
Abstract
DNA mismatch repair proteins play an essential role in maintaining genomic integrity during replication and genetic recombination. We successfully isolated a full length MSH2 and partial MSH7 cDNAs from tomato, based on sequence similarity between MutS and plant MSH homologues. Semi-quantitative RT-PCR reveals higher levels of mRNA expression of both genes in young leaves and floral buds. Genetic mapping placed MSH2 and MSH7 on chromosomes 6 and 7, respectively, and indicates that these genes exist as single copies in the tomato genome. Analysis of protein sequences and phylogeny of the plant MSH gene family show that these proteins are evolutionarily conserved, and follow the classical model of asymmetric protein evolution. Genetic manipulation of the expression of these MSH genes in tomato will provide a potentially useful tool for modifying genetic recombination and hybrid fertility between wide crosses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Advances in genetics and molecular biology provide translational opportunities to facilitate continuous improvement of plant breeding systems. Cultivated tomato (Solanum lycopersicum L., formerly Lycopersicon esculentum Mill.) is an important vegetable crop, both in economic terms and as a source of dietary nutrients. Tomato has relatively low genetic variation as a consequence of its history of migration outside the native area, domestication and selection by early breeders. Thirteen related wild species, (Solanum sect. Lycopersicon) and four more-distantly related nightshade species (Solanum sect. Lycopersicoides and Solanum sect. Juglandifolia) possess many potentially beneficial traits, such as environmental stress tolerances, pest and disease resistance and desirable fruit quality characteristics. However, in order to access germplasm in the wild species, it is necessary (but extremely difficult) to overcome strong breeding barriers such as highly suppressed genetic recombination and low hybrid fertility.
It is well established that the mismatch repair system (MMR) plays key roles in maintaining genomic integrity, by correcting DNA mismatches arising during DNA replication and antagonizing genetic recombination between diverged sequences (Modrich 1991; Harfe and Jinks-Robertson 2000; Surtees et al. 2004; Bray and West 2005; Iyer et al. 2006). Tomato is a convenient crop model to study and manipulate the functions of the MMR system, and the potential to control important biological processes such as meiotic recombination and rapid accumulation of somatic mutations (mutagenesis) could have a major impact in plant breeding. However, our understanding of MMR is mainly based on the well characterized MutHLS system of Escherichia coli, whereby MutS homodimers recognize and bind to insertion/deletion loops (1–4 base pairs, bp) and repair mismatches. In the presence of ATP, MutS recruits MutL (an ATPase), and activates MutH (methylation sensitive endonuclease) that cleaves the transiently unmethylated DNA strand, targeting MMR to the newly synthesized DNA strand (Modrich 1991; Modrich and Lahue 1996; Schofield and Hsieh 2003; Iyer et al. 2006).
In the eukaryotic MMR system, homologues of MutS and MutL have both been found, but not MutH. MutS has seven eukaryotic homolog proteins, namely MSH1 to MSH7, with MSH7 being unique to plants (Culligan and Hays 1997; Adé et al. 1999; Her et al. 1999; Culligan and Hays 2000; Abdelnoor et al. 2003; Higgins et al. 2004). Four MutL homologues (MLH1, MLH2 or hPMS1, MLH3, and PMS1 or hPMS2) have also been identified (Jean et al. 1999; Jiricny 2000; Harfe and Jinks-Robertson 2000; Alou et al. 2004). Heterodimers of these proteins provide substrate specificity: MSH2·MSH6 (MutSα) repair base-base mismatches; MutSα and MSH2·MSH3 (MutSβ) repair + 1 insertion/deletion loops (IDLs); MutSβ also repair larger loops of 2–8 bp (Modrich 1991; Modrich and Lahue 1996, Marti et al. 2002). MSH1 is required for mitochondrial stability (Reenan and Kolodner 1992; Sandhu et al. 2007), while MSH4 and MSH5 function in meiosis (Ross-Macdonald and Roeder 1994; Sym and Roeder 1994; Hollingsworth et al. 1995; Schofield and Hsieh 2003) and recently, it was reported that expression of MSH7 is required for wild-type level of fertility in barley (Lloyd et al. 2007).
In this study, we report the isolation and characterization of the first nuclear MutS homolog from tomato, MSH2, and partial cDNA sequences of the plant specific homolog, MSH7. Characterization of protein sequences and predicted secondary structures confirm that the isolated tomato MSH2 and MSH7 cDNA sequences are homologous to the MSH/MutS genes. Comparative sequence analysis shows that plant MSH genes are evolutionarily conserved and highly concordant with the proposed classical model of asymmetric protein evolution.
Materials and methods
Molecular cloning of tomato MSH2
A tomato MSH2 cDNA was cloned using PCR primers designed on conserved domains in MutS homologs (Varlet et al. 1994), which amplified a partial MutS-like sequence from an immature tomato fruit cDNA library (made from cv. VFNT Cherry). A single PCR product was cloned into pZero vector (pZTmutS-1) and its sequence showed significant similarity to several MutS proteins and was therefore used to further screen the fruit cDNA library. A 2.8 kb cDNA was identified, which is nearly full length, lacking only 66 bp of the 5′ end. The missing 5′ sequence was obtained using RACE-PCR (Rapidly Amplified cDNA ends) according to specifications of the Gene Racer kit (Invitrogen).
Isolation of partial tomato MSH7 cDNA sequence
The cDNA of tomato (cv. VF36), isolated from young leaves (5 mm length at axillary buds) was used as template to amplify MSH7. Primers were designed initially to span the entire length of the MSH7 gene according to conserved regions found in the alignment of MSH7 genes of Arabidopsis thaliana (AF193018, NM180299, AJ007792), Triticum aestivum (AF354709), and Zea mays (AJ238786, AJ238787). However, only four primer sets were successful in PCR amplifications, resulting in isolation of partial MSH7 sequences: 7e3F (5′ TGAGCTSTATGARSTAGATGC 3′), 7R3 (5′ GACCAACATTTTCAG CAAGTGG 3′), and internal primers e12bF (5′ CTGTGTTACATTACCTGGGAAGC 3′) and e12R (5′ ACCCAAACACTTTGACCCGCTG 3′). PCR conditions were: one cycle of 94°C for 5 min; then 40 cycles of 94°C denaturation for 45 s, 52–54°C annealing for 45 s and 72°C extension for 1 min 30 s, with a final extension cycle of 72°C for 7 min. PCR products were visualized by agarose gel electrophoresis, strong bands of expected size were extracted and cleaned using the Qiaquick Gel extraction kit (Qiagen) and sequenced by the DBS Sequencing Facility, UC Davis (http://dnaseq.ucdavis.edu). Sequence files were manually edited and aligned using the program Sequence Navigator (Applied Biosystems).
Phylogenetic analysis
We searched NCBI to obtain MSH protein sequences available for plants. Accession numbers for each homolog used in this study are listed in Table 1. Multiple sequence alignments of the MSH sequences were carried out using the program Clustal W2 (http://www.ebi.ac.uk/Tools/clustalw2/index.html) with default values for gap opening (10) and extension (0.2) penalties, and the GONNET 250 protein similarity matrix. A second multiple sequence alignment was performed using the program EXPRESSO (http://tcoffee.vital-it.ch/cgi-bin/Tcoffee/tcoffee_cgi/index.cgi). Three PDB files were included together with the MSH sequences, namely 1E3M (E. coli MutS), 1EWQ (Thermus. aquaticus Mut S) and 2GFU (Homo sapiens MSH6). EXPRESSO used the three PDB structures as templates to guide the alignment of the original sequences and the final result is a multiple sequence alignment based on the structural information of the templates. Phylogenetic trees were constructed using the distance based method Neighbor-Joining (Saitou and Nei 1987) using mean character difference as implemented in the program PAUP* 4.0 beta 10 (Swofford 2002). Bootstrap support was conducted with 1,000 replicates for Neighbor-Joining analysis. In addition, the PROTDIST program (http://mobyle.pasteur.fr/cgi-bin/MobylePortal/portal.py?form=protdist) was used to compute distance matrices for specific groups of MSH2 and MSH7 protein sequences, using the Jones-Taylor-Thornton (J-T-T) model (default model) (Jones et al. 1992) .
Protein sequence analysis
The tomato MSH2 and MSH7 protein sequences were analyzed on the integrated protein signature databases website, or InterPro (http://www.ebi.ac.uk/interpro/). InterPro is a comprehensive database of protein families, domains, repeats and sites in which identifiable features found in known proteins can be applied to new protein sequences. Member databases include PANTHER, Pfam, PIRSF, PRINTS, Prodom, PROSITE patterns and profiles, SMART, TIGRFAMS, GENE3D and SUPERFAMILY. In addition, the MOTIF metasite (http://motif.genome.jp/) was also used, which included the BLOCKS database.
Predictions of protein structures based on homology modeling were performed using the SAM-T06 program (http://compbio.soe.ucsc.edu/SAM_T06/T06-query.html). This program finds and aligns similar protein sequences, provides sequence logos showing relative conservations of amino acids and secondary structures at different positions. Local structure predictions are done with neural nets for several different local structure alphabets, and hidden Markov models are created (Karplus et al. 2005).
mRNA isolation and transcription analyses by semi-quantitative RT-PCR
Tissues excised from tomato plants (cv. Moneymaker, cv. Gold Nugget) were immediately frozen in liquid nitrogen. Various tissue types were examined: stem, young leaves, mature leaves, floral buds, sepal, petal, anther, pistil and root. Floral bud samples comprised of immature flowers approximately 2–4 mm in length. Mature flowers collected at anthesis were separated into sepal, petal, anther and pistil. Stem samples included the top 1 cm of the shoot apical meristem. Young leaves were sampled at approximately 5 mm in length, obtained from axillary buds. Leaf lamina of mature leaves was sampled approximately at 8 cm in length. Root samples were secondary roots about 5 cm from the root tips. Total RNA was extracted from 200 to 300 mg of frozen tissues using TRIzol Reagent (Invitrogen) following the manufacturer’s protocol. RNA pellets were dissolved in sterile RNAse-free water (Mediatech). DNAse I (Fermentas) was used to eliminate any DNA contamination from the samples.
MSH2
A one-step semi-quantitative RT-PCR method (Superscript One-Step RT-PCR with Platinum Taq, Invitrogen) was used to compare relative levels of MSH2 mRNA expression. Intron positions in MSH2 were predicted from the alignment of tomato and Arabidopsis MSH2 cDNA and genomic DNA sequences. PCR primers were designed to flank introns 5–9. The primer set, U1732 (5′ GTAGTTCAAACAGTTGCGAGTT 3′) and L2146 (5′ ATAAAAGTAGAAACCCCCTTC 3′) produced a predicted 434 bp amplicon from cDNA (or 913 bp from genomic DNA). For each reaction, 100 ng of total RNA from each tissue type was used. The reverse transcription for cDNA synthesis was done at 50°C for 30 min, after which the samples immediately went into the amplification reaction. PCR conditions were: one cycle of 94°C for 2 min; then 34 cycles of 94°C denaturation for 1 min, 52°C annealing for 40 s and 70°C polymerization for 1 min, with an extension cycle of 72°C for 6 min. PCR products were analyzed by agarose gel electrophoresis to verify size and expression levels. Ribosomal RNAs were used as controls.
MSH7
First-strand cDNA synthesis was carried out according to manufacturer’s instructions, using up to 5 μg of template RNA per reaction, 0.5 μg of Oligo(dT)18 primer (Fermentas) and 40 units of M-MLV Reverse transcriptase enzyme (Promega). For the PCR reaction, 500 ng of template cDNA was used with the primer pair msh7RNAiF (5′ CCTCGAGTCTAGATCTTGCCGTCAAGGAGAC 3′) and msh7RNAiR (5′ GGAATTCGGATCCACAAGTGTCTGTCCATCC 3′) to amplify 510 bp of MSH7. As a control, primers were designed for aldolaseA (aldolaseF: 5′ GCTGCTTGCTACAAGGCTCT 3′ and aldolaseR: 5′ GCCTTGAGGGTACTCTGCTG 3′; amplicon length 305 bp). PCR conditions were: one cycle of 95°C for 7 min; then 30 cycles of 94°C denaturation for 30 s, 54°C annealing for 30 s and 72°C extension of 45 s, with a final extension cycle of 72°C for 5 min. PCR products were analyzed by agarose gel electrophoresis to verify size and expression levels.
Genetic mapping of tomato MSH2 and MSH7 genes
Chromosome locations of the tomato MSH2 and MSH7 genes were determined using a set of Solanum pennellii (formerly L. pennellii) introgression lines containing single introgressed chromosome segments from this wild species in a constant genetic background of S. lycopersicum cv. M-82 (Eshed et al. 1992; Eshed and Zamir 1995). The RFLP technique was used in the genetic mapping of MSH2. DNA isolation, restriction enzyme digestion, Southern hybridization and radioactive labeling were carried out according to protocols described previously by Chetelat and Meglic (2000).
The probe was amplified from the MSH2 cDNA clone using primers specific for MSH2: LEstartB3 (5′ GACTACTTCGAAATGACCCTACCCAAGGATGTTAGG 3′) and LEstopB (5′ TAAGCCGCTAGCTAATTTGAAGAACTAAAGAACTGCTG 3′). PCR amplification conditions were: 95°C for 1 min, 30 cycles of 95°C for 30 s, 63°C for 1 min, and 68°C for 2 min. The MSH2 radio-labeled probe was hybridized to genomic DNA. For MSH7, the primers 7F1 (5′ TCT ACCGCCTAACCTGTGGAGC 3′) and 7R3 were used to amplify approx. 324 bp of the MSH7 gene, and a CAPS assay was used. PCR amplification conditions were: 95°C for 5 min, 40 cycles of 94°C for 30 s, 54°C for 30 s, 72°C for 45 s, and 72°C for 5 min. Restriction enzyme digestion of the amplified product with MseI produced band polymorphisms between tomato (M-82) and S. pennellii (accession LA0716) that permitted the localization of MSH7.
Results
Isolation and characterization of tomato MSH2 and MSH7 cDNAs
A tomato fruit cDNA library was screened with a MutS-specific probe resulting in one putative tomato MSH2 clone (pLEMSH2E). Sequencing of this clone revealed a partial reading frame of 2,766 bp, but missing about 66 bp at the 5′ end of the coding sequence. We used 5′ RACE on tomato RNA to obtain the full-length cDNA and 103 bp of 5′ UTR. The 2,832 bp reading frame of the putative tomato MSH2 yields a predicted protein of 943 amino acids, which is very similar in length to other reported eukaryotic MSH2 sequences: A. thaliana (937 aa), P. hybrida (942 aa), Z. mays (942 aa), S. cerevisiae (964 aa); MutS of E. coli (853 aa) and T. aquaticus (791 aa). Analysis of this cDNA sequence using BLASTn shows that it is most similar to a Petunia MSH2 gene: PhMSH2 with maximum 91% sequence identity and approximately 78% identical to the Arabidopsis AtMSH2 cDNA. Amino acid sequence alignment and comparisons of the tomato MSH2 to orthologues in other plants confirm that the tomato cDNA is full length (Fig. 1a). Amino acid sequence distance matrix calculated based on the J-T-T model between tomato MSH2 and seven other MSH2 proteins, shows it is highly similar to MSH2 from Petunia and Vitis with 89.2 and 78.5% levels of identity, respectively.


a Alignment of MSH2 protein sequences. The sequence prefixes Tom, Ath, Osa, Eco, and Taq represent tomato, A. thaliana, O. sativa, E. coli, and T. aquaticus; b alignment of MSH7 protein sequences. The sequence prefixes Tom, Ath, Vvf, Osa, and Hsa represent tomato, A. thaliana, V. vinifera, O. sativa and the PDB sequence file 2GFU (human MSH6). Black boxes denote identical amino acids, grey boxes highlight similar amino acids according to Blosum 62 matrix. Dashes denote gaps. Amino acid positions are shown at right. Boxed lines show conserved regions found in MSH proteins: A = Walker A, B = Walker B, C & D = motifs C and D, H-T-H = helix-turn-helix. I = N-terminal mismatch recognition domain; II = connector domain; III = core domain; IV = clamp domain; V = C-terminal conserved domain. Hatched box denote newly recognized conserved region. Line above the alignment denote the N-terminal PCNA/RPA interaction domain
Primers designed from alignment of conserved regions of MSH7 successfully amplified cDNA of tomato MSH7. However, due to the hypervariable region at the N-termini of the MSH7 gene (Culligan and Hays 2000; Fig. 1b), only partial cDNA sequence of the tomato MSH7 was obtained, for a total of 2,360 bp amplified from primers anchored on exon 3 and exon 17 (based on the gene structure of AtMSH7). Predicted ORF of the partial tomato MSH7 sequence encodes 782 aa. BLASTn analysis of this sequence shows good similarity to AtMSH7 with 67% maximum sequence identity (spanning 95% of the query coverage). It is also very similar to an un-annotated V. vinifera accession (AM477397.2), with maximum identity at 83% (for 80% of the query coverage). Alignment of the predicted partial MSH7 protein sequence with the fully annotated AtMSH7 confirms that the tomato sequence spans from exon 3 to exon 17 (Fig. 1b). J-T-T model based similarity matrix of amino acid sequences of tomato MSH7 shows that it shares high similarities to V. vinifera and A. thaliana with 63.4 and 53.7% identity, respectively.
Phylogenetic relationships of tomato MSH2 and MSH7
Evolutionary relationships of the tomato MSH2 and MSH7 with other MutS/MSH homologues were examined through a phylogenetic study of available plant MSH protein sequences on NCBI. Two sets of multiple aligned sequences were generated, the first comprising 46 accessions from ClustalW2 totaling 2,872 characters, and the second, of 45 accessions and 3PDB files from EXPRESSO, with a total of 2,327 characters. One accession was excluded from the EXPRESSO multiple sequence alignment (CAJ86300, Oryza sativa var. indica) because it exceeded the sequence limit for analysis.
Distance based trees constructed by the Neighbor-Joining (NJ) method using alignments from both methods are very similar, hence the EXPRESSO based tree is presented (Fig. 2). The NJ tree rooted at midpoint shows seven distinct groups representing each class of MSH protein, from MSH1 to MSH7, all with high bootstrap support levels. For each gene cluster, the monocot and dicot MSH proteins separated out easily as two sister groups. The tomato MSH2 and MSH7 resolved clearly within their respective protein groups. Tomato MSH2 is sister to P. hybrida MSH2, and together they are closely related to the MSH2 orthologues of V. vinifera and A. thaliana, all with strongly supported bootstrap values (100%). Tomato MSH7 is sister to its ortholog in V. vinifera, and both are closely related to A. thaliana, with all groups showing 100% bootstrap values.

Phylogram of MSH subfamily from representative plant species. Phylogenetic tree was constructed from full-length aligned protein sequences from EXPRESSO using the Neighbor-Joining method. Bootstrap values are given above the branches
The midpoint rooting function further demonstrates that the group of mitochondrial targeted MSH1 proteins is the most distant from the other MSH proteins, and this relationship has 100% bootstrap support (Fig. 2). Two major groups can be determined, the first consists of MSH3, closely related to the sister groups of MSH6 and MSH7 (99% bootstrap value). The second group consists of either sister groups MSH2 and MSH4 (EXPRESSO alignment) or MSH2 and MSH5 (ClustalW2 alignment). Thus, the placements of MSH4 and MSH5 are unstable, either one resolve in the position between MSH1 and the remaining MSH proteins. In addition, NJ trees were also obtained by restricting the characters to only those in the highly conserved C-terminal regions. These trees are generally consistent in topology and groups resolved with the fully aligned sequences (results not shown).
Protein sequence analysis of tomato MSH2 and partial MSH7
Further analysis of the tomato MSH2 and MSH7 protein sequences on the integrated protein signature databases, or InterPro and the MOTIF metasite, indicates that the tomato MSH2 and partial MSH7 sequences are likely to be functional homologues of the DNA mismatch repair proteins. Protein database searches returned multiple significant hits from Pfam, Prodom and BLOCKS, showing that both sequences contain the conserved domains and motifs recognizable for a MutS/MSH protein. For tomato MSH2 (Fig. 1a), the five major conserved characteristic domains are present, which include the N-terminal mismatch recognition domain (I), middle conserved domain, divided as the connector (II), core (III) and clamp (IV) domains, and the conserved C-terminal domain (V). BLOCKS identified a total of seven possible signature motifs from conserved multiple aligned sequences. The partial cDNA sequence of MHS7 covers part of the N-terminal mismatch recognition domain (I), the middle conserved domain with the connector (II) and core (III), and the highly conserved C-terminal domain (V). No clamp domain (IV) was identified for the MSH7 sequence (Fig. 1b). Six conserved sequence regions corresponding to signature motifs for the N-terminal, core, and C-terminal conserved domains were also identified.
Predicted protein secondary structures
To gain insight on protein structural features of the MSH genes, comparisons were made between the tomato MSH2 and MSH7 sequences with that of the E. coli MutS (Lamers et al. 2000) for which the crystal structure has been resolved. The crystal structure of the T. aquaticus MutS protein is also available (Obmolova et al. 2000), but with more differences in protein sequence alignment. The tomato MSH2 and MSH7 protein sequences were analyzed in three parts: consisting of sequences from the N-terminal, middle core and C-terminal domains (Supplementary Figs. 1a, b and 2a, b). The predicted secondary structure of tomato MSH2 was found to be remarkably similar in structure to the MutS protein, in the core, clamp and C-terminal domains. Differences detected in secondary structures involve the mismatch recognition domain–missing of one beta strand (β3) and one 310 helix (η3) with an additional alpha helix located towards the end of this domain, just after β6. The connector domain is also missing a 310 helix (η6) at the junction in the core domain. The most apparent difference for the MSH7 protein sequence (and hence, predicted secondary structure) is the absence of the entire clamp domain (α19, β14, β15, η7, β16). In the core domain, it is also missing β13 and α18, but has two additional beta sheets at the junction leading to the C-terminal domain. In the mismatch recognition (partial) and connector domains, the secondary structure of tomato MSH7 is missing two beta strands, β4, β11 and one 310 helix (η6). All predicted secondary structures are similar in the C-terminal domain. Thus, both tomato MSH2 and MSH7 lack the 310 helix (η6), and more differences are observed between MSH2 and MSH7 than between either of these when compared with MutS.
Chromosome locations of MSH2 and MSH7 in the tomato genome
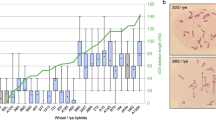
The MSH2 and MSH7 genes were mapped using a set of introgression lines (ILs) containing single overlapping chromosome segments from S. pennellii in the genetic background of cultivated tomato (Eshed and Zamir 1995). For MSH2, genotyping the primary set of 50 ILs revealed the S. pennellii-specific polymorphism only in IL 6-2, thus placing the gene in bins 6C or 6D of chromosome 6 (Fig. 3). MSH7 was mapped in similar fashion to IL 7-4 on chromosome 7. A set of recombinant IL lines for chromosome 7 further narrowed the location of MSH7 to IL7-4-1. Since this gene was not polymorphic in IL7-5 or IL7-5-5, which span bins 7B and 7C, we infer that MSH7 must lie in the region of either bin 7A or bin 7D (Fig. 3). Genetic mapping results also suggest that these genes exist as single copies in the tomato genome.

Map locations of tomato MSH2 and MSH7 on tomato chromosomes 6 and 7, respectively, based on the introgression lines of S. pennellii in the background of S. lycopersicum cv. M82 (Eshed and Zamir 1995). This IL map is based on markers of the F2-2000 map
Tomato MSH2 and MSH7 mRNA expression
Expression of RNA transcripts of MSH2 and MSH7 in various tissues was investigated by semi-quantitative RT-PCR. Primers designed specifically for detecting MSH2 and MSH7 show that mRNA for both genes are detectable at different levels across various tissue types (Fig. 4). Levels of MSH2 are highest in young leaves, followed by slightly lower expression in floral buds and young stems. Sepals, anthers, petals and mature leaves all show a lower level of MSH2 mRNA, with expression not detected in root tissue. Similarly, MSH7 also showed the highest levels of expression in floral buds and young leaves. This is followed by moderate expression in sepals, with slightly lower expression in petal, pistil, stem and anther tissue. Semi-quantitative RT-PCR of MSH7 (and aldolaseA) was not successful in the root tissue even after multiple rounds of RNA extractions.

Gene expression of tomato MSH2 and MSH7 from various tissue types; a one-step RT-PCR of MSH2 (434 bp). Lower panel shows control ribosomal RNAs; b semi-quantitative RT-PCR of MSH7 (510 bp, upper sized bands). Lower sized bands are control, AldolaseA (305 bp)
Discussion
Isolation and characterization of MSH2 and partial MSH7 cDNA sequences
The main objective of our study was to identify and characterize tomato homologues of the mismatch repair gene MutS/MSH. Molecular cloning of MSH genes will subsequently enable their manipulation using recombinant technology to alter gene expression and allow study of their function(s) in tomato. Sequence conservation among previously identified MutS homologues allowed us to isolate a full-length tomato MSH2 and partial MSH7 cDNA sequences, both unambiguously identified as MMR homologues. Knowledge of protein structure provides understanding of detailed function and pathology, and bioinformatics resources are now available for comprehensive analysis of protein sequences (Stein 2001; Cole et al. 2008). Multiple alignment of protein sequences also generate useful predictions for conserved amino acid residues, motifs and domains that have known functional roles in mismatch repair.
Conservation of known important motifs
The mismatch detection motif, Phe36-Tyr37-Glu38 (F-Y-E of E. coli) is responsible for specific mismatch-binding contacts and this F-Y-E motif is conserved for plant MSH7, MSH1 and MSH6, but variable for MSH3 and, missing for MSH4 and MSH5, consistent with the evolution of functional diversification of these proteins. For example, MSH4 and MSH5 are key proteins in meiosis but do not have a role in error correction (Snowden et al. 2004; Franklin et al. 2006), whilst MSH3 specializes in binding a broad range of loop-out DNA strands, as opposed to mostly base mispairs (or very short loop-outs) in the case of MSH6 and MSH7 (Culligan and Hays 2000; Culligan et al. 2000; Wu et al. 2003). Based on this, MSH7 should possess mismatch recognition specificity similar to MSH6 or MSH1.
In the highly conserved C-terminal domain, four known important motifs include the Walker A (P-loop), Walker B, motifs C, D and the helix-turn-helix subdomain characteristic of NTP-binding domains (Ohlendorf et al. 1983; Gorbalenya and Koonin 1990). Our alignment and modeling results show six very conserved amino acids in the classic Walker A motif, GPN-XXX-GKS, identical in the seven plant MSH proteins. It is noted that for MSH7, the Phe596 (large, aromatic) underwent a major change to Proline (small, aliphatic) and Ile597 to a Valine, perhaps contributing to the subfunctionalization of MSH7. The Walker B motif is also conserved in both tomato MSH2 and MSH7 sequences with modeling results indicating three conserved residues, L-XXX-DE, and in our alignment, the residues SL-XXX-DE are identical for plant MSH proteins. Similarly, for motif C (=disordered loop 659–668 of E. coli), residues ST are conserved (STF identical from MSH2 through MSH7). For motif D, the residues TH are conserved, with Histidine recognized as a possible catalytic residue. A non-conservative change is detected in MSH5, from A to C (H-bonding, disulfide) and might be important for MSH5 specific function. Located at the end of the C-terminal is the helix-turn-helix subdomain, important for dimer interface and three amino acids are shown to be conserved, the Y (Y760), G (G765) and A (A789). The nearby motif F-L-Y, conserved for MSH5, 6 and MSH7, differed for MSH4 (F-K-F), and K (H-bonding, positive charged) is a significant substitution that might be definitive for MSH4 function.
Newly identified conserved motifs
Protein sequence analyses of both tomato MSH2 and MSH7 cDNAs identified a newly conserved motif in the middle core domain that includes Arginine R305 (E. coli), whereby a previously shown mutation of this residue conferred a dominant negative phenotype (Wu and Marinus 1994). MSH2 has an additional motif recognized in the C-terminal domain, with conservation of residues Phe (F596), Asn (N599) and Asp (D600), the Asn residue being identical among the seven plant MSH proteins and E. coli (N599). For MSH7, a conserved motif is located in the N-terminal domain, corresponding to β6 at the junction of domains I and II, and may signal the importance of a “transmitter” function (see below). Identification of conserved residues and correlation to specific functions should be useful for future transformation work in tomatoes, e.g., site-directed mutagenesis to generate mutants.
Protein secondary structures
With protein databases and structural analysis methods continually being improved, we were able to compare predicted secondary structures for the two isolated tomato MSH genes with the E. coli MutS homolog. The tomato MSH2 shows only minor differences in secondary structures predicted by homology-based modeling when compared to the MutS non-mismatch binding monomer. In the mismatch recognition domain, the tomato MSH2 predicted secondary structure is missing β3 and η3, changes not unexpected since they involve mismatch DNA contact, especially β3, which has six DNA contact sites. A similar deletion of 12-14 residues corresponding to the region encoding the β3 and β4 hairpin was seen in T. aquaticus for subunit B (Obmolova et al. 2000). Other differences include an additional α helix located in between domains I and II; and domain II is also missing η6, as is in T. aquaticus, at the junction before domain III. Therefore, the minimal changes observed in tomato MSH2 seem concentrated at junctures between structures. Strong conservation of MSH2 clearly reflects its important role as the major subunit in the eukaryotic pattern of heterodimerization with other MSH polypeptides.
Sequence comparisons and secondary structural predictions for MSH7 show loss of the clamp domain (IV) for MSH7 (α19, β14, β15, η7, β16). It has been previously discovered that MSH7, which is unique to plants, is missing this particular domain (Wu et al. 2003) involved in making non-specific DNA contacts. In E. coli, the clamp domain (about 100 residues 432–537) might function in initial recognition of homoduplex DNA by MutS (Lamers et al. 2000). In the core domain, β13 and α18 are also missing, but two additional β sheets are detected, leading into the C-terminal domain. Domains I and IV are known to share similar folding topology, with two pairs of β hairpins linked by a helical segment to form an anti-parallel β sheet (Obmolova et al. 2000). For tomato MSH7, the predicted appearance of an additional two β sheets, followed by α helix (α21) and two β sheets (β18, β19) may somewhat replace the DNA binding function of the clamp domain. Also, as β4 is actively involved in recognizing the mismatch by van der Waals contacts (Lamers et al. 2000), it may be that the missing β4 in MSH7 might have altered its recognition specificity.
Study of the MutS crystal structure of T. aquaticus led to the proposal that domain junctions (especially between II, III and V) are significantly important to facilitate inter-domain contacts, serving as a transmitter for information exchange between the ATP- and DNA binding sites (Obmolova et al. 2000). This might partially explain the changes located at junctions between domains seen in MSH2 and MSH7 of tomato. An additional N-terminal PCNA/RPA interaction domain was also identified for MSH6 and MSH7, and in Arabidopsis, interaction between MSH2 and MSH7 proteins is similar to that of MSH2 and MSH6, and in fact, observably better than MSH2 and MSH3 (Culligan and Hays 2000). The AtMSH2-MSH7 heterodimer did show novel substrate specificity, a preference for (T/G) base/base mispairs and recognized several base mismatches better than MSH2-MSH6 (Wu et al. 2003). It was proposed that AtMutSγ may have specialized recognition of DNA lesions (e.g., UV irradiation), (T/G) mispairs in mC-containing contexts (Culligan and Hays 2000) or is involved in antagonizing homeologous recombination (Dong et al. 2002). TaMSH7 reportedly affects fertility in barley (Lloyd et al. 2007) but to date, no definitive special role is yet found for MSH7 befitting its significant change in structure.
mRNA transcription and genomic locations of tomato MSH2 and MSH7 genes
In order to obtain more information on expression of MMR genes in tomatoes, we performed a simple investigation of MSH2 and MSH7 mRNA expression in different tissues of tomato. Using semi-quantitative RT-PCR, transcriptional differences are visually detectable when comparing different tissue types, with considerably higher levels in young leaves and floral buds. This is consistent with previous studies reporting higher levels of MSH activity in actively dividing cells compared to cells in mature tissues. Adé et al. (1999) had reported poor expression of AtMSH2, 3 and 6-2 genes in plant tissues, being undetectable using Northern analysis. Instead, only by replacing the plant tissues with mitotically dividing Arabidopsis cell suspensions did they manage to identify mRNAs for MSH2, 3 and 6-2, with high levels of MSH6-2 transcripts in the early exponential growth phase of the cell culture. Similarly, in maize, it was reported that MUS1 (MSH2) and MUS2 (MSH6-like) RNA expressions were only successfully detected in young maize seedlings (at low levels) using RNA gel-blot analyses (Horwath et al. 2002). The tissues of young leaves and floral buds used in our study would contain a source of more actively dividing cells, when compared to mature leaves or other parts of the plant.
Floral buds are especially interesting since they consist of two types of tissues, mitotically dividing cells (calyx, corolla, pistils and stamens), and meiotically dividing cells (pollen mother cells and megaspore mother cells). Mixtures of these two types of tissues may explain the high MSH2 and MSH7 expression levels, but further study is required to determine if the genes are expressed at similar levels in mitotic and meiotic cells. From a study of MutS and MutL transcriptions in yeast, it is known that all MutS homologues (MSH1-6) are induced during meiosis, with MSH2, MSH4 and MSH5 being strongly regulated, and MSH2 showing co-regulation with Spo11 (Meyer et al. 2001). In a study by Crismani et al. (2006), both microarray and Q-PCR data for MSH4 and MSH6 showed that both genes are expressed during meiosis (pre-meiosis to immature pollen) in wheat T. aestivum but expression fell sharply at the mature anther stage. Also in wheat, Northern hybridization successfully detected gene expression of MSH7 in mitotic tissues of root tip, shoot meristem and young meiotic flower tissues, with notably higher expression levels in early meiotic tissues, suggestive of MSH7 playing a specific role during meiosis (Dong et al. 2002). This may partially explain the higher expressions of MSH2 (and MSH7) seen in floral buds. It is also known that MSH proteins strongly antagonize spontaneous mutations in floral cells and meristematic precursors (plant equivalents of reserved germ lines) since strong microsatellite instability (MSI) was detected in AtMSH2 defective progenies (Hoffman et al. 2004), providing additional support for spatial and temporal regulations of MSH genes.
Genetic mapping placed MSH2 on the long arm of chromosome 6 and MSH7 on chromosome 7. Knowledge of the map locations of MSH genes might be helpful for interpretation of their functions by association with other mapped traits or loci. For species in which the locations of genetic recombination or pairing modifier genes are known, such as the Ph genes (Ph1, Ph2) controlling homeologous recombination in wheat (Sears 1982; Dong et al. 2002), or isolated meiotic mutants in maize (Golubovskaya et al. 2002), comparisons of MSH gene locations could indicate candidate genes responsible for the phenotypes and facilitate gene cloning. For example, the location of TaMSH7 on the short chromosome arm of 3A, 3B and 3D coincided with a minor suppressor of homeologous pairing, Ph2 (chromosome 3D, Sears 1982), this, coupled with results showing reduction of TaMSH7 gene expression in the ph2a mutant led to the proposal that MSH7 might be a candidate for the Ph2 gene (Dong et al. 2002). However, recent results from further characterization of MSH7 in wheat and Ph2 mutants revealed that MSH7 is probably not responsible for the Ph2 phenotype (Lloyd et al. 2007). Two segregation distorter loci are located near the positions of MSH2 and MSH7 in tomato (sd6.1 and sd7.1, Canady et al. 2005), but to date, no Ph-like genes or meiotic mutants have been identified in tomato.
Asymmetric protein sequence evolution of plant MSH homologues
Phylogenetic analyses of a subset of MSH protein sequences from plants support the identities of the isolated tomato MSH sequences as MSH2 and MSH7 genes. More extensive phylogenetic analyses detailing the origin and evolution of DNA mismatch repair genes have been performed (Eisen 1998; Culligan et al. 2000; Lin et al. 2007). In a previous study of eukaryotic MutS proteins, Culligan et al. (2000) reported tree instabilities with sequence analyses using only the C-terminal regions, and deducted that the C-terminal region alone is insufficient to resolve critical relationships between MutS-like sequences. In this study, the NJ trees obtained using restricted C-terminal sequences are consistent in the groups resolved with minor changes in tree topology compared with full sequences, thus we present here the final NJ tree based on the full sequence alignments.
The NJ tree rooted at midpoint clearly shows well-defined clusters corresponding to respective families of MSH genes (MSH1 to MSH7) with high bootstrap supports, and tree topology in general agreement with those of other studies (Eisen 1998; Culligan et al. 2000; Lin et al. 2007). As expected for gene phylogenies, orthologous proteins across species are more similar than paralogues within the same species. The considerable divergence between the mitochondrial MSH1 and other MSH genes has been noted previously (Eisen 1998; Lin et al. 2007), and is indicated by our study as well. It was reported that MSH1 genes are likely to be the most primitive eukaryotic MutS1 members, with relatively strong support indicating the origins of other eukaryotic MSH genes from MSH1 due to multiple rounds of gene duplication events (Lin et al. 2007). Both the tomato MSH2 and MSH7 genes resolved clearly within their respective protein classes. The NJ tree shows the MSH2 cluster with relatively shorter terminal branch lengths, denoting fewer changes between orthologues. This is compatible with the biochemical function of MSH2 as the core dimer in the center of a complex protein network, thus severely restricting permissible changes. In contrast, both MSH7 and MSH3 classes show longer terminal branch lengths reflecting a higher number of modifications in these protein sequences.
In our analysis, two major groups are apparent with the first consisting of MSH3 and sister groups of MSH6 and MSH7. The second group, however, comprises either MSH4 or MSH5, with MSH2. The positions of MSH4 and MSH5 are unstable, and low bootstrap values suggest that this branching pattern is not robust. In an earlier study, MSH2, MSH4 and MSH5 formed an unresolved polytomy (Lin et al. 2007). Branching patterns for the MSH genes inferred here and reported from the other studies mentioned clearly distinguish the evolution of the two major groups of paralogues (MSH2/MSH4/MSH5 and MSH3/MHS6/MSH7). For the latter group, two rounds of gene duplication and subsequent specialization were postulated (Culligan et al. 2000; Lin et al. 2007). Evolutionary processes operating in the former (MSH2 et al.) group, however, are not so clear since relationships among these genes remain unresolved. It was suggested that the most recent common ancestor of MSH4 and MSH5 diverged from MSH2 and evolved to specialized meiotic functions (Culligan et al. 2000). However, earlier phylogenomic analyses had proposed the division of the MutS family into two main lineages, namely MutS-I with proteins involved in MMR (MutS1, MSH1, 2, 3 and 6) and MutS-II, consisting of MutS2, MSH4 and 5, involved in meiotic crossing over and chromosome segregation (Eisen 1998; Malik and Henikoff 2000). Additionally, it is also very likely that the basal positions of MSH4 and 5 could be attributed to long-branch attraction (Lin et al. 2007), providing an alternative explanation for their unstable positions on the NJ tree.
The MSH gene family is evolutionarily conserved, with homologues recognizable from archaea and bacteria to higher plants and animals. Duplicated MMR genes are maintained as single copies over vast evolutionary distances and across the divergence of major eukaryotic lineages (Lin et al. 2007). Therefore, the notable difference in evolutionary rates between the two major groups of MSH genes is of much interest. Generation of the two orthologous groups is accompanied by different scales of functional divergence, such as significant rearrangements (complete loss/gain of novel function) leading to neofunctionalization as seen between MSH2 and MSH4/MSH5 but with MSH2 itself under very strong evolutionary constraint; whereas differences in MSH3, MSH6 and MSH7 are suggestive of more gradual diversification, or subfunctionalization, since these proteins all retain similar and even overlapping functions in mismatch repair. It has been observed that duplicated genes may exhibit asymmetric protein sequence evolution, with the slow copy maintaining an ancestral role and rate of change; and the fast copy evolving to optimize novel function(s) (Ohno 1970; Van de Peer et al. 2001; Conant and Wagner 2003). The evolution of these plant MSH genes is highly concordant with the proposed classical model of asymmetric protein evolution.
Our characterization of MSH2 and partial MSH7 will now permit further study of these MSH genes in the model crop tomato. Significant insights gained from experimental manipulations of MMR functions will provide more efficient ways to develop novel genetic material and accomplish genetic transfer of beneficial traits. Results from tomato might also be applicable for the improvement of other crop species.
References
Abdelnoor RV, Yule R et al (2003) Substoichiometric shifting in the plant mitochondrial genome is influenced by a gene homologous to MutS. Proc Natl Acad Sci USA 100:5968–5973
Adé J, Belzile FJ et al (1999) Four mismatch repair paralogues coexist in Arabidopsis thaliana: AtMSH2, AtMSH3, AtMSH6–1 and AtMSH6–2. Molecular Genomics Genet 262:239–249
Alou AH, Azaiez A et al (2004) Involvement of the Arabidopsis thaliana AtPMS1 gene in somatic repeat instability. Plant Mol Biol 56:339–349
Bray CM, West CE (2005) DNA repair mechanisms in plants: crucial sensors and effectors for the maintenance of genome integrity. New Phytol 168:511–528
Canady MA, Meglic V et al (2005) A library of Solanum lycopersicoides introgression lines in cultivated tomato. Genome 48:685–697
Chetelat RT, Meglic V (2000) Molecular mapping of chromosome segments introgressed from Solanum lycopersicoides into cultivated tomato (Lycopersicon esculentum). Theor Appl Genet 100:232–241
Cole C, Barber JD et al (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res 36:197–201
Conant GC, Wagner A (2003) Asymmetric sequence divergence of duplicate genes. Genome Res 13:2052–2058
Crismani W, Baumann U et al (2006) Microarray expression analysis of meiosis and microsporogenesis in hexaploid bread wheat. BMC Genomics 7:267
Culligan KM, Hays JB (1997) DNA mismatch repair in plants. An Arabidopsis thaliana gene that predicts a protein belonging to the MSH2 subfamily of eukaryotic MutS homologs. Plant Physiol 115:833–839
Culligan KM, Hays JB (2000) Arabidopsis MutS homologs-AtMSH2, AtMSH3, AtMSH6, and a novel AtMSH7-form three distinct protein heterodimers with different specificities for mismatched DNA. Plant Cell 12:991–1002
Culligan KM, Meyer-Gauen G et al (2000) Evolutionary origin, diversification and specialization of eukaryotic MutS homolog mismatch repair proteins. Nucleic Acids Res 28:463–471
Dong C, Whitford R et al (2002) A DNA mismatch repair gene links to the Ph2 locus in wheat. Genome 45:116–124
Eisen JA (1998) A phylogenomic study of the MutS family of proteins. Nucleic Acids Res 26:4291–4300
Eshed Y, Zamir D (1995) An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics 141:1147–1162
Eshed Y, Abu-Abied M et al (1992) Lycopersicon esculentum lines containing small overlapping introgressions from L. pennellii. Theor Appl Genet 83:1027–1034
Franklin FCH, Higgins JD et al (2006) Control of meiotic recombination in Arabidopsis: role of the MutL and MutS homologues. Biochem Soc Trans 34:542–544
Golubovskaya IN, Harper LC et al (2002) The pam1 gene is required for meiotic bouquet formation and efficient homologous synapsis in maize (Zea mays L.). Genetics 162:1979–1993
Gorbalenya AE, Koonin EV (1990) Superfamily of UvrA-related NTP binding proteins implication for rational classification of recombination/repair systems. J Mol Biol 213:583–591
Harfe BD, Jinks-Robertson S (2000) DNA mismatch repair and genetic instability. Annu Rev Genet 34:359–399
Her C, Wu X et al (1999) Identification and characterization of the mouse MutS homolog 5: Msh5. Mamm Genome 10:1054–1061
Higgins JD, Armstrong SJ et al (2004) The Arabidopsis MutS homolog AtMSH4 functions at an early step in recombination: evidence for two classes of recombination in Arabidopsis. Genes Dev 18:2557–2570
Hoffman PD, Leonard JM et al (2004) Rapid accumulation of mutations during seed-to-seed propagation of mismatch-repair-defective Arabidopsis. Genes Dev 18:2676–2685
Hollingsworth NM, Ponte L et al (1995) MSH5, a novel MutS homolog, facilitates meiotic reciprocal recombination between homologs in Saccharomyces cerevisiae but not mismatch repair. Genes Dev 9:1728–1739
Horwath M, Kramer W et al (2002) Structure and expression of the Zea mays mutS-homologs Mus1 and Mus2. Theor Appl Genet 105:423–430
Iyer RR, Pluciennik A et al (2006) DNA mismatch repair: functions and mechanisms. Chem Rev 106:302–323
Jean M, Pelletier J et al (1999) Isolation and characterization of AtMLH1, a MutL homologue from Arabidopsis thaliana. Mol Genomics Genet 262:633–642
Jiricny J (2000) Mediating mismatch repair. Nat Genet 24:6–8
Jones DT, Taylor WR et al (1992) The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8:275–282
Karplus K, Katzman S et al (2005) SAM-T04: what’s new in protein-structure prediction for CASP6. Proteins Struct Funct Bioinformat 61:135–142
Lamers MH, Perrakis A et al (2000) The crystal structure of DNA mismatch repair protein MutS binding to a G.T mismatch. Nature 407:711–717
Lin Z, Nei M et al (2007) The origins and early evolution of DNA mismatch repair genes–multiple horizontal gene transfers and co-evolution. Nucleic Acids Res 35:7591–7603
Lloyd AH, Milligan AS et al (2007) TaMSH7: A cereal mismatch repair gene that affects fertility in transgenic barley (Hordeum vulgare L.). BMC Plant Biol 7:67
Malik HS, Henikoff S (2000) Dual recognition-incision enzymes might be involved in mismatch repair and meiosis. Trends Biochem Sci 25:414–418
Marti TM, Kunz C et al (2002) DNA mismatch repair and mutation avoidance pathways. J Cell Physiol 191:28–41
Meyer C, Scheller J et al (2001) Transcription of mutS- and mutL- homologous genes during meiosis in Saccharomyces cerevisiae and identification of a regulatory cis-element for meiotic induction of MSH2. Mol Gen Genomics 265:826–836
Modrich P (1991) Mechanisms and biological effects of mismatch repair. Annu Rev Genet 25:229–253
Modrich P, Lahue R (1996) Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem 65:101–133
Obmolova G, Ban C et al (2000) Crystal structure of mismatch repair protein MutS and its complex with a substrate DNA. Nature 407:703–710
Ohlendorf DH, Anderson WF et al (1983) Many gene-regulatory proteins appear to have similar alpha-helical fold that binds DNA and evolved from a common precursor. J Mol Evol 19:109–114
Ohno S (1970) Evolution by gene duplication. George Allen and Unwin, London
Reenan RA, Kolodner RD (1992) Characterization of insertion mutations in the Saccharomyces cerevisiae MSH1 and MSH2 genes: evidence for separate mitochondrial and nuclear functions. Genetics 132:975–985
Ross-Macdonald P, Roeder GS (1994) Mutation of a meiosis-specific MutS homolog decreases crossing over but not mismatch correction. Cell 79:1069–1080
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sandhu AP, Abdelnoor RV et al (2007) Transgenic induction of mitochondrial rearrangements for cytoplasmic male sterility in crop plants. Proc Natl Acad Sci USA 104:1766–1770
Schofield MJ, Hsieh P (2003) DNA mismatch repair: molecular mechanisms and biological function. Annu Rev Microbiol 57:579–608
Sears ER (1982) A wheat mutation conditioning an intermediate level of homeologous chromosome pairing. Can J Genet Cytol 24:715–719
Snowden T, Acharya S et al (2004) hMSH4-hMSH5 recognizes Holliday junctions and forms a meiosis-specific sliding clamp that embraces homologous chromosomes. Mol Cell 15:437–451
Stein L (2001) Genome annotation: from sequence to biology. Nature Rev Genet 2:493–503
Surtees JA, Argueso JL et al (2004) Mismatch repair proteins: key regulators of genetic recombination. Cytogenet Genome Res 107:146–159
Swofford DL (2002) PAUP* 4.0 beta 10. Phylogenetic analysis using parsimony (and other methods). Sinauer Associates, Sunderland
Sym M, Roeder GS (1994) Crossover interference is abolished in the absence of a synaptonemal complex protein. Cell 79:283–292
Van de Peer Y, Taylor JS et al (2001) The ghost of selection past: rates of evolution and functional divergence of anciently duplicated genes. J Mol Evol 53:436–446
Varlet I, Pallard C et al (1994) Cloning and expression of the Xenopus and mouse Msh2 DNA mismatch repair genes. Nucleic Acids Res 22:5723–5728
Wu TH, Marinus MG (1994) Dominant negative mutator mutations in the mutS gene of Escherichia coli. J Bacteriol 176:5393–5400
Wu SY, Culligan K et al (2003) Dissimilar mispair-recognition spectra of Arabidopsis DNA-mismatch-repair proteins MSH2*MSH6 (MutSα) and MSH2*MSH7 (MutSγ). Nucleic Acids Res 31:6027–6034
Acknowledgments
The authors gratefully acknowledge the assistance of Jeff Peralta and James Hatteroth with the cloning of the MSH2 gene, and Prof. John B. Hays for sharing the Arabidopsis gene sequence. This work was supported by grants from the UC-Biotech Program (#99-13) and the USDA-NRI (#2005-35301-15736 and #1999-35300-7683).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Sheh May Tam and Sompid Samipak have equally contributed to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Tam, S.M., Samipak, S., Britt, A. et al. Characterization and comparative sequence analysis of the DNA mismatch repair MSH2 and MSH7 genes from tomato. Genetica 137, 341–354 (2009). https://doi.org/10.1007/s10709-009-9398-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-009-9398-3