
Abstract
To understand the evolution of Puccinia triticina, we used single nucleotide polymorphisms (SNPs) to evaluate the P. triticina population genetic structure in five Chinese provinces, Gansu, Henan, Hubei, Shaanxi and Sichuan from 2013 to 2015. The populations exhibited characteristics of high haplotype diversity but low nuclear diversity. The gene flow was frequent between Gansu and Henan, Gansu and Sichuan and Sichuan and Henan. Genetic differentiation was significant among populations except between Henan and Gansu and between Henan and Sichuan (P < 0.05). The P. triticina isolates from the five provinces were grouped into two clusters: Gansu/Henan/Shaanxi/Sichuan and Hubei clusters. AMOVA analysis showed that the populations exhibited genetic variation; the variation percentage reached 87.45%, and the genetic variation within populations was the major source of variation. Our results suggest that the genetic diversity of P. triticina was high and that gene flow was frequent between populations. P. triticina population of China had experienced a rapid expansion, which caused some differences between populations. The Hubei population was different from other population due to genetic variation within populations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Wheat leaf rust caused by Puccinia triticina is the most common and widely distributed among the three wheat rusts worldwide (Huerta-Espino et al. 2011). Sometimes, leaf rust may cause greater yield losses in an epidemic year due to its widespread occurrence than a normal year (Singh et al. 2004; Kolmer et al. 2009). Annual yield losses are estimated to be 3 million tons in China (Huerta-Espino et al. 2011). Wheat leaf rust generally affects 10–30% of wheat fields but can exceed 60% in some years. Gansu, Henan, Shaanxi, Hubei and Sichuan are main wheat producing regions in China and wheat leaf rust is a frequently damaging disease. Understanding both the population structure and evolutionary process of the leaf rust pathogen allows determining the epidemic areas and sources of inoculum, epidemic rules and dispersal routes, which are essential for disease control.
The study of the population genetics of plant pathogenic fungi involves two aspects (Li et al. 2003). Identifying the pathogen and detecting its pathogenicity by traditional methods; however, researchers can also use molecular biology technology to understand population structure. Race identification and virulence of P. triticina detection and patterns have provided valuable information for the study of leaf rust epidemics in China. Chen et al. (1998) proved that P. triticina of the population virulence detection was proficient at dividing epidemic areas in China from 1992 to 1996, and some spatial structures were also determined. However, traditional methods focused on studying the dynamic changes of the population. The research method based on virulence phenotypes that were affected by regional cultivar preferences rather than genotypes does not contribute to explore population structure. To further understand the evolutionary process of wheat leaf rust in China, we intended to use the molecular marker technique as a way to determine the population structures of P. triticina.
Molecular marker techniques are widely applied in studying population structures of P. triticina (Xu et al. 2002; Gao and Liu 2002; Kolmer et al. 1995). We have determined population genetic diversity and genetic relationship between populations; however, the evolutionary processes of P. triticina were still unknown. Single nucleotide polymorphisms (SNPs), which are third-generation molecular markers characterized by high stability and the ability to detect the most frequent type of DNA variations (Tang et al. 2012), can be used to infer historical processes in more complex population genetic models. Parks et al. (2009) identified four housekeeping genes and used SNP markers to identify isolates of the wheat powdery mildew pathogen that originated from the southern United States and were subdivided into two distinct regions: northern and southern subpopulations. In addition, those authors reported that, due to its physical isolation and genetic drift, the wheat powdery mildew population in the eastern United States shared a common ancestor with the British and Irish populations. By using SNP markers, Liu et al. (2014) suggested an evolutionary history of P. triticina races virulent on three wheat varieties.
It is possible to deduce or detect population structures of P. triticina by RAPD technique and the results showed that the DNA polymorphisms were correlated with the pathogens’ geographic origins (Xu et al. 2002). Strong phylo-geographical distributions were revealed by Morgado et al. (2013) based on ITS (internal transcribed spacer, ITS), LSU (nuclear large ribosomal subunit, LSU), RPB2 (nuclear RNA polymerase II second largest subunit, RPB2) and mtSSU (mitochondrial ribosomal small subunit gene, mtSSU) and provided insights into the evolutionary history for Entoloma systematics. IGS (Intergenic Spacer of rDNA units, IGS) region and the elongation factor EF-1α were interesting tools for phylogenetic analysis and evaluated the genetic variability in this species (Mirete et al. 2004). Understanding the variations among populations, it will contribute to define population time series of occurrence and dispersal routes. In the present study, we used SNPs to analyze the population genetic structures of P. triticina in Gansu, Henan, Shaanxi, Hubei and Sichuan provinces from 2013 to 2015. We aimed to understand the genetic variation and evolution, and the results could provide information for defining dispersal pathways and for disease management.
Materials and methods
Isolates of P. triticina
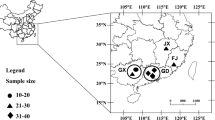
The samples were collected from Gansu (27), Henan (25), Shaanxi (15), Hubei (28) and Sichuan (26) provinces from 2013 to 2015 (Fig. 1). A total of 121 samples of propagated P. triticina spores were stored at the Cereal Fungal Disease Laboratory of the Institute of Plant Protection, Chinese Academy of Agricultural Sciences.

Colored provinces where P. triticina isolates were collected in 2013–2015: Gansu (blue), Henan (yellow), Shaanxi (red), Hubei (green) and Sichuan (pink) provinces
Genomic DNA extraction
Genomic DNA was isolated in accordance with the methods of Zheng et al. (2008). First, 10 mg of fresh spores, 0.4 g of 1-mm-diameter glass beads and 0.2 g of 0.1-mm-diameter glass beads were mixed together and vigorously shaken in an oscillator (MiniBeadbeater-96, Biospec, USA) at 6.5 m/s for 1 min (twice) to disrupt fungal spore cell walls. Six hundred microliters of cetyl-trimethylammonium bromide (1.998 g of Tris, 1.928 g of EDTA, 9 g of NaCl, 1.1 g of CTAB, dH2O adjusted to 100 mL, pH = 8.0), which was preheated to 65 °C; 60 μL of 20% SDS; and 10 μL of proteinase K (20 μg/mL) were then added to the mixture. The mixture was subsequently incubated in a water bath at 65 °C for 1 h, and it was shaken every 10 min during the incubation. Afterward, the aqueous phase was extracted with a phenol: chloroform: isoamyl alcohol solution (25:24:1). Each mixture was vigorously shaken and then centrifuged (5417R, Eppendorf, Hamburg, Germany) at 15000 rpm for 10 min at 4 °C to remove the aqueous phase, which was placed into a new micro centrifuge tube. The aqueous phase was subsequently extracted with an equal volume of chloroform: isoamyl alcohol (24:1) solution. Each tube was centrifuged again at 15000 rpm for 10 min, and each supernatant was subsequently incubated with 10 μL of RNase (100 mg/mL) at 37 °C for 1 h to remove any RNA. The solutions added to isoamyl alcohol (24:1) were then centrifuged at 15000 rpm for 10 min at 4 °C. To each tube, 0.6 times the volume of precooled isopropyl alcohol and incubated overnight at −20 °C. To discard the aqueous phase, the tube was then centrifuged at 15000 rpm for 10 min at 4 °C. The DNA was rinsed twice with 70% ethanol, air-dried and dissolved in a 30-μL ddH2O solution. The genomic DNA was stored at −20 °C.
Primer information
In this study, five conserved genes (Table 1), including those for 28S ribosomal DNA (28S rDNA), intergenic spacer ribosomal DNA (IGS rDNA), RPB2 (the second largest RNA polymerase subunit), elongation factor-1α (EF-1α), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), were used to analyze the population genetic structure because they were useful to diffentiate the diversities and phylogeny (Morgado et al. 2013; Mirete et al. 2004). The PCR primers used were synthesized by Sangon Biotech (Shanghai, China).
PCR conditions
PCR was performed using an Eppendorf PCR system. PCR was carried out in 25 μL solutions consisting of 12.5 μL of Easypfu PCR Supermix (TransGen Biotech, Beijing, China), 1 μL (100 ng/ μL) of genomic DNA, 1 μL (10 ng/ μL) of forward primers, 1 μL (10 ng/ μL) of reverse primers, and 9.5 μL of ddH2O. The PCR conditions were as follows: denaturation at 94 °C for 4 min; 35 amplification cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 60 s; and a final extension step at 72 °C for 10 min.
Cloning and sequencing
The PCR products were determined by agarose gel electrophoresis (150 V, 30 min). The concentration of the agarose gels was 1.2%. DNA was purified by EasyPure Quick Gel Extraction Kit (TransGen Biotech, Beijing, China).
One microliter of pEASY-Blunt vectors (TransGen Biotech, Beijing, China) and 2 μL of DNA were ligated at 23–28 °C for 10 min. Fifty microliters of Trans-T1 competent cells were added to the mixture, which were chilled on ice for 20 min, heated to 42 °C for 45 s, and chilled on ice for 2 min. The mixtures were transferred to 5-mL centrifuge tubes that contained 800 μL of Luria-Bertani (LB) medium, and the tubes were kept at 37 °C rotating at <200 rpm for 1 h. Two hundred microliters of the bacterial solution was added to solid LB media that contained ampicillin (Amp), isopropyl β-D-1-thiogalactopyranoside (IPTG) and 5-bromo-4-chloro-3-indolyl-D-galactoside (X-gal); the media were cultured at 37 °C overnight. Eight promising colonies were picked and cultured in 1 mL of liquid LB medium supplemented with 1 μL of Amp at 37 °C and 200 rpm for 5 h. After PCR detection, 5 positive clones were selected in each sample for sequencing by Sanger sequencing (Sangon Biotech, Shanghai, China).
Population structure analysis
The sequences were compared with those in the NCBI database to confirm that they were the target fragments. If the sequences were the target fragments, the five repeats were compared using MEGA 6.0 software, after which we identified the consensus target sequence. DnaSP 5.0 software (Librado and Rozas 2009) to calculate the polymorphism indexes including the number of haplotypes (H), haplotype diversity (Hd), and nucleotide diversity (Pi). Tajima’s D and Fu and Li’s D and F were calculated with DnaSP 5.0 software, and Fu’s FS was calculated with Arlequin 3.11 (Excoffier et al. 2007).
Arlequin 3.11 was used to analyze the genetic variation and gene flow of five populations; the genetic differentiation coefficient (Fst) and the number of migrants per generation (Nm) were calculated by the equation Nm = (1-Fst)/4 Fst (Jimenez et al. 1999). AMOVA based on Arlequin 3.11 was used to detect population differentiation between regions, within populations and between populations within regions.
The Snn (nearest-neighbors statistic) method in the Hudson software (Hudson et al. 1992) was used to subdivide the population structures. We tested the zero distribution by 1000 permutations to assess the significance of the results. The software Structure 2.3.4 (Falush et al. 2003) was used to predict the possible population structures. The parameter of Length of Burn-in Period and Number of MCMC Reps after Burn-in were 100,000 and 500,000. The K value from 1 to 10 was repeated 10 times, and ΔK value was used to determine the optimal population. Finally, we used Distruct software (Rosenberg 2004) to generate a histogram based on the Structure’s result for the population structures.
HapStar software (Teacher and Griffiths 2011) was used to explore the evolutionary relationships based on the haplotypes between the five provinces used the Euclidean distance matrix generated by Arlequin 3.11. An SVG file was saved and optimized using InkScape software.
Results
Nucleotide sequence polymorphisms
The sequences of 116 samples were manually assembled to a total length of 4885 bp by the order of 28S rDNA, IGS, RPB2, EF-1α, and GAPDH genes. There were 71 polymorphic nucleotides, including 14 parsimony-informative nucleotides, four base-transition nucleotides and 10 transformation nucleotides. Five populations consisted of a total of 42 haplotypes and that H1 was a haplotype shared by all five populations. The overall population had the characteristics of high haplotype diversity but low nucleotide diversity (Table 2). The difference among the populations was not significant (Table 2).
The results of this study depended on the manual sequence assembly at an order of 28S rDNA, IGS, RPB2, EF-1α, and GAPDH genes. However, we did not know whether the results would differ between the different manual assembled sequences. Therefore, to further understand the differences in the results, we analyzed the manual assembled sequences at the order of RPB2, IGS, EF-1α, GAPDH, and 28S rDNA genes and GAPDH, IGS, 28S rDNA, RPB2, and EF-1α genes. The results showed that these sequences had no effect on the haplotype networks, population structures and the molecular variance and genetic diversity analyses had little effects on the results.
Neutrality tests
The neutral theory means that, at the molecular level, most of the evolution and variation of species are not a natural choice, but due to the selection of neutral or near neutral mutation caused by genetic drift phenomenon (Kimura 1983). The neutral parameters of Tajima’s D, Fu and Li’s D and F, and Fu’s Fs were calculated for neutrality tests (Table 3). The results showed that the Tajiama’ D, Fu and Li′ D, Fu and Li′ F were not significant, indicating that sequence followed neutral evolutionary model. Only Fu’s Fs were significant indicating an expanding population, genetic hitchhiking or sexual recombination (Fu 1997). The single peak of mismatch distributions of P. triticina (Fig. 2) indicated that the population had gone a rapid expansion (Mousset et al. 2004).

Mismatch distributions based on pairwise differences between DNA sequences of P. triticina
Gene flow and genetic divergence
The result of gene flow showed that genetic exchange occurred among populations and the Nm values of the Gansu/Henan, Sichuan/Henan and Sichuan/Gansu populations were greater than 4, which indicated frequent gene flow among these populations (Table 4). The genetic differentiation fixation coefficient showed that genetic differentiation was significant among populations except Henan/Gansu and Henan/Sichuan, especially the differences between Hubei and Shaanxi were the largest (Table 4). The result indicated that gene exchange between populations was frequent and the P. triticina populations experienced long-distance transmission, while independent evolutionary differentiations were occurred in local populations.
Haplotype networks
To analyze the evolutionary relationships, HapStar software was used to construct a haplotype network according to the Euclidean distance matrix obtained from Arlequin 3.11. As shown in Fig. 3, the haplotype network was centered on H1 and H38. H1 was from five populations, and the H38 haplotype was shared except the Shaanxi population. The haplotypes close to the center are older, and the surrounding haplotypes are younger. So the centrally located haplotypes were shared haplotypes, while those on the edge were relatively young and unique. What’s more, the Hubei population was significantly different from other four populations. Shared haplotypes (H1, H38, H42, etc.) were common and widely distributed among populations, so they were more stable during the evolution of the population. The special haplotypes (H2, H3, H4, etc.) indicated that there were genetic differentiations within a population.

HapStar was used to construct the network map based on the distance matrix results of Arlequin 3.11. Black node: haplotypes may exist but may not occur in nature
Population structure
We predicted the population structure using Structure 2.3.4 software. When K was 2, the five subpopulations can be grouped into two clusters including Gansu/Henan/Shaanxi/Sichuan and Hubei populations (Fig. 4). When K was 3, they were grouped three clusters, Gansu/Sichuan, Henan/Shaanxi and Hubei populations. When K was 4, the population structure was similar and there were non-significant difference. However, the broken line graph (Fig. 5) of predicting the best population structure showed that △K was largest while K was equal to 2 (Zhang et al. 2014). When △K was largest, K was the most optimum population. Therefore, Gansu/Henan/Shaanxi/Sichuan represented one cluster and the Hubei population represented the other cluster.

Distruct software divided the five populations based on the Structure results. The results are shown when K = 2. Each individual is represented by a vertical line; each population partitioned into two colors, and the different population are divided by a black line

Clusters of Puccinia triticina isolates using Distruct software based on the Structure results. The figure is similar with Fig.4 except K = 3
The Snn of Hudson program analyzed the possible population structure (Table 5). With respect to the Snn, when P was greater than 0.05, no significant differences occurred. There were significant differences between the Gansu/Henan populations and the Hubei/Sichuan populations, but there were no significant differences between the Shaanxi/Henan populations and the Gansu/Sichuan populations. However, there were significant differences between the Hubei, Sichuan and Hubei populations. Therefore, the five populations could be subdivided into three or two clusters: Gansu/Shaanxi/Henan, Sichuan, and Hubei clusters or Gansu/Shaanxi/Henan/Sichuan and Hubei clusters. To further differentiate the clusters, we continued to analyze both possibilities by Snn. There were no significant differences between the Gansu/Shaanxi/Henan and Sichuan clusters. The five populations of P. triticina could be grouped into two clusters, namely, a Gansu/Shaanxi/Henan/Sichuan cluster and a Hubei cluster.
Analysis of AMOVA
The results of the AMOVA analysis performed using Arlequin 3.11 software based on the potential population structure showed that approximately 87. 5% of the total variation occurred within populations and that 14.2% of the total variation occurred between populations (Table 6). This indicated that genetic variation within populations was the major source of variation. The genetic differentiation coefficient among the populations was large, Fst = 0.12552 (P < 0.05), indicating genetic differentiation among the populations.
Discussion and conclusion
Gene flow and genetic divergence
Wheat leaf rust is a wind-dispersed pandemic disease that is spread by clonal urediniospores. When their numbers are enough, these urediniospores can be dispersed by atmospheric circulation up to 1500~5000 m, spreading to 800~2000 km to infect wheat plants (Guo et al. 2014). That means that urediniospores of different regions are often exchanged. Gene flow (Nm) can reveal genetic infiltration between populations and eliminate genetic differences in populations (Rousset 1997). The result showed that gene exchange between populations was frequent and extensive. Fst can explicit the level of variation among different populations and is an important parameter studying the evolutionary history of the population. Hedrick (2005) reported that if Fst are larger, the level of genetic differentiation between populations is greater. Here, our results showed that significant genetic differentiation occurred, except for the Henan/Gansu and Henan/Sichuan populations. Although gene exchange between populations was frequent and the pathogen populations experienced long-distance transmission, independent evolutionary pathways were still followed. What’s more, the special haplotypes indicated that there were genetic differentiations within populations.
The genetic diversity of the five populations was high, but there were differences between populations, which were caused by many factors. Wheat leaf rust of China is distributed widely in all wheat planting areas. Resistance of cultivars, environment of farmlands and variation of races are different in different regions. Meanwhile, urediniospores of P. triticina experienced long-distance dissemination. So populations of P. triticina in China have a rich diversity and complicated population structure.
Population structure
The structure analysis and Hudson test both indicated that the five populations could be grouped into two clusters: the Gansu/Henan/Shaanxi/Sichuan cluster and the Hubei cluster. The haplotype network map revealed the Hubei population was significantly different from other populations. By using SSR molecular markers to explore genetic structure, Xu et al. (2013) reported that the Sichuan and Henan populations were closely related. Kolmer et al. (1995) has found that Shaanxi, Hubei, Sichuan and Henan populations were closely related to each other and likely form a single epidemiological zone for P. triticina. However, only four isolates were used for the Hubei population.
Mcdermott and McDonald (1993) have reported that genetic drift, gene mutation, selection gene flow and reproduction are the important forces that affect the divergence of lineages. The results of the AMOVA analysis revealed that the genetic variation within populations was the major source of variation. Kolmer and Ordoñez (2007) used SSR markers to analyze the population structure of P. triticina in Central Asia and the Caucasus; and authors reported that population variation occurred mostly within populations. Our results agreed with what they delivered in their documentation. To our knowledge, the reason why they differed about the variation within populations could be caused by unintentionally used of different cultivars or Lr genes since tens of cultivars were released each year. However, the reason that the Hubei population was different from other populations needs further studies using larger populations.
Phylogeny and evolution
Haplotype networks revealed that some of the shared ancestral haplotypes, such as H1 and H38, were widely distributed and were relatively stable during the evolution of the population. This result suggested that P. triticina in China may have a common origin. In this study, neutrality tests were not significant, indicating that the sequences followed the neutral evolutionary model and only Fu’s Fs was significant, indicating that population expansion, genetic drift or sexual recombination occurred (Fu 1997). The mismatch distribution of the P. triticina populations in the five provinces had a single peak, and, the haplotype diversity was high and nuclear diversity was low, which also suggested that the population experienced a rapid expansion (Mousset et al. 2004) which was accordance with the increasing occurrence of wheat leaf rust in the last 5 years. Zhao et al. (1994) confirmed that Thalictrum aquilegiifolium is an alternate host of P. triticina under conditions of artificial inoculation, but sexual recombination was low in the natural environment. Therefore, the P. triticina population in China is pandemic, which is also consistent with the occurrences of wheat leaf rust epidemics throughout the history (Li and Zeng 2002).
References
Chen, W. Q., Qin, Q. M., & Chen, Y. L. (1998). Virulence of Puccinia triticina in China from 1992 to 1996. Acta Phytophylacica Sinica, 28, 6–11.
Du, D. D., Zhang, Y. M., Zhang, H. S., Yang, W. X., & Liu, D. Q. (2015). Primary study on polymorphism of wheat leaf rust based IGS. Acta Agriculturae Boreali-Sinica, 30, 51–56.
Excoffier, L., Laval, G., & Schneider, S. (2007). Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online, 1, 47–50.
Falush, D., Stephens, M., & Pritchard, J. K. (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics, 164, 1567–1587.
Fu, Y. X. (1997). Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics, 147, 915–925.
Gao, J., & Liu, D. Q. (2002). The application of AFLP on population genetics of fungal phytopathogen. Journal of Agricultural University of Hebei, 25(z1), 155–159.
Guo, Y. Y., Wu, K. M., & Chen, W. Q. (2014). Crop disease and insect pests in China. Beijing: China Agriculture Press.
Hedrick, P. W. (2005). A standardized genetic differentiation measure. Evolution, 59, 1633–1638.
Hudson, R. R., Boos, D. D., & Kaplan, N. L. (1992). A statistical test for detecting geographic subdivision. Molecular Biology and Evolution, 9, 138–151.
Huerta-Espino, J., Singh, R. P., Germán, S., McCallum, B. D., Park, R. F., Chen, W. Q., et al. (2011). Global status of wheat leaf rust caused by Puccinia triticina. Euphytica, 179, 143–160.
Jeffreys, A. J., Royle, N. J., Wilson, V., & Wong, Z. (1988). Spontaneous mutation rates to new length alleles at tandem-repetitive hypervariable loci in human DNA. Nature, 332, 278–281.
Jimenez, P., Agundez, D., Alia, R., & Gil, L. (1999). Genetic variation in central and marginal populations of Quercus suber L. Silvae Genetica, 48(6), 278–284.
Kimura, M. (1983). The neutral theory of molecular evolution. Genetics, 154, 956–958.
Kolmer, J. A., & Ordoñez, M. E. (2007). Genetic differentiation of Puccinia triticina populations in Central Asia and the Caucasus. Phytopathology, 97, 1141–1149.
Kolmer, J. A., Liu, J. Q., & Sies, M. (1995). Virulence and molecular polymorphism in Puccinia recondita f. sp. tritici in Canada. Phytopathology, 85, 276–285.
Kolmer, J. A., Long, D. L., & Hughes, M. E. (2009). Physiologic specialization of Puccinia triticina on wheat in the United States in 2007. Plant Disease, 93, 538–544.
Li, Z. Q., & Zeng, S. M. (2002). Wheat leaf rust in China. Beijing: China Agriculture Press.
Li, C., Liu, H. Y., Fan, Y. S., & Li, C. W. (2003). Advance on population genetics of plant pathogenic fungi. Journal of Agricultural University of Hebei, 26, 177–179.
Librado, P., & Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25, 1451–1452.
Liu, M., Rodrigue, N., & Kolmer, J. (2014). Population divergence in the wheat leaf rust fungus Puccinia triticina is correlated with wheat evolution. Heredity, 112, 443–453.
McDermott, J. M., & McDonald, B. A. (1993). Gene flow in plant pathosystems. Annual Review of Phytopathology, 31(1), 353–373.
Mirete, S., Vazquez, C., Mule, G., Jurado, M., & Gonzalezjaen, M. T. (2004). Differentiation of Fusarium verticillioides from banana fruits by IGS and EF-1 alpha sequence analyses. European Journal of Plant Pathology, 110(5-6), 515–523.
Morgado, L. N., Noordeloos, M. E., Lamoureux, Y., & Geml, J. (2013). Multi-gene phylogenetic analyses reveal species limits, phylogeographic patterns, and evolutionary histories of key morphological traits in Entoloma (agaricales, basidiomycota). Persoonia Molecular Phylogeny & Evolution of Fungi, 31(8), 159–178.
Mousset, S., Derome, N., & Veuille, M. (2004). A test of neutrality and constant population size based on the mismatch distribution. Molecular Biology and Evolution, 21(4), 724–731.
Parks, R., Carbone, I., Murphy, J. P., & Cowger, C. (2009). Population genetic analysis of an eastern U.S. wheat powdery mildew population reveals geographic subdivision and recent common ancestry with U.K. and Israeli populations. Phytopathology, 99, 840–849.
Rosenberg, N. A. (2004). Distruct: A program for the graphical display of population structure. Molecular Ecology Resources, 4, 137–138.
Rousset, F. (1997). Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics, 145, 1219–1228.
Singh, R. P., Huerta-Espino, J., Pfeiffer, W., & Figueroa-Lopez, P. (2004). Occurrence and impact of a new leaf rust race on durum wheat in northwestern Mexico from 2001 to 2003. Plant Disease, 88, 703–708.
Stielow, J. B., Lévesque, C. A., Seifert, K. A., Meyer, W., Iriny, L., Smits, D., et al. (2015). One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia, 35, 242–263.
Tang, L. Q., Xiao, C. L., & Wang, W. P. (2012). Research and application progress of SNP markers. Chinese Agricultural Science Bulletin, 28, 154–158.
Teacher, A. G. F., & Griffiths, D. J. (2011). HapStar: automated haplotype network layout and visualization. Molecular Ecology Resources, 11, 151–153.
Xu, D. Q., Huang, G. H., Yang, W. X., & Liu, D. Q. (2002). Analysis of the viurlence and molecular polymoprhism of Puccinia recondita f.sp tritici. Joumalof Agricultural Biotechnology, 10(1), 41–45.
Xu, M. Q., Wang, S., Meng, Q. F., & Liu, D. Q. (2013). Genetic diversity of Puccinia triticina by SSR in some regions of China. Journal of Agricultural Biotechnology, 21, 89–96.
Zhang, J., Wang, J. Y., Chen, Y., Feng, H. M., Wu, Y. Y., & Wang, P. Z. (2014). Clustering analysis and genetic relationship analysis of Banana Germplasm resources based on the model. Chinese Journal of Tropical Crops, 35(2), 232–238.
Zhao, L. B., Yuan, J. S., Cao, K. Q., Wang, H. R., & Zhang, Z. (1994). Thalictrum Aquilegifolium of wheat leaf rust’s alternate host. Acta Phytopatholgica Sinica, 24, 219–222.
Zheng, D. Y., Zhang, M., Liu, T. G., Gao, L., & Chen, W. Q. (2008). DNA extraction of urediniospores of wheat leaf rust in: Annual meeting, Chinese Society of Plant Pathology. China: Chongqing.
Acknowledgments
This work was funded by grants from the National Natural Science Foundation of China (No. 31671967), National Key Research and Development Program of China (No. 2016YFD0300705), and China Agriculture Research System (CARS-3). We thank Shouqin Yang for reproducing and maintaining the P. triticina spores in the greenhouse.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human and animal rights and informed consent
No human participants and/or animals are involved in this research.
Ethical approval
The authors declare that they have followed the guidelines of Committee on Publication Ethics (COPE) and obeyed all the Ethical Standards requested by EJPP.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, Y., Liu, T., Liu, B. et al. Population genetic structures of Puccinia triticina in five provinces of China. Eur J Plant Pathol 156, 1135–1145 (2020). https://doi.org/10.1007/s10658-020-01956-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-020-01956-4