
Abstract
Understanding the actual distribution of different Legionella species in water networks would help prevent outbreaks. Culture investigations followed by serological agglutination tests, with poly/monovalent antisera, still represent the gold standard for isolation and identification of Legionella strains. However, also MALDI-TOF and mip-gene sequencing are currently used. This study was conducted to genetically correlate strains of Legionella non pneumophila (L-np) isolated during environmental surveillance comparing different molecular techniques. Overall, 346 water samples were collected from the water system of four pavilions located in a hospital of the Apulia Region of Italy. Strains isolated from the samples were then identified by serological tests, MALDI-TOF, and mip-gene sequencing. Overall, 24.9% of water samples were positive for Legionella, among which the majority were Legionella pneumophila (Lpn) 1 (52.3%), followed by Lpn2-15 (20.9%), L-np (17.4%), Lpn1 + Lpn2-15 (7.1%), and L-np + Lpn1 (2.3%). Initially, L-np strains were identified as L. bozemanii by monovalent antiserum, while MALDI-TOF and mip-gene sequencing assigned them to L. anisa. More cold water than hot water samples were contaminated by L. anisa (p < 0.001). PFGE, RAPD, Rep-PCR, and SAU-PCR were performed to correlate L. anisa strains. Eleven out of 14 strains identified in all four pavilions showed 100% of similarity upon PFGE analysis. RAPD, Rep-PCR, and SAU-PCR showed greater discriminative power than PFGE.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Legionella are Gram-negative bacteria that can be found in natural (Atkinson et al., 2022; Zhan et al., 2022) and artificial aquatic environments (Prussin et al., 2017; Volker et al., 2016). The genus includes 64 species and more than 70 different serogroups (LPSN, 2022). Legionella are responsible for legionellosis, an infectious disease acquired by inhalation or aspiration of aerosols from contaminated water (De Giglio et al., 2019). The presence of amoebas, biofilms, and stagnant water and a temperature between 37 and 42 °C promote the growth of this microorganism (Van der Koiji et al., 2016). However, it is able to adapt to a wide range of water parameters, including temperatures of 5.7–63 °C, pH values of 5.5–8.1, and oxygen concentrations of 0.3–8.2 mg/L (Fliermans, 1996; Schwake et al., 2021; De Giglio et al., 2021).
Recently, the new European Drinking Water Directive (Directive EU 2020/2184) has inserted Legionella as mandatory among microbiological parameters to detect in water intended for human consumption for the risk assessment of domestic distribution systems in health and community facilities.
In Europe, notification rates of Legionnaires’ disease vary from fewer than 0.5 cases to 5.7 cases per 100,000 of the population (ECDC, 2022). Although over 80% of human cases are caused by L. pneumophila (Lpn) serogroup 1 (Lpn1), the real number of Lpn non-serogroup 1 and Legionella non-pneumophila (L-np) cases (e.g., L. micdadei, L. bozemanii, L. longbeachae, L. dumofii, L. feeleii, and L. anisa) is poorly documented (Beauté et al., 2013; Craun et al., 2010; ESGLI, 2017; Girolamini et al., 2021; Head et al., 2019; Vaccaro et al., 2016; Yang et al., 2010). In particular, L. anisa is the most commonly detected L-np in aquatic environments, and rarely causes infections in humans (Doebelling et al., 1989; Akermi et al., 2006). L. bozemanii and L. micdadei which are not frequently found in the environment are the most common causes of culture-verified L-np infections (Doebelling et al., 1989; Svarrer & Uldum., 2012; Neiderud et al., 2013; Miller et al., 2007).
The risk of Legionella transmission with severe outcomes has been found to be affected by the complexity of hospital water systems and the vulnerability of hospitalized patients (De Giglio et al., 2021). Two thousand seven hundred and twenty-six cases of legionellosis were reported in Italy in 2021 with an incidence rate of 46 cases per 1 million of which 3.7% was of nosocomial origin (Istituto Superiore di Sanità, 2022). When it was possible to use culture-based methods, Lpn was identified in 100% of cases by Istituto Superiore di Sanità (2022). However, data describing the distribution of L-np in water systems of healthcare facilities are scarce (Arrigo et al., 2022; Girolamini et al., 2019, 2020, 2021; Mazzotta et al., 2021; Napoli et al., 2010). To our knowledge, there have been no reports of Italian legionellosis cases of nosocomial origin associated with L-np. However, it is likely that cases due to L-np are underestimated because the diagnostic methods commonly used in routine microbiological investigations are not sufficient for identification of species other than Lpn. Although culture-based method is considered the “gold standard” for microbiological surveillance of Legionella, it has some limitations in the identification of L-np (Scaturro et al., 2020). In recent years, many laboratories use a rapid identification method through the analysis of ribosomal protein pattern based on array-assisted laser desorption ionization mass spectrometry (MALDI-TOF MS) (Dilger et al., 2016; Pascale et al., 2020). Additionally, various genotyping techniques such as total genomic DNA analysis and specific (mip gene) and internal gene sequencing are available for identification of Legionella (Svarrer and Uldum 2012).
However, the use of different molecular techniques (e.g., pulsed-field gel electrophoresis (PFGE), RAPD, Rep-PCR, and SAU-PCR) is required to assess whether clinical patient isolates correspond to environmental isolates (Linee Guida per la prevenzione e il Controllo della Legionellosi, 2015; Haroon et al., 2010; Corich et al., 2005; Yamamoto et al., 2003).
In this study, L-np strains isolated during environmental surveillance conducted for 13 months in four pavilions of a large hospital in Apulia region (southern Italy) were identified using traditional methods (i.e., culture and latex agglutination) and spectrometry (MALDI-TOF MS) in conjunction with mip-gene sequencing. The aim of the study was to genetically correlate strains isolated from the water networks of the different hospital pavilions using traditional molecular methods as PFGE compared with alternative techniques (RAPD, Rep-PCR, and SAU-PCR) not usually applied to L-np strains.
Materials and methods
Our Environmental and Food Hygiene Laboratory (ACCREDIA n.1683), which is recognized as a Regional Reference Center in Apulia (Southern Italy), has conducted clinical and environmental surveillance of legionellosis in nosocomial and community facilities since 2001.
In January 2020, systematic monitoring of the water network of a large Apulian public hospital was started as part of the implementation program of a Water Safety Plan. The surveillance plan covered 33 separate buildings and provided for the quarterly microbiological control of 50% of the water supply points uniformly distributed in each pavilion.
Sampling and culture-based investigation
From 1 March 2021 to 30 March 2022, 346 water samples (1 L) were collected from taps of water system of four pavilions: pI (81), pII (109), pIII (128), and pIV (28). In detail, 277 samples were collected from the hot water systems and 69 from the cold water networks when it was not possible to obtain hot water samples (e.g., due to exhaustion of hot water in the boilers). The temperature was recorded for each water sample. According to Italian Guideline (Linee guida per la Prevenzione e il Controllo della Legionellosi, 2015), the collected samples were stored in sterile dark glass bottles containing sodium thiosulfate pentahydrate (0.01%, w/v) to neutralize chloride present in water. Samples were then transported, by an isothermal container, to the laboratory at room temperature (19.2 °C; range 18.5–24.2 °C).
According to ISO 11731:2017 (ISO 11731:2017), each sample was first filtered through isopore polycarbonate membranes (47 mm in diameter with a pore size of 0.22 µm) (Millipore Corporation, Bedford, MA, USA) then resuspended in 10 mL of the non-filtered water sample. After vortexing, an aliquot of each sample (200 µL) was seeded onto selective culture medium GVPC plates added with glycine, vancomycin, polymyxin, and cycloheximide B (Liofilchem Srl, Teramo, Italy) followed by incubation at 36 ± 2 °C for 7–10 days in a humid environment (to prevent desiccation of the plates) (ISO 11731:2017). Presumptive colonies (at least three different colonies for each plate) of Legionella were then inoculated in buffered charcoal yeast extract (BCYE) agar plates (BioMérieux, Marcy l'Etoile, France) with and without l-cysteine. The identification of the colonies of Legionella spp. grown only on BCYE agar plates added with cysteine was performed using a latex agglutination test with polyvalent (Biolife Italiana Srl, Milan, Italy) and monovalent antisera (Biogenetics Srl, Tokyo, Japan) (Linee guida per la Prevenzione e il Controllo della Legionellosi, 2015). In particular, regarding Legionella species group, the polyvalent antisera provided by the manufacturer (Biolife Italiana Srl, Milan, Italy) identify only few species implicated in clinical cases: L. anisa, L. micdadei, L. bozemanii 1 and 2, L. gormanii, L. longbeachae 1 and 2, L. dumoffii, and L. jordanis. Moreover, the monovalent sera commercially available allow to identify all Legionella species mentioned above except L. anisa, L. longbeachae 1 and 2, and L. jordanis (Biogenetics Srl, Tokyo, Japan).
All the strains identified as Legionella spp. were viewed under a long-wave ultraviolet light (360 ± 20 nm) emitted by a wood lamp to evaluate the emission of autofluorescence due to the presence of an intracellular pigment. The results of Legionella contamination in water samples were expressed as colony-forming units per liter (CFU/L), and the detection limit was 50 CFU/L.
Identification by MALDI-TOF MS analysis and mip-gene sequencing
Legionella species strains were identified by MALDI-TOF using the Vitek MS system (BioMérieux, Italy) at level serogroup according to instruction of manufacturer. Also the mip gene sequence analysis was performed as reported by Federici et al. (2017). Briefly, thermal shock was used to extract genomic DNA from colonies of Legionella grown on BCYE medium, followed by PCR amplification of the mip gene. PCR reactions were set up on a total volume of 20 µL containing FIREPol 5 × Master Mix (Solis BioDyne, Tartu, Estonia), 0.4 µM of Legmip-f and Legmip-r primers (Ratcliff et al., 1998), and 4 µl of template DNA. PCR amplification protocol consisted of 34 cycles at 95 °C for 30 s of denaturation, 58 °C for 1 min of annealing, and 72 °C for 1 min of extension. At the end, amplicons were purified with the EuroSAP PCR Enzymatic Clean-up kit (Euroclone SpA, Pero, MI, IT) and sequenced by the Sanger method (Eurofins Genomics, Ebersberg; Germany). Sequencing results were analyzed and processed with Geneious Prime 2022.1.1 (Biomatters Inc., San Diego, CA, USA) running the BLAST (Basic Local Alignment Search Tool) tool to find similar mip genes belonging to Legionella reference strains in the GenBank database. A phylogenetic tree was constructed by the unweighted pair group method with arithmetic mean (UPGMA) method using Jukes-Cantor as a genetic distance model after aligning the obtained mip gene sequences with those of the culture collections strains downloaded from GenBank. Sequence alignment was performed using the multiple sequence comparison by log-expectation (MUSCLE) program with default settings (Edgar, 2004).
Genetic correlation of Legionella non-pneumophila strains
Only 14 (C, F, G1, G2, G3, G4, H, I, L, M, P, R, S, and T) strains isolated from the water networks of the four different pavilions (pI, pII, pIII, and pIV) were possible to analyze establishing a possible genetic correlation. Strains L, M, P, R, S, and T were isolated from pavilion pI; C from pavilion pII; H and I from pavilion pIII; and F, G1, G2, G3, and G4 from pavilion pIV.
Pulsed-field gel electrophoresis
PFGE of the 14 strains was performed using a modification of the CDC PulseNet standardized PFGE protocol (Ribot et al., 2006; Sabrià et al., 2001).
Briefly, isolates were grown on BCYE agar plates at 37 °C for 48 h, after which they were suspended in cell suspension buffer (10 mM Tris, 1 M NaCl, pH 8.0). The cell suspensions were then adjusted to an optical density of 0.5–0.6 at a wavelength of 610 nm and mixed with equal volumes of 1.0% pulsed-field certified agarose (BioRad, Milan, Italy) and 10 µL of proteinase K (20 mg/mL). Next, the agarose plugs were transferred into cell lysis buffer (0.5 M EDTA, pH 8.0, 1% sarcosine, and 200 µg/ml of proteinase K) and incubated at 50 °C overnight. DNA embedded in the plugs was digested with 40 U of AscI or SfiI (New England Biolabs, Schwahlbach, Germany) at 37 °C for 4 h. Plugs of Salmonella braenderup strain H9812 were digested with 40 U XbaI (TaKaRa Bio, Dalian, China) and used in each gel as universal size standards. Fragments of DNA were separated in a CHEF DR III System (BioRad Laboratories, Richmond, CA, USA) with a constant voltage of 6 V cm−1, an included angle of 120°, and increasing pulse times (5.6–50 s) at 14 °C for 21 h. All PFGE profiles were digitalized and the phylogenetic relationship was assessed through the fingerprinting software GelJ (Heras et al., 2015). The phylogenetic relationship was represented through a phylogenetic tree obtained using the Dice coefficient with clustering by the unweighted pair-group method with arithmetic mean (UPGMA). A 2% tolerance in band position differences was applied (Martínez-Puchol et al., 2021).
RAPD, Rep-PCR, and SAU-PCR analyses
DNA extraction for PCR-based genetic fingerprint of the strains was performed using the GenElute™ Bacterial Genomic DNA Kit (Sigma-Aldrich, Milan, Italy) following the manufacturer’s instruction. The initial step was modified as follows: isolates were collected from Legionella agar plates (Sigma-Aldrich, Milan, Italy) using a 10-µL sterile loop, then resuspended thoroughly in 180 μL of lysis solution and the protocol was regularly followed. DNA was then quantified and standardized at 50 ng/µL (Nanodrop One system, Thermo Scientific, Marietta, OH, USA).
PCRs were performed using the amplification condition using a C1000 Touch Thermal Cycler (BioRad, Milan, Italy). The following primers were used for the reactions: M13 (5′-GAG GGT GGC GGT TCT-30′) (Huey & Hall, 1989), (GTG) 5 (5′-GTGGTGGTGGTGGTG-3′) (Gevers et al., 2001), and SAG1 (5′-CCGCCGCGATCAG-3′) for RAPD, Rep-PCR, and SAU-PCR, respectively. Conditions for PCR reaction, gel electrophoresis of the amplicons, and cluster analysis were performed according to Iacumin et al. (2006).
The 14 strains of L-np species were subjected to fingerprint analysis at least three times to confirm the reproducibility of the obtained profiles.
Statistical analysis
The data were presented as numbers or percentages for the categorical variables. To determine if the distribution of load (cfu /L) of Legionella spp. was normal, a Shapiro–Wilk normality test was performed. Chi-square or Fisher’s exact test was performed to compare two or more mutually exclusive proportions or percentages in groups. The Kruskal–Wallis rank sum test and the Wilcoxon rank sum test were used to determine if water temperature influenced the presence of Legionella spp. and L-np species. To accomplish this, the water samples were grouped into negative and positive for L-np and L. pneumophila in the hot and cold water networks. R version 3.6.3 (The R Project for Statistical Computing, Vienna, Austria) was used for analysis. A p-value < 0.05 was considered to indicate statistical significance.
Results
Identification by agglutination test, MALDI-TOF MS technique, and mip-gene sequencing
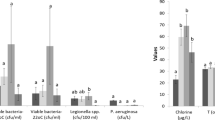
Overall, 86/346 (24.9%) water samples were positive for Legionella spp. Analysis of positive samples using the polyvalent antiserum revealed that 45/86 (52.3%) samples were positive solely for Lpn1 (median value = 1,200 cfu/L; range 50–83,000 cfu/L), 18/86 (20.9%) for Lpn 2–15 (median value = 1,150 cfu/L; range 50–22,500 cfu/L), 6/86 (7.1%) for Lpn1 + Lpn 2–15 (median value = 980 cfu/L; range 450–4,400 cfu/L), 15/86 (17.4%) solely for L-np (median value = 200 cfu/L; range 50–13,000 cfu/L), and 2/86 (2.3%) for L-np + Lpn1 (median value = 905 cfu/L; range 410–1,400 cfu/L).
All colonies of L-np showed blue-white autofluorescence under UV light at 365 nm and were identified by monovalent antiserum as Legionella bozemanii. Subsequent MALDI-TOF analysis and mip-gene sequencing indicated that they were all L. anisa (Fig. 1). To assess the relationship between L. anisa isolates and the closest Legionella spp., a UPGMA-phylogenetic tree was built (Fig. 1) (Pascale et al., 2021). All isolates grouped into a single clade with the L. anisa ATCC strain, indicating 100% homology both among isolates and with the reference sequence. However, the remaining reference sequences were located in other clades, confirming the phylogenetic distance between L. anisa isolates and L. bozemanii (95.19% sequence identity).

UPGMA phylogenetic tree showing similarities between the mip gene sequences of Legionella strains isolated in this study and those of the culture collections strains reported by the GenBank database
Table 1 summarizes the distribution of isolates in the four pavilions. In particular, it shows two water samples revealed the presence of L. anisa mixed with Lpn1 in pII (Lpn1 200 cfu/L + L. anisa 210 cfu/L) and pIV (Lpn1 200 cfu/L + L. anisa 1,200 cfu/L).
Temperature analysis
The cold water network was found to have more Legionella contamination than the hot water network (39.1% vs 21.3%; χ2 = 9.40, p-value = 0.002). The same result was observed for L. anisa (44.5% cold water vs 5.1% hot water; Fisher’s exact test, p-value < 0.001). Conversely, more Lpn were present in the hot water system than the cold water system (94.9% vs 48.1%, χ2 = 22.68, p-value < 0.0001). No significant differences in the distribution of Lpn + L. anisa were observed between the hot and cold water systems (Fisher’s exact test, p-value = 0.09) (Table 2).
Table 2 Distribution of Legionella in hot and cold water samples collected from water networks in the four investigated pavilions (March 2021–March 2022).
As shown in Table 3, Legionella distribution differed significantly among temperature ranges. Specifically, L. anisa was more often isolated from hot water samples with lower median temperatures than from other samples (Kruskal-Wallis χ2=12.545, p-value=0.001), i.e., positive for Lpn (pairwise comparisons using Wilcoxon rank sum test, p-value=0.05) and negative samples (pairwise comparisons using Wilcoxon rank sum test, p-value=0.04).
Correlation among Legionella species strains
PFGE typing
Figure 2 shows the PFGE patterns after restriction with AscI. PFGE patterns differing by 1 or more bands were classified into single clusters. Eleven out of fourteen analyzed strains showed an indistinguishable PFGE profile and were clustered within the same group (termed A). Single strains assigned to groups B and C had > 90% similarity to group A and differed from the latter for 2 and 3 bands, respectively. Group D (also composed of a single strain) had 86% similarity to group A from which it differed for 3 bands. Analysis of PFGE profiles generated with SfiI showed data comparable to that observed with AscI. In particular, strains clustered in group A for AscI showed an indistinguishable PFGE profile with SfiI too. Likewise, single strains that composed groups B, C, and D with AscI differed among them and were assigned to single groups. Clonal relatedness was determined by criteria for interpreting PFGE profiles published by Tenover and colleagues (Tenover et al., 1995). Strains of group A were found to be clonal for both restriction patterns), while strains of groups B and D (according to Tenover’s criteria and percentage of similarity) might be closely related to group prevailing (Tenover et al., 1995).

Dendrogram and PFGE patterns (generated by AscI) of 14 Legionella anisa strains. White arrows highlight different bands in profiles of groups B, C, and D compared with group A
RAPD, Rep-PCR, and SAU-PCR typing
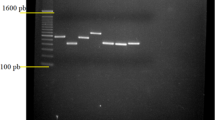
Three other genetic fingerprint techniques based on different genetic targets and with different degrees of stringency were applied to confirm the PFGE results and evaluate strain diversity. RAPD, Rep-PCR, and SAU-PCR effectively produced fingerprints for each individual isolate (Fig. 3). Moreover, these techniques showed greater discriminative power than PFGE with restriction enzyme AscI, allowing confirmation that the isolates were individual strains and not clones. However, the use of all three techniques was critical in establishing that the strains were different, but genetically related.

Example of fingerprints obtained using RAPD (panel A, lines 1–6 and 8–16, L. anisa isolates; line 7, negative control; line 17, molecular marker), Rep-PCR (panel B, lines 1–14, L. anisa isolates; line 15, negative control; line 16, molecular marker), and SAU-PCR (panel C, line 1, negative control; lines 2–10, L. anisa isolates; line 11, molecular marker)
Calculation of similarities in the profiles of bands based on Pearson product-moment correlation coefficients allowed production of dendrograms using UPGMA clustering algorithms (Fig. 4). The RAPD technique allowed discrimination of the strains according to the place of origin. In fact, cutting the cluster at a similarity of 90% resulted in the majority of the strains being grouped into two main clusters, B and C. Cluster B contained all of the strains isolated from pavilion pIV while cluster C included five strains isolated from pavilion pI and two from pavilion pIII. It should be noted that although strain H belonged to group C, it differed greatly from the others. This finding was confirmed by the SAU dendrogram. Using SAU-PCR technique, a higher degree of discrimination among strains emerged. Using a similarity cutoff of 70% resulted in the formation of two large clusters, A and B. Cluster A contained all of the strains isolated from pavilion pIV and a single outlier from pavilion pI (strain M). Although present within cluster A, strain M differed from the others strains within the cluster (82% similarity). This finding agreed with the PFGE results and clearly demonstrated that this strain was one of three that differed from the other strains. Cluster B contained all remaining strains. However, when a cutoff of 88% similarity was used, cluster B broke up into two clusters; namely, cluster C, which contained all strains from pavilion pI, and cluster D, which contained all of the strains isolated from pavilion pIII in one group and a strain isolated from pavilion pII. The Rep-PCR technique confirmed this genetic variability between strains, albeit with different clusters. Specifically, this method denoted a differentiation based on the site of isolation, although there were some strains that were more similar to other isolates at distant points.

Dendrograms obtained from analysis of the strains’ fingerprints obtained by RAPD, SAU-PCR, and Rep-PCR. The analysis was based on Pearson product-moment correlation coefficients determined using UPGMA clustering algorithms. Clusters (indicated by letters) were reported by the same color of the coefficient of similarity used to define them. Clusters (indicated by letters) were reported by the same color of the coefficient of similarity used to define them. Strains L, M, P, R, S, and T were isolated from pavilion pI
 ; C from pavilion pII
; C from pavilion pII
 ; H and I from pavilion pIII
; H and I from pavilion pIII
 ; and F, G1, G2, G3, G4 from pavilion pIV
; and F, G1, G2, G3, G4 from pavilion pIV

Discussion
The best approach to prevent waterborne diseases is systematic environmental monitoring and accurate identification of strains circulating in water distribution systems coupled with implementation of a risk assessment plan. Over the years, this approach has improved in hospitals, especially for the control and prevention of legionellosis (Mazzotta et al., 2021).
Culture-based investigations still represent the most used method for the isolation of Legionella strains, followed by serological agglutination tests with poly/monovalent antisera (Linee guida per la Prevenzione e il Controllo della Legionellosi, 2015; ISO 11731:2017). However, while serological tests can identify Lpn and its serogroups, they cannot identify all of the L-np species. Particularly problematic characteristics of these tests are that the monovalent antiserum for L. anisa is currently not commercially available and anti-L. bozemanii antiserum shows cross-reaction between L. bozemanii and L. anisa (Tateyama, 1992).
Over 13 months, we repeatedly isolated L-np, which were initially identified as L. bozemanii, by the slide agglutination test. Because these strains had not previously been isolated frequently in our hospital, we confirmed their identification by MALDI-TOF and mip-gene sequencing (Dilger et al., 2016; Ratcliff et al., 1998). This approach allowed elucidation of the preliminary misidentification of the strains, which resulted as L. anisa. Both species showed blue-white fluorescence when exposed to a wood lamp.
Alternative methods to slide agglutination tests (i.e., molecular methods) are generally only used during epidemiological investigations, not in routine environmental surveillance (Ratcliff et al., 1998).
The misleading identification of L. bozemani in place of L. anisa demonstrates the importance of correct identification of environmental strains, particularly those belonging to L-np species. In addition to ecological relevance, accurate phylogenetic characterization of Legionella contamination is essential to assessing its potential impact on the etiology of nosocomial pneumonia. Indeed, the importance of such information is widely recognized to acquire correct and complete epidemiological data and, consequently, design effective strategies to control legionellosis (Rota et al., 2020).
Although the pathogenicity of L. anisa is considered low, it has been reported that Lpn serogroup (sg) 1 is not the only species or serogroup responsible for clinical cases and other species can cause human pathologies (Cross et al., 2016; Montagna et al., 2007). In several countries, including Italy, L. anisa has been often identified as one of the most abundant contaminating species (Federici et al., 2017; Mazzotta et al., 2021). Furthermore, L. anisa can mask Lpn contamination in the water supply (Orsini et al., 2011; van der Mee-Marquet et al., 2006). In our study, the concentration of L. anisa was higher than that of Lpn sg 1 when mixed cultures were detected. Our findings also confirmed that higher water temperatures affect the load of Lpn, with lower temperatures favoring the presence of L-np (e.g., L. anisa) (Girolamini et al., 2022). This phenomenon suggests the need for new diagnostic approaches capable of widespread identification even of L-np. Currently, such identification can be performed by MALDI-TOF analysis and mip-gene sequencing (Dilger et al., 2016; Ratcliff et al., 1998) displaying a high concordance level (Pascale et al., 2020).
Epidemiological investigations uncover different aspects of Legionella contamination because the goal of such studies is identification of the source of infection to enable its control (Mazzotta et al., 2021). PFGE is one of the molecular techniques indicated for comparison of the genomic profiles among Legionella strains of different origin by the National Guidelines (Linee Guida per la Prevenzione e il Controllo della Legionellosi, 2015). However, our results suggest that the use of other molecular techniques could be a useful tool that more accurately characterizes the identity of circulating Legionella strains, according to the results found in other studies (Haroon et al., 2010; Yamamoto et al., 2003; Yan et al., 2008).
The unusual presence of L. anisa strains in the water supply of the four investigated pavilions indicated that understanding the genetic variability of the isolated strains was necessary to determine if the hospital was contaminated with one or more different L. anisa strains. PFGE, which is known as a laborious and time-consuming technique (Dilger et al., 2016), was initially used. This was followed by RAPD, rep-PCR, and SAU-PCR analysis, which are rapid, simple, reproducible, and inexpensive (Haroon et al., 2010). Although these techniques are not usually applied to L-np (Haroon et al., 2010; Corich et al., 2005; Yamamoto et al., 2003), they demonstrated a greater discriminative power than PFGE, allowing us to establish that not all of the isolated strains belonged to the same clone. In addition, genetic variability between the strains denoting a differentiation based on the site of origin was highlighted.
The reason about genetic differences among the strains from different pavilions is still unclear. Diverse eras of pavilion construction, different entry points of the water network, water temperatures, disinfection systems, or pipe materials could be responsible for the genetic variability (Girolamini et al., 2022). The presence of pavilion-specific clusters indicates the rooting of strains that have become characteristic for particular isolation sites. This phenomenon may depend on the natural selection of strains adapted to the specific environmental conditions in which they have been isolated (Akermi et al., 2006).
If extended to the entire hospital water network, our study could allow development of a map of Legionella distribution, which would enable investigation of variability in terms of strain diversity between environments. The study of Legionella belonging to L-np could improve knowledge regarding less-documented species in hospitals and other surveillance protocols (Mazzotta et al., 2021). Moreover, a regional map of Legionella will support control and prevention of Legionella and it will allow to follow the evolution of the strains over time which is affected by different factors such as mutations, resistance, and pathogenicity (Mazzotta et al., 2021).
Further investigations are still necessary to investigate the role of molecular typing in the environmental surveillance of a water network contaminated by L-np.
Conclusions
Understanding the distribution and interaction between different Legionella species is valuable for obtaining and programming the correct strategies to control these bacteria (e.g., temperature values or concentrations of disinfectants), as well as for evaluation of the efficacy of preventive strategies applied. Moreover, accurate information regarding the relationships among isolates over time and during the disinfection treatment is helpful in the comprehension of the dynamics of Legionella contamination, in order to promptly take effective corrective measures.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Akermi, M., Doleans, A., Forey, F., Reyrolle, M., Meugnier, H., Freney, J., Vandenesch, F., Etienne, J., & Jarraud, S. (2006). Characterization of the Legionella anisa population structure by pulsed-field gel electrophoresis. FEMS Microbiology Letters, 258(2), 204–207. https://doi.org/10.1111/j.1574-6968.2006.00216.x
Arrigo, I., Galia, E., Fasciana, T., Diquattro, O., Tricoli, M. R., Serra, N., Palermo, M., & Giammanco, A. (2022). Four-year environmental surveillance program of Legionella spp. in one of Palermo's largest hospitals. Microorganisms, 10, 764. https://doi.org/10.3390/microorganisms10040764
Atkinson, A. J., Morrison, C. M., Frehner, W., Gerrity, D., & Wert, E. C. (2022). Design and operational considerations in response to Legionella occurrence in Las Vegas Valley groundwater. Water Research, 220, 118615. https://doi.org/10.1016/j.watres.2022.118615
Beauté, J., Zucs, P., & de Jong, B. (2013). Legionnaires disease in Europe, 2009–2010. Eur Surveill, 18, 20417. https://doi.org/10.2807/ese.18.10.20417-en
Corich, V., Mattiazzi, A., Soldati, E., Carraro, A., & Giacomini, A. (2005). Sau-PCR, a novel amplification technique for genetic fingerprinting of microorganisms. Applied and Environment Microbiology, 71, 6401–6406. https://doi.org/10.1128/aem.71.10.6401-6406.2005
Craun, G. F., Brunkard, J. M., Yoder, J. S., Roberts, V. A., Carpenter, J., Wade, T., Calderon, R. L., Roberts, J. M., Beach, M. J., & Roy, S. L. (2010). Causes of outbreaks associated with drinking water in the United States from 1971 to 2006. Clinical Microbiology Reviews, 23, 507–528. https://doi.org/10.1128/cmr.00077-09
Cross, K. E., Mercante, J. W., Benitez, A. J., Brown, E. W., Diaz, M. H., & Winchell, J. M. (2016). Simultaneous detection of Legionella species and L. anisa, L. bozemanii, L. longbeachae and L. micdadei using conserved primers and multiple probes in a multiplex real-time PCR assay. Diagnostic Microbiology and Infectious Disease, 85(3), 295–301. https://doi.org/10.1016/j.diagmicrobio.2016.03.022
De Giglio, O., Diella, G., Lopuzzo, M., Triggiano, F., Calia, C., Pousis, C., Fasano, F., Calabrese, G., Rafaschieri, V., Carpagnano, L. F., Carlucci, M., Gesualdo, L., Ricci, M. L., Scaturro, M., Rota, M. C., Bonadonna, L., Lucentini, L., & Montagna, M. T. (2021). Management of microbiological contamination of the water network of a newly built hospital pavilion. Pathogens, 10, 75. https://doi.org/10.3390/pathogens10010075
De Giglio, O., Fasano, F., Diella, G., Lopuzzo, M., Napoli, C., Apollonio, F., Brigida, S., Calia, C., Campanale, C., Marzella, A., Pousis, C., Rutigliano, S., Triggiano, F., Caggiano, G., & Montagna, M. T. (2019). Legionella and legionellosis in touristic-recreational facilities: Influence of climate factors and geostatistical analysis in Southern Italy (2001–2017). Environmental research, 178, 108721. https://doi.org/10.1016/j.envres.2019.108721
Dilger, T., Melzl, H., & Gessner, A. (2016). Rapid and reliable identification of waterborne Legionella species by MALDI-TOF mass spectrometry. Journal of Microbiol Methods, 12, 154–159. https://doi.org/10.1016/j.mimet.2016.05.028
Directive (EU) 2020/2184 of the European Parliament and of the Council of 16 December 2020 on the quality of water intended for human consumption (recast). Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32020L2184 (Accessed 7 July 2022), https://doi.org/10.1016/j.mimet.2016.05.028
Doebbeling, B. N., Ishak, M. A., Wade, B. H., Pasquale, M. A., Gerszten, R. E., Gröschel, D. H., Kadner, R. J., & Wenzel, R. P. (1989). Nosocomial Legionella micdadei pneumonia: 10 years experience and a case-control study. Journal of Hospital Infection, 13, 289–298. https://doi.org/10.1016/0195-6701(89)90010-8
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32, 1792–1797. https://doi.org/10.1093/nar/gkh340
ESGLI. (2017). Prevention, control and investigation, of infections caused by Legionella species; European Working Group for Legionella Infections: Solna, Stockholm, Sweden, 2017.
European Centre for Disease Prevention and Control. Legionnaires' disease. In: ECDC. Annual epidemiological report for 2020. Stockholm: ECDC; 2022. Available online: https://www.ecdc.europa.eu/en/publications-data/european-technical-guidelines-prevention-control-and-investigation-infections (Accessed 7 July 2022).
Federici, E., Meniconi, S., Ceci, E., Mazzetti, E., Casagrande, C., Montalbani, E., Businelli, S., Mariani, T., Mugnaioli, P., Cenci, G., & Brunone, B. (2017). Legionella survey in the plumbing system of a sparse academic campus: A case study at the University of Perugia. Water, 9, 662. https://doi.org/10.3390/w9090662
Fliermans, C. B. (1996). Ecology of Legionella: From data to knowledge with a little wisdom. Microbial Ecology, 32(2), 203–228. https://doi.org/10.1007/bf00185888
Gevers, D., Huys, G., & Swings, J. (2001). Applicability of rep-PCR fingerprinting for identification of Lactobacillus species. FEMS Microbiology Letters, 205, 31–36. https://doi.org/10.1111/j.1574-6968.2001.tb10921.x
Girolamini, L., Dormi, A., Pellati, T., Somaroli, P., Montanari, D., Costa, A., Savelli, F., Martelli, A., Grottola, A., Fregni Serpini, G., & Cristino, S. (2019). Advances in Legionella control by a new formulation of hydrogen peroxide and silver salts in a hospital hot water network. Pathogens, 8, 4–209. https://doi.org/10.3390/pathogens8040209
Girolamini, L., Salaris, S., Lizzadro, J., Mazzotta, M., Pascale, M. R., Pellati, T., & Cristino, S. (2020). How molecular typing can support Legionella environmental surveillance in hot water distribution systems: A hospital experience. International Journal of Environmental Research and Public Health, 17, 8662. https://doi.org/10.3390/ijerph17228662
Girolamini, L., Salaris, S., Orsini, M., Pascale, M. R., Mazzotta, M., Grottola, A., & Cristino, S. (2021). Draft genome sequences of Legionella presumptive novel species isolated during environmental surveillance in artificial water systems. Microbiol Resour Announc, 10(19), e00307-e321. https://doi.org/10.1128/mra.00307-21
Girolamini, L., Salaris, S., Pascale, M. R., Mazzotta, M., & Cristino, S. (2022). Dynamics of Legionella community interactions in response to temperature and disinfection treatment: 7 years of investigation. Microbial Ecology, 83(2), 353–362. https://doi.org/10.1007/s00248-021-01778-9
Haroon, A., Koide, M., Higa, F., Hibiya, K., Tateyama, M., & Fujita, J. (2010). Repetitive element-polymerase chain reaction for genotyping of clinical and environmental isolates of Legionella spp. Diagnostic Microbiology and Infectious Disease, 68(1), 7–12. https://doi.org/10.1016/j.diagmicrobio.2010.04.001
Head, B. M., Trajtman, A., Bernard, K., Burdz, T., Vélez, L., Herrera, M., Rueda, Z. V., & Keynan, Y. (2019). Legionella co-infection in HIV-associated pneumonia. Diagnostic Microbiology and Infectious Disease, 95(1), 71–76. https://doi.org/10.1016/j.diagmicrobio.2019.03.005
Heras, J., Domínguez, C., Mata, E., Pascual, V., Lozano, C., Torres, C., & Zarazaga, M. (2015). GelJ- a tool for analyzing DNA fingerprint gel images. BMC Bioinform., 16, 270. https://doi.org/10.1186/s12859-015-0703-0
Huey, B., & Hall, J. (1989). Hypervariable DNA fingerprinting in Escherichia coli Minisatellite probe from bacteriophage M13. Journal of Bacteriology, 171, 2528–2532. https://doi.org/10.1128/jb.171.5.2528-2532.1989
Iacumin, L., Comi, G., Cantoni, C., & Cocolin, L. (2006). Molecular and technological characterization of Staphylococcus xylosus isolated from naturally fermented Italian sausages by RAPD. Rep-PCR and Sau-PCR Analysis. Meat Sci, 74(2), 281–288. https://doi.org/10.1016/j.meatsci.2006.03.020
ISO 11731:2017. Water quality-enumeration of Legionella. Available online: https://www.iso.org/standard/61782.html (Accessed 4 July 2022).
Istituto Superiore di Sanità. (2022). I risultati del sistema di sorveglianza della legionellosi nel 2021. Boll Epidemiol Naz 2022, 3(2), 30–37.
Linee Guida per la Prevenzione e il Controllo della Legionellosi. (2015). Available online: http://www.legionellaonline.it (Accessed 4 July 2022).
List of Prokaryotic Names with Standing in Nomenclature (LPSN). Available online: http://www.bacterio.net (Accessed 4 July 2022).
Martínez-Puchol, S., Riveros, M., Ruidias, K., Granda, A., Ruiz-Roldán, L, Zapata-Cachay, C., Ochoa, T. J., Pons, M. J., & Ruiz J. (2021). Dissemination of a multidrug resistant CTX-M-65 producer Salmonella enterica serovar Infantis clone between marketed chicken meat and children. International Journal of Food Microbiology, 344, 109109. https://doi.org/10.1016/j.ijfoodmicro.2021.109109
Mazzotta, M., Salaris, S., Pascale, M. R., Girolamini, L., & Cristino, S. (2021). Occurrence of Legionella spp. in man-made water sources: Isolates distribution and phylogenetic characterization in the Emilia-Romagna region. Pathogens, 10, 552. https://doi.org/10.3390/pathogens10050552
Miller, M., Hayden, R., & Gaur, A. (2007). Legionella bozemanii pulmonary abscess in a pediatric allogeneic stem cell transplant recipient. The Pediatric Infectious Disease Journal, 26, 760–762. https://doi.org/10.1097/inf.0b013e318054e338
Montagna, M. T., Ricci, M., Napoli, C., Tato’, D., Scaturro, M., Barbuti, G., Pierucci, G., & Castellani Pastoris, M. (2007). Legionella pneumophila serogroup 5 infection in the presence of multiple environmental contamination. The importance of a bacteriological diagnosis. Italian Journal of Public Health, 4, 71–74.
Napoli, C., Fasano, F., Iatta, R., Barbuti, G., Cuna, T., & Montagna, M. T. (2010). Legionella spp. and legionellosis in southeastern Italy: Disease epidemiology and environmental surveillance in community and health care facilities. BMC Public Health, 10, 660. https://doi.org/10.1186/1471-2458-10-660
Neiderud, C. J., Vidh, A. L., & Salaneck, E. (2013). Soft tissue infection caused by Legionella bozemanii in a patient with ongoing immunosuppressive treatment. Infect Ecol Epidemiol, 3, 20739. https://doi.org/10.3402/iee.v3i0.20739
Orsini, M., Cristino, S., Grottola, A., & Romano-Spica, V. (2011). Bacteria misagglutination in Legionella surveillance programmes. Journal of Hospital Infection, 79, 179–180. https://doi.org/10.1016/j.jhin.2011.05.021
Pascale, M.R., Mazzotta, M., Salaris, S., Girolamini, L., Grottola, A., Simone, M.L., Cordovana, M., Bisognin, F., Dal Monte, P., Bucci Sabattini, M.A., Viggiani, M. & Cristino, S. (2020). Evaluation of MALDI–TOF mass spectrometry in diagnostic and environmental surveillance of Legionella species: A comparison with culture and mip-gene sequencing technique. Frontiers in Microbiology, 11, 589369. https://doi.org/10.3389/fmicb.2020.589369
Pascale, M. R., Salaris, S., Mazzotta, M., Girolamini, L., Fregni Serpini, G., Manni, L., Grottola, A., & Cristino, S. (2021). New insight regarding Legionella non-pneumophila species identification: Comparison between the traditional mip gene classification scheme and a newly proposed scheme targeting the rpob gene. Microbiology Spectrum, 9(3), e0116121. https://doi.org/10.1128/spectrum.01161-21
Prussin, A. J., Schwake, D. O., & Marr, L. C. (2017). Ten questions concerning the aerosolization and transmission of Legionella in the built environment. Building and Environment, 123, 684–695. https://doi.org/10.1016/j.buildenv.2017.06.024
Ratcliff, R. M., Lanser, J. A., Manning, P. A., & Heuzenroeder, M. W. (1998). Sequence-based classification scheme for the genus Legionella targeting the mip gene. Journal of Clinical Microbiology, 36, 1560–1567. https://doi.org/10.1128/jcm.36.6.1560-1567.1998
Ribot, E. M., Fair, M. A., Gautom, R., Cameron, D. N., Hunter, S. B., Swaminathan, B., & Barrett, T. J. (2006). Standardization of pulsed-field gel electrophoresis protocols for the subtyping of Escherichia coli O157:H7, Salmonella, and Shigella for PulseNet. Foodbourne Pathog Dis, 3, 59–67. https://doi.org/10.1089/fpd.2006.3.59
Rota, M. C., Caporali, M. G., Bella, A., Scaturro, M., Giannitelli, S., & Ricci, M. L. (2020). Il sistema di Sorveglianza della Legionellosi in Italia: i risultati del 2019. Boll Epidemiol Naz, 1(2), 32–38. https://doi.org/10.53225/BEN_005
Sabrià, M., García-Nuñez, M., Pedro-Botet, M. L., Sopena, N., Gimeno, J. M., Reynaga, E., Morera, J., & Rey-Joly, C. (2001). Presence and chromosomal subtyping of Legionella species in potable water systems in 20 hospitals of Catalonia. Spain. Infection Control & Hospital Epidemiology, 22(11), 673–676. https://doi.org/10.1086/501843
Scaturro, M., Buffoni, M., Girolamo, A., Cristino, S., Girolamini, L., Mazzotta, M., Bucci Sabattini, M. A., Zaccaro, C. M., Chetti, L., Laboratory, M. A. N., Bella, A., Rota, M. C., & Ricci, M. L. (2020). Performance of Legiolert Test vs. ISO 11731 to Confirm Legionella pneumophila Contamination in Potable Water Samples. Pathogens, 9(9), 690. https://doi.org/10.3390/pathogens9090690
Schwake, D. O., Alum, A., & Abbaszadegan, M. (2021). Legionella occurrence beyond cooling towers and premise plumbing. Microorganisms, 9, 2543. https://doi.org/10.3390/microorganisms9122543
Svarrer, C. W., & Uldum, S. A. (2012). The occurrence of Legionella species other than Legionella pneumophila in clinical and environmental samples in Denmark identified by mip gene sequencing and matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clinical Microbiology & Infection, 18(10), 1004–1009. https://doi.org/10.1111/j.1469-0691.2011.03698.x
Tateyama, M. (1992). Misleading serological identification of Legionella anisa as Legionella bozemanii. Kansenshogaku Zasshi, 66(2), 149–155. https://doi.org/10.11150/kansenshogakuzasshi1970.66.149
Tenover, F. C., Arbeit, R. D., Goering, R. V., Mickelsen, P. A., Murray, B. E., Persing, D. H., & Swaminathan, B. (1995). Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: Criteria for bacterial strain typing. Journal of Clinical Microbiology, 33(9), 2233–2239. https://doi.org/10.1128/jcm.33.9.2233-2239.1995
Vaccaro, L., Izquierdo, F., Magnet, A., Hurtado, C., Salinas, M. B., Gomes, T. S., Angulo, S., Salso, S., Pelaez, J., Tejeda, M. I., Alhambra, A., Gómez, C., Enríquez, A., Estirado, E., Fenoy, S., & Del Aguila, C. (2016). First case of legionnaire’s disease caused by Legionella anisa in Spain and the limitations on the diagnosis of Legionella non-pneumophila infections. PLoS ONE, 11, 1–12. https://doi.org/10.1371/journal.pone.0159726
Van der Kooij, D., Brouwer-Hanzens, A. J., Veenendaal, H. R., & Wullings, B. A. (2016). Multiplication of Legionella pneumophila sequence types 1, 47, and 62 in buffered yeast extract broth and biofilms exposed to flowing tap water at temperatures of 38 ◦C to 42 ◦C. Applied and Environment Microbiology, 82, 6691–6700. https://doi.org/10.1128/aem.01107-16
van der Mee-Marquet, N., Domelier, A. S., Arnault, L., Bloc, D., Laudat, P., Hartemann, P., & Quentin, R. (2006). Legionella anisa, a possible indicator of water contamination by Legionella pneumophila. Journal of Clinical Microbiology, 44(1), 56–59. https://doi.org/10.1128/jcm.44.1.56-59.2006
Völker, S., Schreiber, C., & Kistemann, T. (2016). Modelling characteristics to predict Legionella contamination risk—Surveillance of drinking water plumbing systems and identification of risk areas. Int. J. Hyg. Environ. Heal., 219, 101–109. https://doi.org/10.1016/j.ijheh.2015.09.007
Yamamoto, N., Kubota, T., Tateyama, M., Koide, M., Nakasone, C., Tohyama, M., Shinzato, T., Higa, F., Kusano, N., Kawakami, K., & Saito, A. (2003). Isolation of Legionella anisa from multiple sites of a hospital water system: The eradication of Legionella contamination. Journal of Infection and Chemotherapy, 9, 122–125. https://doi.org/10.1007/s10156-002-0228-0
Yan, H., Shi, L., Alam, M. J., Li, L., Yang, L., & Yamasaki, S. (2008). Usefulness of Sau-PCR for molecular epidemiology of nosocomial outbreaks due to Burkholderia cepacia which occurred in a local hospital in Guangzhou. China. Microbiol Immunol., 52(5), 283–286. https://doi.org/10.1111/j.1348-0421.2008.00035.x
Yang, G., Benson, R., Pelish, T., Brown, E., Winchell, J. M., & Fields, B. (2010). Dual detection of Legionella pneumophila and Legionella species by real-time PCR targeting the 23S–5S rRNA gene spacer region. Clinical Microbiology & Infection, 16(3), 255–261. https://doi.org/10.1111/j.1469-0691.2009.02766.x
Zhan, X. Y., Yang, J. L., Sun, H., Zou, X., Qian, Y. C., Huang, K., Leng, Y., Huang, B., & He, Y. (2022). Presence of viable, clinically relevant Legionella bacteria in environmental water and soil sources of China. Microbiology Spectrum, 10(3), e0114021. https://doi.org/10.1128/spectrum.01140-21qt
Acknowledgements
We greatly appreciate Maria Elena Caringella, Caterina Mele, Maria Cicerale, Emanuela Luciani, Antonella Savino, and Sofia Denicolò for providing technical support during the experiments. We thank Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Funding
Open access funding provided by Università degli Studi di Bari Aldo Moro within the CRUI-CARE Agreement. This research was funded by the Apulia Regional Government, Bari, Italy (D.G.R. n.1001 del 19/07/2022).
Author information
Authors and Affiliations
Contributions
Conceptualization: M.T.M., O.D.G., M.D.A; methodology: O.D.G., C.P., L.I., E.F., M.T.M.; software and data curation: F.F., S.B., M.P., M.O., C.P., L.I; validation: O.D.G., F.F., C.P., L.I., E.F., M.T.M; investigation: M.D.A, V.S., G.D., C.M.L., M.L., V.T., C.C., M.O., L.I., S.B., M.P.; writing—original draft preparation: O.D.G., C.P., L.I., E.F., M.T.M.; supervision: M.T.M.; O.D.G; writing—review and editing: M.T.M., F.P.L; A.M.M.; funding acquisition: M.T.M. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
All authors have read, understood, and complied as applicable with the statement on “Ethical responsibilities of Authors” as found in the “Instructions for Authors.”
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
De Giglio, O., D’Ambrosio, M., Spagnuolo, V. et al. Legionella anisa or Legionella bozemanii? Traditional and molecular techniques as support in the environmental surveillance of a hospital water network. Environ Monit Assess 195, 496 (2023). https://doi.org/10.1007/s10661-023-11078-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10661-023-11078-z