
Abstract
Hybridization technology has proven valuable in enhancing yields in many crops, but was only recently adopted in the small grain cereals. Hybrid varieties in barley (Hordeum vulgare) rely on the cytoplasmic male sterility (CMS) system msm1 derived from Hordeum vulgare ssp. spontaneum. The major restorer gene described for the msm1 system is known as Rfm1 and maps to the top of chromosome 6H. To gain further insight into mechanisms underlying male fertility restoration in barley, we used a map-based cloning approach to identify the nuclear gene involved in the restoration mechanism of this hybridization system. Taking advantage of the available genomic resources in barley in combination with a custom-made non-gridded BAC library developed from a restorer line, we cloned and sequenced the Rfm1 restorer locus. The characterization and annotation of the nucleotide sequence for the Rfm1 restorer allele allowed for the identification of the candidate gene for Rfm1. The Rfm1 locus carries a tandem repeat of a gene encoding a pentatricopeptide repeat (PPR) protein. Surprisingly, Rfm1 belongs to the PLS-DYW subfamily of PPR genes known for their involvement in RNA editing in plants organelles, but that to date have not been identified as restorer genes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Barley (Hordeum vulgare) ranks amongst the most important small grain cereals grown worldwide and is used for both human and animal feeding. Because of its diploid genome, barley is considered a model crop for the Triticeae tribe, which includes wheat (Triticum aestivum). Recently, the International Barley Genome Sequencing Consortium (IBGS) published a high-quality reference genome sequence of the elite cultivar Morex (Mascher et al. 2017). Approximately 84% of the genome is composed of mobile elements or other repeated sequences (Mayer et al. 2012; Mascher et al. 2017). The majority of these consists of retrotransposons, 99.6% of which are long terminal repeat (LTR) retrotransposons.
The number of gene loci including protein-coding genes, non-coding RNAs, pseudogenes and transcribed transposons is estimated at approximately eighty thousand, 39,734 of which are considered high-confidence genes.
Despite the important position of barley in modern agriculture with approximately 50 Mha cultivated worldwide, a reliable hybridization system has only lately become available. Recent years have seen the market introduction of hybrid cultivars for winter barley based on the msm1 sterile cytoplasm described by Ahokas (1979, 1982, 1983), following the pioneer work by Paul Bury and his team at New Farm Crops, Lincolnshire UK. The conversion of elite European winter barley material into male and female parental lines culminated into the commercialization of the first ever barley hybrid, named Colossus under the Hyvido trademark.
The CMS system used in barley is very similar in its modus operandi to hybridization systems described in other crops (Hu et al. 2014; Islam et al. 2014). Despite the fact that the molecular mechanisms underlying CMS systems are diverse, the resulting phenotype manifests itself by a common failure to produce viable pollen, whereas the fertility of the female organ remains intact. CMS is caused by the expression and accumulation of male sterility-inducing proteins in the mitochondria (Horn et al. 2014). In most of the CMS systems studied so far, the male sterility genes encode poorly conserved hydrophobic polypeptides frequently showing partial sequence homology with conserved subunits of the respiratory chain. The male sterile phenotype can be suppressed by nuclear-encoded restorer genes (Rf). Most of the Rf genes known to date share common characteristics, even across highly divergent species including both monocots and dicots. Apart from some notable exceptions (Liu et al. 2001; Klein et al. 2005; Matsuhira et al. 2012), most Rf genes identified so far encode pentatricopeptide repeat (PPR) proteins (Dahan and Mireau 2013; Gaborieau and Brown 2016). PPR proteins are characterized by tandem repetitions of the PPR motif, a 35 amino acid degenerate motif that forms a helix-turn-helix structure (Howard et al. 2012; Ban et al. 2013; Ke et al. 2013; Shen et al. 2016). The PPR family is divided in two large classes. The P class proteins only contain successions of the initially-recognized 35-amino-acid (P) motif to as many as 30 repeats. The PLS class proteins are composed of P repeats interspersed with variant repeats that are slightly longer (L) or shorter (S) and often comprise characteristic C-terminal extensions named E and DYW domains. By virtue of their RNA-binding activity, PPR proteins are involved in a range of essential RNA processing steps in organelles, including editing, splicing, cleavage and translation (Barkan and Small 2014; Hammani and Giegé 2014). Restorer genes typically belong to the P-class PPR proteins and are present in small clusters of highly homologous gene copies (Schmitz-Linneweber and Small 2008; Dahan and Mireau 2013) generated by local gene duplication and illegitimate recombination events. As a result of their fast evolving structure, restoration loci show an extreme allelic diversity with a highly complex and diverse gene content (Fujii et al. 2011a; Dahan and Mireau 2013; Melonek et al. 2016).
The msm1 cytoplasm and the nuclear restorer locus Rfm1 used in the barley CMS system were originally derived from wild barley accessions (Hordeum vulgare ssp. spontaneum). In practice the msm1 CMS system proved sufficiently reliable to convert a strictly autogamous species like barley into a hybrid crop relying on wind pollination only. Most of the elite European barley germplasm carrying the msm1 cytoplasm revealed perfectly male sterile without showing any further phenotypic aberrations. The restoration of the msm1 CMS requires the action of the restorer allele Rfm1 that is not natively present in cultivated barley. Rfm1 was mapped to the short arm of chromosome 6H (Matsui 2001; Murakami et al. 2005). Recently, Ui et al. reported the fine mapping of the Rfm1 locus, and the identification of BAC clones from the Morex cultivar that encompass the Rfm1 locus (Ui et al. 2015). However, Morex does not restore the male sterility induced by the msm1 cytoplasm and therefore does not carry the restorer allele at the Rfm1 locus. The final identification of the restorer gene necessitates the sequence analysis of the restorer allele of the Rfm1 locus as originally introgressed from Hordeum vulgare ssp. spontaneum.
Here, we report on the map-based cloning and sequence analysis of the restorer allele of the Rfm1 locus and the comparative analysis of the restorer and non-restorer alleles, taking advantage of the genomic tools available in barley and the syntenic relationships between small grain cereals, including Brachypodium and rice. The identification and characterization of the Rfm1 restorer gene will improve our understanding of the molecular basis of CMS restoration in cereals and provide support to the breeding and production of hybrid barley.
Materials and methods
Plant materials
A mapping population of 2184 F2 individuals was derived from a cross between restorer line Re08 (CMS; Rfm1/Rfm1) and the cultivar Laverda (N; rfm1/rfm1), using Laverda as pollinator following the emasculation of Re08 mother plants. The resulting F1 plants were self-pollinated to produce the F2 population. Male fertility restoration of the F2 plants was assessed by scoring for visual anther extrusion and by measuring seed set on bagged ears.
Selected fertile recombinant plants were self-pollinated to yield F2:3 progenies, sterile recombinants were backcrossed to Laverda to yield F2BC1 progenies. Progeny populations were phenotyped for male fertility restoration in order to confirm the results obtained for the recombinant F2 plants.
Marker development and genotyping
PCR primers were designed against barley ESTs, unigenes and BAC-end sequences using the DS Gene software (Accelrys, version 1.5). Insertion site-based polymorphism markers (ISBP) were identified using the ISBPfinder pipeline and amplified using the proposed primers (Paux et al. 2010). Sanger sequencing of the PCR products obtained for both parents of the mapping population allowed for the identification of sequence polymorphisms by comparing the obtained nucleotide sequences using the Seqman software from the Lasergene package (DNASTAR). Taqman assays targeting polymorphic SNPs were designed using the Primer Express software (Applied Biosystems). Genotyping of the fine-mapping population and recombinant plant individuals using the TaqMan assays was performed on an ABI7900 instrument using standard protocols.
Genetic map of the Rfm1 locus
Recombination frequencies were calculated as the number of recombination events between two markers divided by the number of gametes screened, multiplied by 100, and subsequently converted to genetic distance in centimorgan (cM) by using the Kosambi mapping function (Kosambi 1944). An independent corroboration of the mapping results was obtained by genotyping 8 individuals of the F2:3 and F2BC1 progenies derived from the recombinant F2 plants.
BAC library construction, screening and sequencing
Custom BAC libraries of restorer line Re08 were created and screened at the Centre National de Ressources Génomiques Végétales (CNRGV, Castanet-Tolosan) as described in Santos et al. (2014). Three different restriction enzymes (HindIII, EcoRI and BamHI) were used for the fractionation of the high molecular weight DNA to mitigate the risk of the underrepresentation of the Rfm1 genomic region. Screening of the non-gridded Re08 BAC libraries using markers mapping to the Rfm1 interval was achieved using standard operating procedures at the CNRGV.
Screening of the Minimum Tiling Path (MTP) library derived from the Morex cultivar (Schulte et al. 2011) was achieved by PCR amplification of the BAC clones organized in pools using markers FR56 and FR62.
Positive BAC clones were identified on the barley physical map ‘fpc_10′ (9435 contigs, 507 688 BAC clones), as displayed at the PlantsDB Gbrowse website. Fingerprint contigs were visualized using the CrowsNest tool (http://mips.helmholtz-muenchen.de/plant/barley/fpc/index.jsp).
Selected BAC clones were sequenced using 454 mate-pair sequencing chemistry on a 454 GS-Junior Roche instrument. Sequencing data were cleaned with Pyrocleaner and E.coli sequences removed prior to their assembly using Newbler 2.6 (Roche). PacBio sequence analysis and assembly of selected BAC clones was provided as a service by GATC biotech (Mulhouse, France).
Sequence annotation
Sequence assemblies were submitted for annotation using the TriAnnot pipeline (Leroy et al. 2012) and gene annotation was performed using GreenPhyl (Rouard et al. 2011). Subcellular targeting predictions of protein sequences were obtained using TargetP (Emanuelsson et al. 2007) and Predotar (Small et al. 2004).
The sequence assembly of the rfm1 locus in Morex has been deposited in Genbank under accession number MF443757 and the BAC sequences obtained for the Rfm1 locus in Re08 under numbers MF443751 to MF443756.
Results
Fine mapping of fertility restoration gene Rfm1
Restoration of the CMS induced by the msm1 cytoplasm is controlled by a single locus named Rfm1, located on top of chromosome 6H (Matsui 2001; Murakami et al. 2005; Ui et al. 2015). For the purpose of the map-based cloning of Rfm1, a population of 2184 individuals was developed from a cross between restorer line Re08 (msm1; Rfm1/Rfm1) and Laverda, a regular barley cultivar carrying the normal cytoplasm and the sterile rfm1 allele (normal; rfm1/rfm1). The F2 population was genotyped using TaqMan assays derived from two polymorphic markers: ConsensusGBS0346-1 and 1553-753 that delimit the Rfm1 locus to a genetic interval of 6.5 cM (Fig. 1, our previous internal data). Recombinant individuals were selected, grown to maturity and phenotyped for anther extrusion and seed set (Fig. 2). Fertile and sterile recombinant plants were maintained by self-pollination and backcrossing respectively and their offspring phenotyped for male fertility restoration in order to confirm the results obtained for the F2 recombinant plants.

Genetic map of the Rfm1 locus. The genetic markers spanning the Rfm1 interval were genotyped and mapped in an F2 population of 2184 individuals segregating for male fertility restoration conferred by Rfm1. Genetic markers are indicated on the right, genetic distance in centimorgans on the left

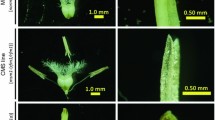
Ear phenotypes of male fertile (left) and male sterile (right) barley plants, both carrying the CMS-inducing msm1 cytoplasm. Fertile plants carry the Rfm1 restorer gene, sterile plants carry the recessive rfm1 allele. Developing ears on fertile plants display profuse anther extrusion and pollen production (black arrows), contrary to the sterile ears that are devoid of anthers and pollen
The polymorphisms underlying the ConsensusGBS0346-1 and 1553-753 markers originate from the Illumina BOPA12 collection of SNPs and were both identified in barley cDNA sequences (Close et al. 2009). Projection of cDNA sequences corresponding to both markers onto the genomes of rice and Brachypodium allowed defining the syntenic region of Rfm1 in both model species. Using the barley genome zipper (Mayer et al. 2011), orthologous barley ESTs and unigenes that were predicted to map within the Rfm1 interval were selected to develop additional markers. The recombinant F2 plants and their corresponding progeny families were genotyped using markers FR42, FR56 and FR62 mapping to the barley homologues of Os02g0107000, Bradi3g00910 and Bradi3g00940 respectively. This allowed refining the genetic interval of Rfm1 to 0.14 cM, between markers FR56 and FR62 (Fig. 1). The genetic order of the relevant markers and the distribution of the recombinant plants across the Rfm1 interval are presented in Fig. 3d.

Physical map and functional annotation of the Rfm1 genetic interval in Morex and restorer line Re08. a Position of the BAC clones spanning the Rfm1 genetic interval in Morex. b, c Functional annotation of the corresponding nucleotide sequence of the Rfm1 locus in Morex; protein-coding genes are shown as black arrows (b), repeated sequences and transposable elements as blue arrows, low complexity sequences as green arrows and black vertical lines (c). d Physical positions of the markers across the Rfm1 genetic interval. The number of recombination events between the respective markers and Rfm1 as observed in the F2 population is indicated below each marker. e Physical map of the Rfm1 locus in restorer line Re08. BAC clones obtained from the custom BAC library of Re08 are indicated as solid lines, gene models are indicated as arrows
Physical map of rfm1 locus in Morex
In order to link the genetic map to the physical map of the barley genome, the two genetic markers defining the Rfm1 interval (FR56, FR62) were used to screen the BAC library representing the minimal tiling path of the Morex cultivar (Schulte et al. 2011). It is noteworthy here that Morex represents a spring barley type that does not carry the restorer allele at the Rfm1 locus. Seven positive BACs were identified that fall into two separate contigs. Contig 6870 contains 31 BAC clones and is 242 Kb long; contig 44,212 contains 225 BACs and is 1641 Kb long. Cross amplifications of markers derived from the BAC ends allowed bridging the gap between the two contigs. Four BACs spanning the entire interval delimited by FR56 and FR62 were subsequently selected for sequence analysis using a combination of 454 mate-pair and PacBio sequencing strategies (Fig. 3).
As depicted in Fig. 3, the sequence obtained for the Rfm1 interval of the Morex allele revealed five potential protein-encoding genes showing significant homology to genes from reference genomes or to Triticeae ESTs. Transposable elements add up to 63% of the sequence, while the annotated genes represent only 7.6%. The sequence also contains five low complexity regions containing a large number of the same repeated element, and two large duplications of 12 kb (Fig. 3).
The putative functions of the five annotated genes found in the interval are summarized in Table 1. Two genes organized in a tandem repeat code for PPR proteins (Barkan and Small 2014) which makes them plausible candidate genes for Rfm1. The two PPR proteins are 754 and 879 amino acids in length, and are 84.1% identical. Both gene copies carry E and DYW sequence motifs at their carboxy terminus and therefore are classified as PLS-type PPR proteins, implying a potential role in RNA editing. The closest homologs of these two genes in rice, Brachypodium and Arabidopsis are Os02g0106300, Bradi3g00900 and At1g68930, respectively. The percentage of identity between the two barley PPR proteins with these three homologs ranged from 56% with At1g68930 to 86% with Bradi3g00900. In the recently released version of the Morex genome sequence, the two gene copies of Bradi3g00900 and Bradi3g00890 have been merged into a single gene model annotated as HORVU6Hr1G004120, probably due to a combination of assembly and annotation errors. Because of these discrepancies, the Rfm1 candidate genes are further referred to as Hv_Bradi3g00900A and B by virtue of their homology to the corresponding gene model in Brachypodium. Like most PPR genes, the gene model for Hv_Bradi3g00900 does not contain any introns. None of the Hv_Bradi3g00900 orthologs in model species have been characterized functionally, although there is some experimental evidence indicating that At1g68930 is targeted to mitochondria (Colcombet et al. 2013). Similarly, both Hv_Bradi3g00900 proteins are predicted to be directed to the mitochondria according to TargetP (Emanuelsson et al. 2007) or Predotar (Small et al. 2004). A full-length cDNA (AK358852) as well as several ESTs corresponding to Hv_Bradi3g00900 could be identified in the barley EST database. The fact that these cDNA fragments were obtained from vegetative tissues at different developmental stages and in different cultivars suggests that Hv_Bradi3g00900 is expressed and exerts a biological function in genetic backgrounds that do not carry the Rfm1 introgression from Hordeum vulgare ssp spontaneum.
Towards the cloning of Rfm1
Restoration loci often show a complex and variable structure containing multiple copies of highly homologous PPR genes resulting from local and recent duplication events (Desloire et al. 2003a; Xu et al. 2009; Hernandez Mora et al. 2010; Dahan and Mireau 2013; Gaborieau and Brown 2016). Since Morex does not restore the msm1 cytoplasm and therefore does not carry the restorer allele of Rfm1, the functional gene supporting the restoration activity is absent from the sequence obtained for Morex, despite the presence of multiple PPR genes at the rfm1 locus.
To obtain and characterize the complete sequence of the restorer allele of Rfm1, a non-gridded BAC library was created from the restorer line Re08. The screening of the library was performed using the same markers as for the Morex BAC library. The availability of the Morex genome sequence proved useful for the development of additional markers across the Rfm1 interval, targeting either genes, including Hv_Bradi3g00900 as well as sequences bridging transposon insertion sites, also known as insertion site based polymorphism or ISBPs (Paux et al. 2010).
In total, six BAC clones positive for one or more markers mapping to the Rfm1 interval were sequenced for comparative analysis to the Morex allele (Fig. 3e). Although the non-coding regions of the Rfm1 allele show substantial differences in sequence content and organization to the Morex reference, the gene content of the Rfm1 interval revealed identical. Like for Morex, the restorer allele harbours two tandem copies of Hv_Bradi3g00900 (that we further refer to as Hv_Bradi3g00900A and Hv_Bradi3g00900B) in addition to the homologue of Bradi3g00890.
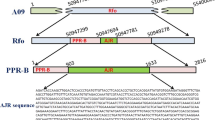
Contrary to the A-copy of Hv_Bradi3g00900, the coding regions of B-copies as present at both alleles are 100% identical and Hv-Bradi3g00900B therefore is probably not responsible for the male fertility restoration conferred by the Re08 allele. Due to a partial duplication of the first two PPR motifs, the protein sequence of the A-copy at the restorer allele in Re08 is slightly longer than the corresponding A-copy in Morex (880 versus 755 amino acid residues respectively). Moreover, the amino-terminus including the signal sequence of the Hv_Bradi3g00900A protein from Re08 is highly divergent from its counterpart in Morex. According to subcellular prediction programs (Small et al. 2004; Emanuelsson et al. 2007), the Hv_Bradi3g00900A protein from Re08 is targeted to the mitochondria with a much higher confidence than the Morex allele, which is in line with its function as a restorer gene. Apart from the polymorphisms at the amino-terminus, the Hv_Bradi3g00900A proteins from Re08 and Morex differ by several amino acid substitutions suggesting that they may display differences in their RNA binding properties as well. The amino acid substitution at position 645 of the Re08 allele (Fig. 4, aspartate replaced by a histidine) is the only position that distinguishes the restorer allele of Hv_Bradi3g00900A from both, the corresponding A-copy at the Morex allele and the B-copy genes. This substitution is of particular interest since it is located at position 3 of the PPR repeat motif that has been shown to play an essential role in the sequence specific RNA-binding of PPR proteins.

Alignment of the deduced amino acid sequences of the Hv_Bradi3g00900 gene copies from both the Morex and the Re08 restorer allele. The A-copy at the restorer allele reveals several unique features in comparison to its counterpart at the Morex allele, contrary to the B-copy genes that are 100% identical between both alleles. The different motifs identified in the protein sequence are indicated on the consensus sequence at the bottom of the alignment, except for the first PPR-S motive that is unique to Re08_Hv_Bradi3g00900A. PPR-P motifs are highlighted in yellow, PPR-S in green, PPR-L in light blue, E in purple, E+ in grey and DYW in dark blue. Consecutive copies of the same sequence motif are distinguished by alternating underlining. (Color figure online)
Discussion
In the present study, we utilized a positional cloning approach in conjunction to the syntenic relationship between barley and Brachypodium to delimit the Rfm1 locus both genetically and physically. Sequence analysis of the 0.3 Mb region enclosed by the nearest flanking markers permitted the identification of a Bradi3g00900 homologue as candidate gene for Rfm1 that is annotated as a PPR protein. Unlike most restorer genes identified so far, Rfm1 does not encode a P-type PPR protein, but a protein belonging to the PLS subfamily of PPR genes characterized by the DYW domain at the carboxy-terminus. The only other known candidate restorer gene encoding a PPR protein belonging to the PLS subclass is Rf1 from Sorghum, but this candidate awaits functional validation as well (Klein et al. 2005). Nevertheless, the combined observations in barley and sorghum indicate that restorer genes can be found outside the Rf-like clade of PPR proteins identified by Fujii (Fujii et al. 2011a, b; Dahan and Mireau 2013). The PLS subfamily of PPR proteins is specific to land plants and is known for its involvement in RNA editing in organelles (Takenaka et al. 2013), suggesting that RNA editing may play a role in male fertility restoration.
The possible link between RNA editing and male fertility restoration raises questions with respect to the nature of the presumed editing sites. Restorer genes of the P-type PPR proteins typically prevent the accumulation of the corresponding CMS-inducing factor by means of post-transcriptional RNA processing (e.g. RNA cleavage) or translation inhibition of the corresponding mRNA (Gaborieau and Brown 2016). In a similar way RNA editing could prevent the accumulation of the sterility factor by either creating a stop codon within the CMS-inducing transcript or an amino acid substitution in the putative sterility protein, but to date there is no precedent for such mechanism. The amino acids specified by codons subject to RNA editing are generally highly conserved across species. RNA editing is alleged to re-establish the default (or conserved) sequence of the predicted protein as compared with other plant species (Castandet and Araya 2011), which is not really in line with the editing of a unique transcript to prevent the accumulation of the foreign sterility factor. Extrapolating the basic principles or RNA editing to male fertility restoration would imply that the cytoplasmic male sterility in barley is not the result of the accumulation of a novel and non-conserved CMS-inducing protein, but the lack of editing of a transcript encoding an essential and conserved protein of the respiratory chain. This hypothesis would corroborate one of the explanations put forward to explain CMS from a physiological perspective (Touzet and Meyer 2014). Since pollen development is known as one of the most energy-demanding developmental processes, the suboptimal performance of mitochondria may result in insufficient energy levels to sustain the production and maturation of functional pollen. This interpretation is further supported by the study of Kurek et al. (1997) showing differential levels of RNA editing in transcripts encoding ATP synthase subunits between wheat plants differing in male fertility levels, the sterile background displaying the lowest efficiency of RNA editing. Moreover, the transgenic expression of a non-edited copy of the wheat atp9 gene in tobacco was shown to induce male sterility, further demonstrating the impact of an impropriately edited mitochondrial transcript on male fertility in plants (Hernould et al. 1993). Despite the fact that the recognition code of PPR proteins is currently being deciphered (Barkan et al. 2012), the target sequence of Rfm1 remains unknown. The models are still unsufficiently precise to allow predicting the target sequence and corresponding editing sites of Rfm1 without ambiguity. Alternatively, the comparative analysis of the editing profile of mitochondrial transcripts between isogenic male-sterile and restored plants could provide insights into the potential targets of Rfm1 and thus enhance our understanding of the mechanism underlying male sterility and fertility restoration in barley.
The chromosomes of the sixth homeologous group in the small grain cereals have been associated with male fertility restoration before. Several studies have mapped loci for restoration on chromosome six in wheat (Martin et al. 2008; Castillo et al. 2014), triticale (Stojałowski et al. 2013) and rye (Miedaner et al. 2000; Stojałowski et al. 2011). The rye restorer genes for the Pampa and the C-type cytoplasms, referred to as Rfp and Rfc respectively, map to the long arm of chromosome 4R, but this region is known to comprise a translocation of the distal end of the short arm of chromosome 6R (Devos et al. 1993). Fine-mapping of Rfp1 and Rfp3 pinpointed to a chromosomal region slightly shifted with respect to the Rfm1 locus (Hackauf et al. 2012, 2017). This observation would suggest that the CMS systems in barley and rye rely on tightly linked but different restorer genes, unless chromosomal rearrangements and gene duplications have resulted in the local duplication or redisposition of the same restorer gene. In contrast to Rfm1, the Rfp1 syntenic interval in Brachypodium (and rice) does not harbor any PPR genes, which makes Bradi3g00900 the major candidate in case of the later scenario. One indication that chromosomal rearrangements may have played a role is illustrated by the fact that several paralogues of Bradi3g00900 are located in its near vicinity. This observation suggests a highly dynamic chromosomal context subject to gene duplication and expansion that has interrupted the local colinearity between the barley and the rye genome. It should also be noted that the local duplication of Hv_Bradi3g00900 as found in Morex and in the restorer line Re08 is specific to barley and is not found in Brachypodium or rice. Such local gene duplications represent a general feature of cereal genomes that show a tendency to increase gene copy number, especially in more distal chromosomal regions as observed in wheat (Choulet et al. 2010), but is also a well-known feature of restorer loci (Desloire et al. 2003; Brown et al. 2003; Akagi et al. 2004; Geddy and Brown 2007).
The question whether the restoration loci described in barley and rye also correspond to those reported on the group 6 chromosomes in wheat and triticale requires more detailed comparative mapping first. The map-based cloning and characterization of the Rfm1 locus now opens avenues for the functional validation of Hv_Bradi3g00900A as restorer gene of the msm1 CMS system in barley and to decipher the molecular mechanism underlying the male fertility restoration. The intriguing suggestion that RNA editing may play a role here sheds a novel light on the biological role of RNA editing and on the relevance of the appropriate interplay between the nuclear genome and the cytoplasm in order to maximize plant performance.
References
Ahokas H (1979) Cytoplasmic male sterility in barley. III. Maintenance of sterility and restoration of fertility in the msml cytoplasm. Euphytica 28:409–419. https://doi.org/10.1007/BF00056599
Ahokas H (1982) Cytoplasmic male sterility in barley: evidence for the involvement of cytokinins in fertility restoration. Proc Natl Acad Sci 79:7605–7608
Ahokas H (1983) Cytoplasmic male sterility in barley. XV. PI296897 as a restorer of fertility in msm1 ad msm2 cytoplasms. Hereditas 99:157–159
Akagi H, Nakamura A, Yokozeki-Misono Y et al (2004) Positional cloning of the rice Rf-1 gene, a restorer of BT-type cytoplasmic male sterility that encodes a mitochondria-targeting PPR protein. Theor Appl Genet 108:1449–1457. https://doi.org/10.1007/s00122-004-1591-2
Ban T, Ke J, Chen R et al (2013) Structure of a PLS-class pentatricopeptide repeat protein provides insights into mechanism of RNA Recognition. J Biol Chem 288:31540–31548. https://doi.org/10.1074/jbc.M113.496828
Barkan A, Small I (2014) Pentatricopeptide repeat proteins in plants. Annu Rev Plant Biol 65:415–442. https://doi.org/10.1146/annurev-arplant-050213-040159
Barkan A, Rojas M, Fujii S et al (2012) A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet 8:e1002910. https://doi.org/10.1371/journal.pgen.1002910
Brown GG, Formanova N, Jin H et al (2003) The radish Rfo restorer gene of Ogura cytoplasmic male sterility encodes a protein with multiple pentatricopeptide repeats. Plant J 35:262–272. https://doi.org/10.1046/j.1365-313X.2003.01799.x
Castandet B, Araya A (2011) RNA editing in plant organelles. Why make it easy ? Biochem 76:924–931. https://doi.org/10.1134/S0006297911080086
Castillo A, Atienza SG, Martin AC (2014) Fertility of CMS wheat is restored by two Rf loci located on a recombined acrocentric chromosome. J Exp Bot 65:6667–6677. https://doi.org/10.1093/jxb/eru388
Choulet F, Wicker T, Rustenholz C et al (2010) Megabase level sequencing reveals contrasted organization and evolution patterns of the wheat gene and transposable element spaces. Plant Cell 22:1686–1701. https://doi.org/10.1105/tpc.110.074187
Close TJ, Bhat PR, Lonardi S et al (2009) Development and implementation of high-throughput SNP genotyping in barley. BMC Genomics 10:582. https://doi.org/10.1186/1471-2164-10-582
Colcombet J, Lopez-Obando M, Heurtevin L et al (2013) Systematic study of subcellular localization of arabidopsis PPR proteins confirms a massive targeting to organelles. RNA Biol 10:1557–1575. https://doi.org/10.4161/rna.26128
Dahan J, Mireau H (2013) The Rf and Rf-like PPR in higher plants, a fast-evolving subclass of PPR genes. RNA Biol 10:1469–1476. https://doi.org/10.4161/rna.25568
Desloire S, Gherbi H, Laloui W et al (2003) Identification of the fertility restoration locus, Rfo, in radish, as a member of the pentatricopeptide-repeat protein family. EMBO Rep 4:588–594. https://doi.org/10.1038/sj.embor.embor848
Devos KM, Atkinson MD, Chinoy CN et al (1993) Chromosomal rearrangements in the rye genome relative to that of wheat. Theor Appl Genet 85:673–680. https://doi.org/10.1007/BF00225004
Emanuelsson O, Brunak S, von Heijne G, Nielsen H (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2:953–971. https://doi.org/10.1038/nprot.2007.131
Fujii S, Bond CS, Small ID (2011) Selection patterns on restorer-like genes reveal a conflict between nuclear and mitochondrial genomes throughout angiosperm evolution. Proc Natl Acad Sci U S A 108:1723–1728. https://doi.org/10.1073/pnas.1007667108
Gaborieau L, Brown GG (2016) Comparative genomic analysis of the compound Brassica napus Rf locus. BMC Genomics 17:834. https://doi.org/10.1186/s12864-016-3117-0
Geddy R, Brown GG (2007) Genes encoding pentatricopeptide repeat (PPR) proteins are not conserved in location in plant genomes and may be subject to diversifying selection. BMC Genomics 8:130. https://doi.org/10.1186/1471-2164-8-130
Hackauf B, Korzun V, Wortmann H et al (2012) Development of conserved ortholog set markers linked to the restorer gene Rfp1 in rye. Mol Breed 30:1507–1518. https://doi.org/10.1007/s11032-012-9736-5
Hackauf B, Bauer E, Korzun V, Miedaner T (2017) Fine mapping of the restorer gene Rfp3 from an Iranian primitive rye (Secale cereale L.). Theor Appl Genet 130:1179–1189. https://doi.org/10.1007/s00122-017-2879-3
Hammani K, Giegé P (2014) RNA metabolism in plant mitochondria. Trends Plant Sci 19:380–389. https://doi.org/10.1016/j.tplants.2013.12.008
Hernandez Mora JR, Rivals E, Mireau H, Budar F (2010) Sequence analysis of two alleles reveals that intra-and intergenic recombination played a role in the evolution of the radish fertility restorer (Rfo). BMC Plant Biol 10:35. https://doi.org/10.1186/1471-2229-10-35
Hernould M, Suharsono S, Litvak S et al (1993) Male-sterility induction in transgenic tobacco plants with an unedited atp9 mitochondrial gene from wheat. Proc Natl Acad Sci U S A 90:2370–2374. https://doi.org/10.1073/pnas.90.6.2370
Horn R, Gupta KJ, Colombo N (2014) Mitochondrion role in molecular basis of cytoplasmic male sterility. Mitochondrion 19:198–205. https://doi.org/10.1016/j.mito.2014.04.004
Howard MJJ, Lim WHH, Fierke CA, Koutmos M (2012) Mitochondrial ribonuclease P structure provides insight into the evolution of catalytic strategies for precursor-tRNA 5′ processing. Proc Natl Acad Sci 109:16149–16154. https://doi.org/10.1073/pnas.1209062109
Hu J, Huang W, Huang Q et al (2014) Mitochondria and cytoplasmic male sterility in plants. Mitochondrion. https://doi.org/10.1016/j.mito.2014.02.008
Islam MS, Studer B, Møller IM, Asp T (2014) Genetics and biology of cytoplasmic male sterility and its applications in forage and turf grass breeding. Plant Breed. https://doi.org/10.1111/pbr.12155
Ke J, Chen R-Z, Ban T et al (2013) Structural basis for RNA recognition by a dimeric PPR-protein complex. Nat Struct Mol Biol 20:1377–1382. https://doi.org/10.1038/nsmb.2710
Klein RR, Klein PE, Mullet JE et al (2005) Fertility restorer locus Rf1 of sorghum (Sorghum bicolor L.) encodes a pentatricopeptide repeat protein not present in the colinear region of rice chromosome 12. Theor Appl Genet 111:994–1012. https://doi.org/10.1007/s00122-005-2011-y
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175. https://doi.org/10.1111/j.1469-1809.1943.tb02321.x
Kurek I, Ezra D, Begu D et al (1997) Studies on the effects of nuclear background and tissue specificity on RNA editing of the mitochondrial ATP synthase subunits α, 6 and 9 in fertile and cytoplasmic male-sterile (CMS) wheat. TAG Theor Appl Genet 95:1305–1311. https://doi.org/10.1007/s001220050697
Leroy P, Guilhot N, Sakai H et al (2012) TriAnnot: a versatile and high performance pipeline for the automated annotation of plant genomes. Front Plant Sci 3:5. https://doi.org/10.3389/fpls.2012.00005
Liu F, Cui X, Horner HT et al (2001) Mitochondrial aldehyde dehydrogenase activity is required for male fertility in maize. Plant Cell 13:1063–1078
Martin AC, Atienza SG, Ramırez MC et al (2008) Male fertility restoration of wheat in Hordeum chilense cytoplasm is associated with 6HchS chromosome addition. Aust J Agric Res 59:206–213
Mascher M, Gundlach H, Himmelbach A et al (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature 544:427–433. https://doi.org/10.1038/nature22043
Matsuhira H, Kagami H, Kurata M et al (2012) Unusual and typical features of a novel restorer-of-fertility gene of sugar beet (Beta vulgaris L.). Genetics 192:1347–1358. https://doi.org/10.1534/genetics.112.145409
Matsui K (2001) Molecular mapping of a fertility restoration locus (Rfm1) for cytoplasmic male sterility in barley (Hordeum vulgare L.). TAG Theor Appl Genet 102:477–482. https://doi.org/10.1007/s001220051670
Mayer KFX, Martis M, Hedley PE et al (2011) Unlocking the barley genome by chromosomal and comparative genomics. Plant Cell 23:1249–1263. https://doi.org/10.1105/tpc.110.082537
Mayer KFX, Waugh R, Langridge P et al (2012) A physical, genetic and functional sequence assembly of the barley genome. Nature 491:711–716. https://doi.org/10.1038/nature11543
Melonek J, Stone JD, Small I (2016) Evolutionary plasticity of restorer-of-fertility-like proteins in rice. Sci Rep 6:35152. https://doi.org/10.1038/srep35152
Miedaner T, Glass C, Dreyer F et al (2000) Mapping of genes for male-fertility restoration in“Pampa” CMS winter rye (Secale cereale L.). TAG Theor Appl Genet 101:1226–1233. https://doi.org/10.1007/s001220051601
Murakami S, Matsui K, Komatsuda T, Furuta Y (2005) AFLP-based STS markers closely linked to a fertility restoration locus (Rfm1) for cytoplasmic male sterility in barley. Plant Breed 136:133–137
Paux E, Faure S, Choulet F et al (2010) Insertion site-based polymorphism markers open new perspectives for genome saturation and marker-assisted selection in wheat. Plant Biotechnol J 8:196–210. https://doi.org/10.1111/j.1467-7652.2009.00477.x
Rouard M, Guignon V, Aluome C et al (2011) Green PhylDB v2.0: comparative and functional genomics in plants. Nucleic Acids Res 39:1095–1102. https://doi.org/10.1093/nar/gkq811
Santos A, Penha H, Bellec A et al (2014) Begin at the beginning: a BAC-end view of the passion fruit (Passiflora) genome. BMC Genomics 15:816. https://doi.org/10.1186/1471-2164-15-816
Schmitz-Linneweber C, Small I (2008) Pentatricopeptide repeat proteins: a socket set for organelle gene expression. Trends Plant Sci 13:663–670. https://doi.org/10.1016/j.tplants.2008.10.001
Schulte D, Ariyadasa R, Shi B et al (2011) BAC library resources for map-based cloning and physical map construction in barley (Hordeum vulgare L.). BMC Genomics 12:247. https://doi.org/10.1186/1471-2164-12-247
Shen C, Zhang D, Guan Z et al (2016) Structural basis for specific single-stranded RNA recognition by designer pentatricopeptide repeat proteins. Nat Commun 7:1–8. https://doi.org/10.1038/ncomms11285
Small I, Peeters N, Legeai F, Lurin C (2004) Predotar: a tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics 4:1581–1590. https://doi.org/10.1002/pmic.200300776
Stojałowski SA, Milczarski P, Hanek M et al (2011) DArT markers tightly linked with the Rfc1 gene controlling restoration of male fertility in the CMS-C system in cultivated rye (Secale cereale L.). J Appl Genet 52:313–318. https://doi.org/10.1007/s13353-011-0049-x
Stojałowski S, Bobrowska A, Hanek M, Myśków B (2013) The importance of chromosomes from the sixth homeologic group in the restoration of male fertility in winter triticale with Triticum timopheevii cytoplasm. J Appl Genet 54:179–184. https://doi.org/10.1007/s13353-013-0144-2
Takenaka M, Zehrmann A, Verbitskiy D et al (2013) RNA editing in plants and its evolution. Annu Rev Genet 47:335–352. https://doi.org/10.1146/annurev-genet-111212-133519
Touzet P, Meyer EH (2014) Cytoplasmic male sterility and mitochondrial metabolism in plants. Mitochondrion. https://doi.org/10.1016/j.mito.2014.04.009
Ui H, Sameri M, Pourkheirandish M et al (2015) High-resolution genetic mapping and physical map construction for the fertility restorer Rfm1 locus in barley. Theor Appl Genet. https://doi.org/10.1007/s00122-014-2428-2
Xu X, Liu Z, Zhang D (2009) Isolation and analysis of rice Rf1-orthologus PPR genes Co-segregating with Rf3 in Maize. Plant Mol Biol Rep. https://doi.org/10.1007/s11105-009-0105-4
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Plant Breeding: the Art of Bringing Science to Life. Highlights of the 20th EUCARPIA General Congress, Zurich, Switzerland, 29 August–1 September 2016
Edited by Roland Kölliker, Richard G. F. Visser, Achim Walter & Beat Boller
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rizzolatti, C., Bury, P., Tatara, E. et al. Map-based cloning of the fertility restoration locus Rfm1 in cultivated barley (Hordeum vulgare). Euphytica 213, 276 (2017). https://doi.org/10.1007/s10681-017-2056-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10681-017-2056-4