs. Beltrán et al. 2002 vs. Fig. 3, above). Nor is it clear that samples of the ingroup taxa from a single locality (and collection date?—this is not specified) adequately represent the genetic diversity present in the entire distributions of the three species. For example, it would be very interesting to compare the current data to the 3′ kinesin sequence from a form of H. melpomene lacking a red forewing band, and also to other putative H. cydno cognates with red wing pattern elements, such as H. timareta and H. tristero.
Stepping back from these details, it is valuable to recall the original H. heurippa hybrid speciation hypothesis (Mavárez et al. 2006) and consider how this evidence bears upon it. It would seem that the traditional mosaic hybrid genome idea is soundly refuted: as Salazar et al. (2010) state of most of their compared gene regions, “these data do not strongly support a hybrid speciation scenario, but are more consistent with either recent gene flow among the three species or shared ancestral polymorphism.” By contrast, they argue, the 3′ kinesin region, with its 14 shared SNPs between H. heurippa and H. melpomene, is “the most convincing molecular evidence to date for homoploid hybrid speciation in animals.” However, they also note that only one of the SNPs represents a nonsynonymous site, and state that “there was no significant evidence for selection on the locus.” “Magic traits,” as stated above, are supposed to facilitate speciation by promoting selective differentiation of polymorphic lineages. It is hard to envision how 13 silent polymorphisms and a neutral amino acid substitution could accomplish that.
Thus, while the shared 3′ kinesin SNPs are consistent with the introgression interpretation, Salazar et al. (2010) present no evidence either that these SNPs have anything to do with color pattern (other that they are located in a region of a chromosome to which a wing pattern gene has been mapped), nor that they are under selection, both fundamental criteria for “hybrid trait speciation” in this system. Interpretations of the various sources of evidence evaluated above are enthusiastically cited as though they were facts in overwhelming support of the HHS scenario, rather than a confection of equivocal, irrelevant or even contradictory data, as I have shown. The efforts of these authors are truly heroic, but it is unfortunate that their enthusiasm to confirm a hypothesis conceived in the Gilbert lab in the 1980s appears to have blinkered them to the consideration of alternative interpretations of the evidence.
“What characterizes the empirical method is its manner of exposing to falsification, in every conceivable way, the system to be tested. Its aim is not to save the lives of untenable systems, but, on the contrary, to select the one which is by comparison the fittest, by exposing them all to the fiercest struggle for survival.” (Popper 1965).
Notes
The genus Heliconius has been anointed with over 2,000 names for subspecies, races, forms and aberrations of its 47 species. Names such as form “gustavi” indicated in quotation marks are infrasubspecific quadrinomina that have no status in zoological nomenclature.
Heliconius melpomene melpomene (L.) is a geographically widespread, “polyphyletic” entity (cf. Brower 1996a) probably deserving of multiple subspecific names. Honey and Scoble (2001) consider the lectotype of H. melpomene (L.) to have originated from the Guianas. The similar-looking but genetically distinct forms from eastern Panama and northern Colombia appear to be unnamed.
Oberthür (1902) illustrates a number of natural hybrids between H. erato hydara Hewitson and H. erato erato from French Guiana reminiscent of the H. heurippa pattern (red proximal-yellow distal forewing band).
I stated above that H. melpomene and H. cydno are not “sister species” and here am saying that they are “sister taxa.” This is not a contradiction, since H. heurippa, H. tristero and H. timareta are viewed as “species” and form a clade with H. cydno. The paraphyletic H. melpomene is sister to that clade. While paraphyletic taxa are undesirable, current nomenclature and ranks are maintained pending a taxonomic revision.
Mavárez et al. (2006) did not indicate the provenance of their microsatellites and whether or not they meet the assumptions of the Structure algorithm (Hardy–Weinberg equilibrium and no linkage), so there is no way to evaluate the meaning or validity of their interpretations of the data. The microsatellite markers may have been selected from among those reported in Mavárez and González (2006), most of which have a significant heterozygote deficiency and thus would not satisfy the Structure assumptions. Structure also assumes that the populations are of equal size, while Mavárez et al. sampled 168 H. melpomene, 165 H. cydno but only 46 H. heurippa. Pritchard et al. (2000) also caution that inference of K (the number of populations) is difficult to infer from a limited number of markers when there is admixture, as in this case.
Aliquots of genomic DNA from the holotype and paratype specimens of H. tristero were sent to Mavárez by the author.
The taxon described as Heliconius timareta florencia Giraldo, Salazar, Jiggins, Bermingham and Linares, 2008 (Giraldo et al. 2008) exhibits a typical Amazonian “dennis-ray” wing pattern like that of the “strandi” form of the polymorphic H. timareta timareta, but it occupies a disjunct distribution (in Caquetá, Colombia) and does not form a clade with other “H. timareta” individuals in the mtDNA, Tpi or microsatellite analyses of Giraldo et al. (2008; see Figs. 3 and 4). Thus, it (and the other Amazonian H. cydno cognates) should either be subsumed as a race of H. cydno, or ranked as a species, as I have done here (see Brower 1996b, 2000b).
It is relevant to observe that most of the putative H. melpomene × H. cydno hybrid specimens compiled by Mallet et al. (2007) or illustrated elsewhere (e.g., Ackery and Smiles 1976; Brown and Fernandez-Yepez 1985; Posla-Fuentes 1993; Salazar 1993) do not appear to be F1 hybrids. Given that interspecific hybridization is a rare event, and the various selective disadvantages of being a hybrid discussed here, one would suppose that most of the “interspecific hybrid” specimens encountered by collectors would be F1 individuals like the one analyzed by Dasmahapatra et al. (2007). Intriguingly, this does not seem to be the case.
Of course, there are black-and-white Heliconius species, such as H. congener Weymer, and H. antiochus (L.) that occur east of the Andes and may be involved in mimicry with H. cydno cordula and H. cydno gadouae Brown & Fernandez-Yepez but perhaps these species are too uncommon to exert a mimetic gravitational pull, or do not overlap in elevational distribution with other H. cydno cognates.
References
Ackery PR, Smiles RL (1976) An illustrated list of the type-specimens of the Heliconiinae (Lepidoptera: Nymphalidae) in the British Museum (Natural History). Bull British Mus (Nat Hist) Entomol 32:171–214
Bates HW (1862) Contributions to an insect fauna of the Amazon Valley. Lepidoptera: Heliconidae. Trans Linn Soc 23:495–566
Baxter SW, Papa R, Chamberlain N, Humphray SJ, Joron M, Morrison C, ffrench-Constant RH, McMillan WO, Jiggins CD (2008) Convergent evolution in the genetic basis of Müllerian mimicry in Heliconius butterflies. Genetics 180:1567–1577
Baxter SW, Nadeau NJ, Maroja LS, Wilkinson P, Counterman BA, Dawson A, Beltrán M, Perez-Espona S, Chamberlain N, Ferguson L, Clark R, Davidson C, Glithero R, Mallet J, McMillan WO, Kronforst MR, Joron M, ffrench-Constant RH, Jiggins CD (2010) Genetic hotspots for adaptation: the population genetics of Müllerian mimicry in the Heliconius melpomene clade. PLoS Genet 6:e1000794
Beldade P, Brakefield PM (2002) The genetics and evo-devo of butterfly wing patterns. Nat Rev Genet 3:442–452
Beltrán M, Jiggins CD, Bull V, Linares M, Mallet J, McMillan WO, Bermingham E (2002) Phylogenetic discordance at the species boundary: comparative gene genealogies among rapidly radiating Heliconius butterflies. Mol Biol Evol 19:2176–2190
Beltrán M, Jiggins CD, Brower AVZ, Bermingham E, Mallet J (2007) Do pollen feeding and pupal-mating have a single origin in Heliconius butterflies? Inferences from multilocus DNA sequence data. Biol J Linn Soc 92:221–239
Blum MJ (2008) Ecological and genetic associations across a Heliconius hybrid zone. J Evol Biol 21:330–341
Brower AVZ (1996a) Parallel race formation and the evolution of mimicry in Heliconius butterflies: a phylogenetic hypothesis from mitochondrial DNA sequences. Evolution 50:195–221
Brower AVZ (1996b) A new mimetic species of Heliconius (Lepidoptera: Nymphalidae), from southeastern Colombia, as revealed by cladistic analysis of mitochondrial DNA sequences. Zool J Linn Soc 116:317–332
Brower AVZ (1999) Delimitation of phylogenetic species with DNA sequences: a critique of Davis and Nixon’s Population Aggregation Analysis. Syst Biol 48:199–213
Brower AVZ (2000a) Phylogenetic relationships among the Nymphalidae (Lepidoptera) inferred from partial sequences of the wingless gene. Proc R Soc Lond B 267:1201–1211
Brower AVZ (2000b) On the validity of Heliconius tristero and Heliconius melpomene mocoa Brower, with notes on species concepts in Heliconius Kluk (Lepidoptera: Nymphalidae). Proc Ent Soc Wash 103:678–687
Brower AVZ (2006) The how and why of branch support and partitioned branch support, with a new index to assess partition incongruence. Cladistics 22:378–386
Brower AVZ, Egan MG (1997) Cladistics of Heliconius butterflies and relatives (Nymphalidae: Heliconiiti): the phylogenetic position of Eueides based on sequences from mtDNA and a nuclear gene. Proc R Soc Lond B 264:969–977
Brown KS Jr (1979) Ecologia geográfica e evolução nas florestas neotropicais. Universidade Estadual de Campinas. Campinas, São Paulo, Brasil
Brown KS Jr (1981) The biology of Heliconius and related genera. Annu Rev Entomol 26:427–456
Brown KS Jr, Fernandez-Yepez F (1985) Los Heliconii (Lepidoptera, Nymphalidae) de Venezuela. Bol Ent Venez NS 3:29–76
Brown KS Jr, Sheppard PM, Turner JRG (1974) Quaternary refugia in tropical America: evidence from race formation in Heliconius butterflies. Proc R Soc Lond B 187:369–378
Bull V, Beltrán M, Jiggins CD, McMillan WO, Bermingham E, Mallet J (2006) Polyphyly and gene flow between non-sibling Heliconius species. BMC Biol 4. doi:10.1186/1741-7007-4-11
Chamberlain NL, Hill RI, Kapan DD, Gilbert LE, Kronforst MR (2009) Polymorphic butterfly reveals the missing link in ecological speciation. Science 326:847–850
Clarke CA, Sheppard PM (1960) The evolution of mimicry in the butterfly Papilio dardanus. Heredity 14:163–173
Counterman BA, Araujo-Perez F, Hines HM, Baxter SW, Morrison CM, Lindstrom DP, Papa R, Ferguson L, Joron M, ffrench-Constant RH, Smith CP, Nielsen DM, Chen R, Jiggins CD, Reed RD, Halder G, Mallet J, McMillan WO (2010) Genetic hotspots for adaptation: the population genetics of Müllerian mimicry in Heliconius erato. PLoS Genet 6:e1000796
Coyne JA, Orr HA (2004) Speciation. Sinauer Associates, Sunderland, MA
Dasmahapatra KK, Silva-Vasquez A, Chung JW, Mallet J (2007) Genetic analysis of a wild-caught hybrid between non-sister Heliconius butterfly species. Biol Lett (Royal Soc) 3:660–663
Descimon H, Mast de Maeght J (1983) Semispecies relationships between Heliconius erato cyrbia Godt. and H. himera Hew. in southwestern Ecuador. J Res Lepid 22:229–237
Duenez-Guzman EA, Mavárez J, Vose MD, Gavrilets S (2009) Case studies and mathematical models of ecological speciation. 4. Hybrid speciation in butterflies in a jungle. Evolution 63:2611–2626
Eltringham H (1916) On specific and mimetic relationships in the genus Heliconius, L. Trans Ent Soc Lond 1916:101–148
Emsley MG (1965) Speciation in Heliconius (Lep., Nymphalidae): morphology and geographic distribution. Zool NY 50:191–254
Endler JA (1982) Problems in distinguishing historical from ecological factors in biogeography. Am Zool 22:441–452
Flanagan NS, Tobler A, Davison A, Pybus OG, Kapan DD, Planas S, Linares M, Heckel DG, McMillan WO (2004) Historical demography of Müllerian mimicry in the neotropical Heliconius butterflies. Proc Natl Acad Sci USA 101:9704–9709
Ford EB (1953) The genetics of polymorphism in the Lepidoptera. Adv Genet 5:43–87
Gavrilets S (2004) Fitness landscapes and the origin of species. Princeton University Press, Princeton, NJ
Gilbert LE (2003) Adaptive novelty through introgression in Heliconius wing patterns: evidence for a shared genetic “tool box” from synthetic hybrid zones and a theory of diversification. In: Boggs CL, Watt WB, Ehrlich PR (eds) Butterflies: ecology and evolution taking flight. University of Chicago Press, Chicago, pp 281–318
Giraldo N, Salazar CA, Jiggins CD, Bermingham E, Linares M (2008) Two sisters in the same dress: Heliconius cryptic species. BMC Evol Biol 8. doi:10.1186/1471-2148-8-324
Goloboff PA, Farris JS, Nixon KC (2003)TNT: tree analysis using new technology. Program and documentation. Available at http://www.zmuc.dk/public/phylogeny/tnt
Gompert Z, Fordyce JA, Forister ML, Shapiro AM, Nice CC (2006) Homoploid hybrid speciation in an extreme habitat. Science 314:1923–1925
Harrison RG (1990) Hybrid zones: windows on evolutionary process. Oxf Surv Evol Biol 7:69–128
Harrison RG (1993) Hybrids and hybrid zones: historical perspective. In: Harrison RG (ed) Hybrid zones and the evolutionary process. Oxford University Press, Oxford, pp 1–12
Hennig W (1966) Phylogenetic systematics. University of Illinois Press, Urbana, IL
Honey MR, Scoble MJ (2001) Linnaeus’s butterflies (Lepidoptera: Papilionoidea and Hesperioidea). Zool J Linn Soc 132:277–399
Jiggins CD (2008) Ecological speciation in mimetic butterflies. Bioscience 58:541–548
Jiggins CD, McMillan WO, King P, Mallet J (1997) The maintenance of species differences across a Heliconius hybrid zone. Heredity 79:495–505
Jiggins CD, Linares M, Naisbit RE, Salazar C, Yang ZH, Mallet J (2001a) Sex-linked hybrid sterility in a butterfly. Evolution 55:1631–1638
Jiggins CD, Naisbit RE, Coe RL, Mallet J (2001b) Reproductive isolation caused by colour pattern mimicry. Nature 411:302–305
Jiggins CD, Mavárez J, Beltrán M, McMillan WO, Johnston JS, Bermingham E (2005) A genetic linkage map of the mimetic butterfly Heliconius melpomene. Genetics 171:557–570
Jiggins CD, Salazar CA, Linares M, Mavárez J (2008) Hybrid trait speciation and Heliconius butterflies. Phil Trans R Soc Lond B 1506:3047–3054
Joron M, Mallet JLB (1998) Diversity in mimicry: paradox or paradigm. Trends Ecol Evol 13:461–466
Joron M, Papa R, Beltrán M, Chamberlain N, Mavárez J, Baxter S, Abanto M, Bermingham E, Humphray SJ, Rogers J, Beasley H, Barlow K, ffrench-Constant RH, Mallet J, McMillan WO, Jiggins CD (2006) A conserved supergene locus controls colour pattern diversity in Heliconius butterflies. PLoS Biol 4:e303. doi:10.1371/journal.pbio.0040303
Kapan DD (2001) Three-butterfly system provides a field test of Müllerian mimicry. Nature 409:338–340
Kapan DD, Flanagan NS, Tobler A, Papa R, Reed RD, Gonzalez JA, Restrepo MR, Martinez L, Maldonado K, Ritschoff C, Heckel DG, McMillan WO (2006) Localization of Müllerian mimicry genes on a dense linkage map of Heliconius erato. Genetics 173:735–757
Kronforst MR (2008) Gene flow persists millions of years after speciation in Heliconius butterflies. BMC Evol Biol 8:98
Kronforst MR, Young LG, Blume LM, Gilbert LE (2006) Multilocus analyses of admixture and introgression among hybridizing Heliconius butterflies. Evolution 60:1254–1268
Kronforst MR, Salazar C, Linares M, Gilbert LE (2007) No genomic mosaicism in a putative hybrid butterfly species. Proc R Soc Lond B 274:1255–1264
Kuusela J, Zietara MS, Lumme J (2007) Hybrid origin of Baltic salmon-specific parasite Gyrodactylus salaris: a model for speciation by host switch for hemiclonal organisms. Mol Ecol 16:5234–5245
Langham GM (2004) Specialized avian predators repeatedly attack novel color morphs of Heliconius butterflies. Evolution 58:2783–2787
Langham GM (2006) Rufous-tailed jacamars and aposematic butterflies: do older birds attack novel prey? Behav Ecol 17:285–290
Linares M (1989) Adaptive microevolution through hybridization and biotic destruction in the Neotropics. Dissertation, University of Texas, Austin
Linares M (1997a) Origin of neotropical mimetic biodiversity from a three-way hybrid zone of Heliconius cydno butterflies. In: Ulrich H (ed) Tropical biodiversity and systematics. Proceedings of the international symposium on biodiversity and systematics in tropical ecosystems, Bonn, 1994. Zoologisches Forschung Institut und Museum Alexander Koenig, Bonn, pp 93–108
Linares M (1997b) The ghost of mimicry past: laboratory reconstitution of an extinct butterfly ‘race’. Heredity 78:628–635
Mallet J (1989) The genetics of warning colour in Peruvian hybrid zones of Heliconius erato and H. melpomene. Proc R Soc Lond B 236:163–185
Mallet J (1993) Speciation, raciation and color pattern evolution in Heliconius butterflies: evidence from hybrid zones. In: Harrison RG (ed) Hybrid zones and the evolutionary process. Oxford University Press, Oxford, pp 226–260
Mallet J (2007) Hybrid speciation. Nature 446:279–283
Mallet J (2009) Rapid speciation, hybridization and adaptive radiation in the Heliconius melpomene group. In: Butlin RK, Bridle JR, Schluter D (eds) Speciation and patterns of diversity. Cambridge University Press, Cambridge, pp 177–194
Mallet J, Barton NH (1989) Strong natural selection in a warning-color hybrid zone. Evolution 43:421–431
Mallet J, Gilbert LE Jr (1995) Why are there so many mimicry rings? Correlations between habitat, behaviour and mimicry in Heliconius butterflies. Biol J Linn Soc 55:159–180
Mallet J, Barton N, Lamas G, Santisteban CJ, Muedas MM, Eeley H (1990) Estimates of selection and gene flow from measures of cline width and linkage disequilibrium in Heliconius hybrid zones. Genetics 124:921–936
Mallet J, McMillan WO, Jiggins CD (1998) Mimicry and warning color at the boundary between races and subspecies. In: Howard JD, Berlocher SH (eds) Endless forms. Oxford University Press, Oxford, pp 390–403
Mallet J, Beltrán M, Neukirchen W, Linares M (2007) Natural hybridization in heliconiine butterflies: the species boundary as a continuum. BMC Evol Biol 7:28. doi:10.1186/1471-2148-7-28
Mavárez J, González M (2006) A set of microsatellite markers for Heliconius melpomene and closely related species. Mol Ecol Notes 6:20–23
Mavárez J, Linares M (2008) Homoploid hybrid speciation in animals. Mol Ecol 17:4181–4185
Mavárez J, Salazar C, Bermingham E, Salcedo C, Jiggins CD, Linares M (2006) Speciation by hybridization in Heliconius butterflies. Nature 441:868–871+11 supplementary pages
Melo MC, Salazar C, Jiggins CD, Linares M (2009) Assortative mating preferences among hybrids offers a route to hybrid speciation. Evolution 63:1660–1665
Monteiro AF, Brakefield PM, French V (1994) The evolutionary genetics and developmental basis of wing pattern variation in the butterfly Bicyclus anynana. Evolution 48:1147–1157
Müller F (1879) Ituna and Thyridia; a remarkable case of mimicry in butterflies. Proc Ent Soc Lond 1879:xx–xxix
Naisbit RE, Jiggins CD, Mallet J (2001) Disruptive sexual selection against hybrids contributes to speciation between Heliconius cydno and H. melpomene. Proc R Soc Lond B 268:1849–1854
Neustetter H (1929) Nymphalididae: Subfam. Heliconiinae. In: Strand E (ed) Lepidopterorum catalogus, vol 36. W. Junk, Berlin, pp 1–136
Nijhout HF, Wray GA, Gilbert LE (1990) An analysis of the phenotypic effects of certain color pattern genes in Heliconius (Lepidoptera: Nymphalidae). Biol J Linn Soc 40:357–372
Nixon KC, Wheeler QD (1990) An amplification of the phylogenetic species concept. Cladistics 6:211–223
Nolte AW, Freyhof J, Stemshorn KC, Tautz D (2005) An invasive lineage of sculpins, Cottus sp. (Pisces, Teleostei) in the Rhine with new habitat adaptations has originated from hybridization between old phylogeographic groups. Proc R Soc Lond B 272:2379–2387
Oberthür C (1902) Observations sur la variation des Heliconia vesta et thelxiope. Etudes d’Entomol 21:1–26, 11 plates
Papa R, Morrison CM, Walters JR, Counterman BA, Rui C, Georg H, Ferguson L, Chamberlain N, french-Constant RH, Kapan DD, Jiggins CD, Reed RD, McMillan WO (2008) Highly conserved gene order and numerous novel repetitive elements in genomic regions linked to wing pattern variation in Heliconius butterflies. BMC Genomics 9:345
Plate L (1928) Ueber Vervollkommnung Anpassung und Unterscheidung von niederen und höheren Tieren. Zool Jahrb 45:745–798
Popper KR (1965) The logic of scientific discovery. Harper Torchbooks, New York
Popper KR (1968) Conjectures and refutations: the growth of scientific knowledge. Harper Torchbooks, New York
Posla-Fuentes M (1993) An unusual form of Heliconius cydno from Costa Rica (Lepidoptera: Nymphalidae: Heliconiinae). Trop Lepid 4:92
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Quek S-P, Counterman BA, Albequerque de Moura P, Cardoso MZ, Marshall CR, McMillan WO, Kronforst MR (2010) Dissecting comimetic radiations in Heliconius reveals divergent histories of convergent butterflies. Proc Natl Acad Sci USA 107:7365–7370
Riffarth H (1901) Die Gattung Heliconius Latr.: Neu bearbeitet und Beschreibung neuer Formen. Berl Entomol Zeit 46:25–183
Salazar JA (1993) Notes on some populations of Heliconius heurippa in Colombia (Lepidoptera: Nymphalidae: Heliconiinae). Trop Lepid 4:119–121
Salazar CA, Jiggins CD, Arias CF, Tobler A, Bermingham E, Linares M (2005) Hybrid incompatibility is consistent with a hybrid origin of Heliconius heurippa Hewitson from its close relatives, Heliconius cydno Doubleday and Heliconius melpomene (Linnaeus). J Evol Biol 18:247–256
Salazar CA, Jiggins CD, Taylor JE, Kronforst MR, Linares M (2008) Gene flow and genealogical history of Heliconius heurippa. BMC Evol Biol 8:132
Salazar CA, Baxter SW, Pardo-Diaz C, Wu G, Surridge A, Linares M, Bermingham E, Jiggins CD (2010) Genetic evidence for hybrid trait speciation in Heliconius butterflies. PLoS Genet 6:e1000930
Schwarz D, Matta BM, Shakir-Botteri NL, McPheron BA (2005) Host shift to an invasive plant triggers rapid animal hybrid speciation. Nature 436:546–549
Sheppard PM (1963) Some genetic studies of Müllerian mimics in butterflies of the genus Heliconius. Zool NY 48:145–154
Sheppard PM, Turner JRG, Brown KS, Benson WW, Singer MC (1985) Genetics and the evolution of muellerian mimicry in Heliconius butterflies. Phil Trans R Soc Lond B 308:433–613
Smiley J (1978) Plant chemistry and the evolution of host specificity: new evidence from Heliconius and Passiflora. Science 201:745–747
Srygley RB, Ellington CP (1999) Discrimination of flying mimetic, passion-vine butterflies Heliconius. Proc R Soc Lond B 266:2137–2140
Stichel H (1906) Lepidoptera Rhopalocera fam. Nymphalidae subfam. Heliconiinae. In: Wytsman P (ed) Genera insectorum. V Verteneuil and L Desmet, Bruxelles, pp 1–74 (6 plates)
Stichel H, Riffarth H. (1905) Heliconiidae. Das Tierreich, vol 22. R Friedländer und Sohn, Berlin
Turner JRG (1971) Two thousand generations of hybridisation in a Heliconius butterfly. Evolution 25:471–482
Turner JRG (1976) Adaptive radiation and convergence in subdivisions of the butterfly genus Heliconius (Lepidoptera: Nymphalidae). Zool J Linn Soc 58:297–308
Turner JRG (1977) Butterfly mimicry: the genetical evolution of an adaptation. In: Hecht MK, Steere WC, Wallace B (eds) Evolutionary Biology, vol 10. Plenum, New York, pp 163–206
Turner JRG (1982) How do refuges produce biological diversity? allopatry and parapatry, extinction and gene flow in mimetic butterflies. In: Prance GT (ed) Biological diversification in the tropics: proceedings, fifth international symposium of the association for tropical biology, Macuto Beach, Caracas, Venezuela, pp 309–335
Turner JRG (1983) Mimetic butterflies and punctuated equilibria: some old light on a new paradigm. Biol J Linn Soc 20:277–300
Turner JRG, Johnson MS, Eanes WF (1979) Contrasted modes of evolution in the same genome: allozymes and adaptive change in Heliconius. Proc Natl Acad Sci USA 76:1924–1928
Wahlberg N, Leneveu J, Kodandaramaiah U, Peña C, Nylin S, Freitas AVL, Brower AVZ (2009) Nymphalid butterflies diversify following near demise at the cretaceous/tertiary boundary. Proc R Soc Lond B 276:4295–4302
Acknowledgments
I thank Rick Grosberg, Dan Howard, Ben Normark and Dave Rand for organizing the symposium and this festschrift issue of Genetica in honor of our mentor Rick Harrison. The latter Rick diagnosed me many years ago as a person who does not suffer fools, and encouraged me to be patient and deliberate in my scholarly endeavors. This paper has been gestating for over 4 years, and I trust it lives up to his exacting standards. In any event, I owe Rick a debt of gratitude for taking me in, way back in 1987, and letting me run amok in his lab for more than 6 years. The paper would have been written save for the enormous amount of work by my friends in the Larry Gilbert Heliconius dynasty, and it is my sincere hope that this review will fan the flames of their enthusiasm. Thanks to Eirik Rindal for help with analyses and figures. The revised version of the manuscript benefited from the mainly constructive criticism of two anonymous reviewers. My research is currently sponsored by NSF DEB 0640301.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Brower, A.V.Z. Hybrid speciation in Heliconius butterflies? A review and critique of the evidence. Genetica 139, 589–609 (2011). https://doi.org/10.1007/s10709-010-9530-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-010-9530-4