
Abstract
Light-at-night triggers the decline of pineal gland melatonin biosynthesis and secretion and is an IARC-classified probable breast-cancer risk factor. We applied a large-scale molecular epidemiology approach to shed light on the putative role of melatonin in breast cancer. We investigated associations between breast-cancer risk and polymorphisms at genes of melatonin biosynthesis/signaling using a study population of 44,405 women from the Breast Cancer Association Consortium (22,992 cases, 21,413 population-based controls). Genotype data of 97 candidate single nucleotide polymorphisms (SNPs) at 18 defined gene regions were investigated for breast-cancer risk effects. We calculated adjusted odds ratios (ORs) and 95% confidence intervals (CI) by logistic regression for the main-effect analysis as well as stratified analyses by estrogen- and progesterone-receptor (ER, PR) status. SNP-SNP interactions were analyzed via a two-step procedure based on logic regression. The Bayesian false-discovery probability (BFDP) was used for all analyses to account for multiple testing. Noteworthy associations (BFDP < 0.8) included 10 linked SNPs in tryptophan hydroxylase 2 (TPH2) (e.g. rs1386492: OR = 1.07, 95% CI 1.02–1.12), and a SNP in the mitogen-activated protein kinase 8 (MAPK8) (rs10857561: OR = 1.11, 95% CI 1.04–1.18). The SNP-SNP interaction analysis revealed noteworthy interaction terms with TPH2- and MAPK-related SNPs (e.g. rs1386483R ∧ rs1473473D ∧ rs3729931D: OR = 1.20, 95% CI 1.09–1.32). In line with the light-at-night hypothesis that links shift work with elevated breast-cancer risks our results point to SNPs in TPH2 and MAPK-genes that may impact the intricate network of circadian regulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the most common cancer and the leading cause of cancer death for women worldwide with a higher incidence among women in developed countries [1]. Besides several reproductive and lifestyle-associated risk factors [2], exposure to light-at-night has been suggested to promote breast cancer [3]. In 2007, the International Agency for Research on Cancer (IARC) classified shift work that includes circadian disruption as probably carcinogenic to humans (group 2A) [4]. In their 2019 re-evaluation the IARC confirmed and specified this classification to night-shift work. Studies on the effects of light in animal bioassays were key to this evaluation [5]. Although risk estimates between epidemiological studies vary due to different exposure assessments and study populations, a large pooled analysis of case–control studies confirmed the association between a high number of night shifts and breast cancer [6].
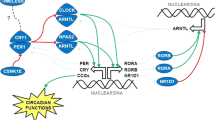
The circadian system is orchestrated by a multisynaptic pathway that is governed by a master clock, the suprachiasmatic nucleus (SCN), located in the hypothalamus. Following photic input, the pathway is set into operation via the retino-hypothalamic tract by intrinsically photosensitive retinal ganglion cells. The light signal is directly projected into the SCN to finally synapse with the pineal gland via complex networks including the sympathetic nervous system, superior cervical ganglions as well as other participating hypothalamic areas (paraventricular nucleus, PVN) [7, 8]. In the circadian clock, mitogen-activated protein kinase (MAPK) pathways function both as input pathways to maintain or reset the oscillator to 24 h environmental cycles, and output pathways that connect the timekeeping oscillator through control of the expression of a large number of functionally related genes [9]. Several variants in circadian genes have been linked to general breast-cancer susceptibility [10, 11].
Melatonin (N-acetyl-5-methoxytryptamine) is the key-player for this synchronization of bodily circadian rhythms. Its biosynthesis follows a multistep process starting with the hydroxylation of the precursor molecule L-tryptophan catalyzed by tryptophan hydroxylase (TPH). Decarboxylation of 5-hydroxy-L-tryptophan by L-aromatic amino acid decarboxylase (AADC) results in the neurotransmitter serotonin, the acetylation of which by aralkylamine N-acetyltransferase (AANAT) and methylation by N-acetylserotonin O-methyltransferase (ASMT, alias HIOMT) finally yields melatonin [12]. During darkness, AANAT activity increases via phosphorylation thereby blocking its proteasomal proteolysis, and its high affinity to serotonin leads to a strong increase in melatonin production [12].
Melatonin is mainly secreted from the pineal gland upon photic neural input, but also produced by other ocular tissues such as photoreceptors and ciliary body epithelium, albeit to a lesser extent, as well as other bodily tissues [13, 14]. With its secretion being affected by the light–dark cycle, melatonin synchronizes bodily circadian rhythms relevant to many endogenous processes including the production of sex hormones [15, 16]. The desynchronization of SCN activity either by day length or timing/phasing of light exposure consequently affects the production of melatonin by the pineal gland and is referred to as circadian disruption [3, 17].
The light-at-night-associated breast-cancer risk has been attributed to a reduced nocturnal biosynthesis and lower secretion of melatonin [3, 17]. In particular, an increased risk for hormone-sensitive breast cancer has been mechanistically accredited to a modified crosstalk between melatonin-receptor and estrogen-receptor pathways triggered upon reduced melatonin and modulated estrogen exposure [18]. Here we investigated the putative contribution of genetic polymorphisms of key enzymes of melatonin biosynthesis and signaling to the risk of developing breast cancer, and highlight a cooperative role in favor of this risk based on a large international association study of more than 44,000 breast-cancer cases and controls.
Material and methods
Study population
We screened 106,621 breast-cancer cases and control subjects with available pheno- and genotype data deposited in the database of the Breast Cancer Association Consortium (BCAC) [19, 20] at the University of Cambridge. Exclusion criteria at the study and individual level are specified in Fig. 1. Studies were not included if reference age (age at diagnosis for cases, age at interview for controls) was missing in > 30% of the study participants, relevant epidemiological variables were not recorded, or controls were not population-based or a case-only design was used. All subjects had to be women as well as of European descent, and cases were required to have a diagnosis of primary breast cancer. Based on these criteria, 44,405 eligible women (22,992 cases and 21,413 controls) from 14 population-based case–control studies were included in the analysis. Individual study descriptions are given in Supplementary table S1. All studies were approved by local ethics committees and all participants gave informed consent.

Flow chart of BCAC data set for the inclusion of case-control studies in the analysis, detailed information on individual studies is provided in Suppl. table S1
Polymorphisms and genotype data
We focused on 97 single nucleotide polymorphisms (SNPs) at 18 genes including melatonin biosynthesis (e.g. TPH1 and TPH2), melatonin receptors (MTNR1A and MTNR1B) as well as various MAP kinases (e.g. MAP2K1, MAP2K2, MAPK1, MAPK8). All 97 SNPs are listed in Supplementary table S2 together with their characteristics in the study population. Corresponding genotypes were retrieved from the BCAC database Cambridge. They were previously generated within the framework of the Collaborative Oncological Gene-environment Study (COGS) using a custom Illumina iSelect array with 211,155 SNPs as described elsewhere [19]. For the SNP selection, all available SNPs on the array at the aforementioned genes were considered.
Statistical analysis
Quality criteria
We checked for Hardy–Weinberg-Equilibrium (HWE) by Χ2-tests and analyzed the heterogeneity between studies by calculating Cochran’s Q for the heterozygous and homozygous rare genotypes for each SNP (Suppl. table S2). To consider multiple testing, we used 0.05 divided by the number of analyzed SNPs as threshold for p values.
Main-effect analysis and confounder selection
For the main-effect analysis of each SNP, we estimated odds ratios (ORs) and 95% confidence intervals (CIs) for each SNP assuming different genetic models (dominant, recessive, and additive model). All models were adjusted for reference age, study, and a set of eight principal components to consider a possible population stratification effect. Furthermore, we adjusted these models for parity (nulliparous/1+ full term pregnancies/unknown), breast-feeding status (never/ever/unknown), smoking status (never/ever/unknown), and current use of estrogen-progesterone combined menopausal hormone therapy (MHT) (no/yes/unknown). Regarding menopausal hormone therapy, current use was defined as ‘use at reference date or within six months prior to the reference date’. Missing values in categorical covariates were coded as ‘unknown’. In a sensitivity analysis, we additionally adjusted the models for menopausal status (pre- and peri-menopausal/post-menopausal/unknown). Post-menopausal was defined as ‘last menstruation more than 12 months before the reference date’. We also calculated ORs separately for estrogen-receptor (ER) positive/negative (ER±) and progesterone-receptor (PR) positive/negative (PR±) cases.
Interaction analyses with logic regression
To analyze interactions between SNPs, we used a two-staged procedure based on logic regression models [21]. In short, a logic regression model is a so-called logic tree embedded in a generalized linear model. The logic tree consists of binary covariates linked by logic expressions with the AND-expression (conjunction) representing interactions (notation of interactions: A \(\wedge\) B (A and B); C \(\wedge\) !D (C and not D)). An optimization algorithm is used to select interactions for the logic tree. Here, we used the logit as link function of the framing generalized linear model with the case–control status as outcome and the simulated annealing algorithm to select interactions for the logic tree as independent variable. To express SNP-SNP interactions in logic regression models, SNP coding in the dominant and recessive genetic model was required [21].
In the first stage of our procedure, we selected interactions for the logic tree by using the logic Feature Selection (logicFS) algorithm to avoid overfitting [21]. Here, we used logicFS to fit 100 logic regression models from bootstrap samples and to calculate a variable importance measure for the multiple tree approach based on the number of correctly classified out-of-bag observations for each bootstrap sample for every interaction consisting of up to six terms included in these models [21]. We ran the algorithm three times with a different random seed and selected the 20 most important interactions each. In the second stage, we fitted individual adjusted logistic regression models with the selected terms.
To account for multiple testing and an increased type I error rate, we calculated the Bayesian false-discovery probability (BFDP) for SNPs/interaction terms with a p value < 0.05 in the adjusted models, assuming a four-fold cost of a false non-discovery compared to a false discovery as suggested by Wakefield [22]. Effects with BFDP < 0.8 are termed noteworthy. We calculated the BFDP for three different prior probabilities (0.1, 0.05, 0.01) for a true association and the OR corresponding to the 97.5% quantile of the prior OR was set to 1.2 for positive associations and to 0.83 for negative associations. Linkage disequilibrium was checked for noteworthy SNPs.
The statistical software R, version 3.4.2, was used for all calculations [23]. The R-packages ‘logicFS’ and ‘LogicReg’ were used for the interaction analysis [24, 25]. All statistical models were fitted as complete-case analyses, including the category ‘unknown’ for missing values in categorical variables, therefore the number of individuals available for calculations varied respectively. This also accounts for slight differences in ORs between the main-effect analysis and the interaction analysis, when an interaction term consists of only one SNP.
Results
The study population of 44,405 women was contributed by 14 case–control studies (Suppl. table S1) of which the smallest study comprised 243 women (NBHS) and the largest 16,746 women (SEARCH). Among the eligible 22,992 cases and 21,413 controls, the mean reference age was 56 years for cases and 57 years for controls with a standard deviation of 10 and 9 years, respectively. Most women had at least one full term pregnancy (76%) and nearly 50% had ever breastfed (Table 1).
Most of the 97 analyzed SNPs had very few missing values, with a maximum of 4% for rs10217741 (RORB). The minor allele frequency (MAF) in controls ranged from 2–49%. Detailed information for all SNPs is provided in Supplementary table S2. HWE was not met for rs10765576 (MTNR1B) and rs14303 (MAP2K1), and therefore, these polymorphisms were not followed up further.
Main-effect analysis
Noteworthy associations (BFDP < 0.8) between individual SNPs and breast-cancer risk have been identified particularly for TPH2 intronic polymorphisms. ORs (95% CIs) of individual SNPs with a p value < 0.05 and respective BFDPs with different priors (0.1, 0.05, and 0.01) for an effect in the adjusted dominant or recessive model are given in Table 2. ORs and CIs for all SNPs and models are given in Supplementary table S3. The largest OR (adjusted for study, reference age, parity, breast feeding, smoking status, current intake of estrogen-progesterone MHT, and principle components) was observed for recessive rs10857561 (MAPK8, OR = 1.11, 95% CI 1.04–1.18, BFDP < 0.8 at prior 0.01). Increasing the prior to 0.05 also revealed eight linked noteworthy dominant TPH2 SNPs (rs7300641, rs1386492, rs1473473, rs4760751, rs1487276, rs1386489, rs1487281, rs7299582) with similar adjusted ORs around 1.07 (95% CIs 1.02–1.12) in the dominant model. At a prior of 0.1, protective effects were revealed for two TPH2 SNPs in the recessive model (rs17110627, OR = 0.91, 95% CI 0.83–0.99; and rs2129575, OR = 0.91, 95% CI 0.83–0.99), as well as recessive rs13515 (MAPK1, OR = 0.89, 95% CI 0.79–0.99). Recessive rs7075976 (MAPK8, OR = 1.06, 95% CI 1.01–1.12) was also noteworthy. The additive model showed similar results (Suppl. table S3). A sensitivity analysis with additional adjustment for menopausal status did not change the results. When we compared our results with those obtained in the meta GWAS analysis, none of the noteworthy SNP associations reported here were identified at the genome wide association level (5E-08) [20, 26, 27].
Analysis by tumor hormone-receptor status
Tumor hormone-receptor status was ER-positive (ER+) for 14,724 patients and ER-negative (ER−) for 3516 patients, as well as PR-positive (PR+) for 10,016 patients and PR-negative (PR−) for 4,768 patients (Table 1). Noteworthy breast-cancer risk associations (BFDP < 0.8) in the ER+ , ER−, PR+ , and PR− subgroups for the dominant and recessive model are listed in Table 3. Results for all SNPs and models are given in Supplementary table S4. None of the four tumor-hormone-receptor subgroups showed noteworthy associations at a prior of 0.01. At a prior of 0.05, six SNPs showed noteworthy associations. These comprised MAPK8 SNP rs10857561 in the ER+ subgroup (OR = 1.10, 95% CI 1.02–1.18; recessive model) and two tightly linked TPH2 SNPs rs7300641 and rs1386492 in the PR+ group (both: OR = 1.07, 95% CI 1.02–1.13; dominant model). In the ER− subgroup the two linked TPH2 SNPs rs1473473 and rs1487276 (both: OR = 1.12, 95% CI 1.04–1.22; dominant model) and the RORA SNP rs17237290 (OR = 0.85, 95% CI 0.73–0.97; dominant model) showed noteworthy associations, while none of the associations in the PR− subgroup were noteworthy at prior 0.05. Besides rs17237290, the variants mentioned above were also noteworthy in the main analysis at prior 0.05 under the identical genetic models. At a prior of 0.1, in total 25 noteworthy associations of 19 SNPs were observed for all four breast cancer subtypes.
Interaction analysis
The 20 most important interaction terms each from three different starting seeds for the logicFS algorithm resulted in 53 unique interactions terms (Suppl. table S5). The adjusted logistic regression models for these terms yielded ten interaction terms with a p-value < 0.05 (Suppl. table S6), hence suitable for BFDP calculation.
With a prior probability for an effect of 0.01, we found three noteworthy interaction terms in the adjusted models (BFDP < 0.8, Table 4): rs10857561R ∧ !rs1347069D (OR = 1.15, 95% CI 1.05–1.25), rs10857561R, (OR = 1.11, 95% CI 1.04–1.18), and rs1386483R ∧ rs1473473D ∧ rs3729931D (OR = 1.20, 95% CI 1.09–1.32). With increased priors of 0.05 and 0.1, in total six and eight interaction terms reached noteworthiness, respectively. All noteworthy interactions (all priors) included at least one SNP that showed noteworthy associations, respectively, in the main effect analysis (TPH2: rs1386489, rs1473473, rs7299582; MAPK8: rs10857561, rs7075976).
Discussion
This hypothesis-based breast-cancer association study focused on the putative role of modulators of the pineal gland hormone melatonin and their potential influence on breast-cancer risk. In line with the light-at-night hypothesis, according to which altered light-induced nocturnal melatonin production and signaling increases the risk of breast cancer [3], our findings point to a cooperative role of genetic variations that may modulate serotonergic brain networks and/or the signaling of melatonin within the context of breast-cancer susceptibility.
The strongest observed risk effects were driven by various interactions of polymorphisms at TPH2 and MAPK genes (MAPK8, MAP2K1, RAF1). The triple interaction of TPH2 rs1386483 and rs1473473 as well as RAF1 rs3729931 increased breast-cancer risk by 20%, a dual interaction of MAPK8 rs10857561 and MAP2K1 rs1347069 by 15%, and MAPK8 rs10857561 alone by 11% (all observed at a prior probability of 0.01). In most instances, risk effects were evident at the individual SNP level both, in main and stratified risk analyses by hormone-receptor status. To the best of our knowledge and based on the combined iCOGs/Oncoarray meta-analysis of the BCAC cohort [20] as well as the Catalogue of Curated Breast Cancer Genes [28], these breast-cancer risk associations are newly described. Yet, some TPH2 polymorphisms have been reported in the literature within the context of psychiatric disorder related endpoints such as antidepressant response and GABA concentration, conditions in which effects of serotonin are underlying biological mechanisms [29, 30].
The involvement of TPH2 as a putative breast cancer susceptibility locus is plausible, as the enzyme is exclusively expressed in neuronal cells of the central nervous system where it catalyzes the rate limiting step in serotonin (5-HT) biosynthesis, the chemical precursor of melatonin [31]. Its pertinent role in females has been inferred from expression studies of post mortem brain tissues in which female thalamic and hypothalamic brain showed higher TPH2 mRNA expression levels compared to male counterparts [32], thereby highlighting the critical role of an intact serotonergic pathway for female neurohormone/neurotransmitter production. In general, TPH2 is present in various brain regions with particular abundance in the major central serotonergic neuronal networks that localize to median and dorsal raphe nuclei in the brainstem known to participate in basal functions such as temperature regulation, feeding and energy balance, as well as mood and sleep [33, 34]. The synthesis and periodical secretion of serotonin from these brain stem regions and neuronal terminal fields are regulated at the level of TPH2 gene expression that was shown to be under the circadian control of melatonin, estrogen, and corticoids in rodents and primates [34, 35]. This underscores the various feedback mechanisms to the serotonergic system that are involved in the entrainment of the hypothalamic SCN [7, 36, 37]. To a smaller extent, TPH2 is also rhythmically expressed in ocular tissues with rhythmical release of melatonin, the levels of which are highest in darkness and lowest in the light [8, 38]. Intrinsic photosensitive retinal ganglion cells by virtue of their concurrent rhythmic melatonin-receptor expression may therefore contribute to the output signal of the retino-thalamic tract to the SCN [39]. Hence, our findings of an association between TPH2 variants and increased breast-cancer risk are well in line with the notion that such variants impact on serotonin formation, thereby disrupting the SCN pacemaker circuitry feedback. In particular, the observed increased breast-cancer risk in TPH2 variant carriers may result from modified brain melatonin levels due to a dysfunctional SCN (together with reduced melatonin levels in the retina) that upon light-at-night periods may reduce pineal melatonin secretion.
If the disruption of pineal gland melatonin biosynthesis during extended light-at-night periods affects nocturnal serotonin levels and transmission to the hypothalamic nuclei SCN and PVN [7, 40], we need to consider further that these nuclei also make up major parts of the hypothalamic-pituitary–gonadal (HPG) axis. In the HPG axis, serotonin modulates the hypothalamic secretion of the gonadotropin releasing hormone (GnRH) as well as the pituitary luteinizing and follicle stimulating hormones (LH, FSH), and prolactin that stimulate the production of sex hormones in peripheral target organs such as the ovaries or testes [41]. Similar to melatonin, prolactin secretion may be driven by the central circadian pacemaker located in the SCN of the hypothalamus [42], and our observed TPH2 variant-associated increased breast-cancer risk may in addition relate to local serotonergic effects accountable for increased prolactin production in the anterior pituitary gland. Of note, circulating prolactin is an established breast-cancer risk factor that has been confirmed in a series of analyses from the prospective Nurses’ Health Study, particularly with respect to ER-positive breast cancer [43], and a large analysis from the prospective EPIC cohort [44] with emphasis on users of hormone replacement therapy. This is underpinned by a vast body of evidence from animal and in vitro studies. Together with estradiol and progesterone, it exerts effects on normal epithelial cell expansion, ductal side branching of the breast during puberty, and formation of lobuloalveolar structures during pregnancy [45]. As prolactin and progesterone have synergic roles to induce cell growth and proliferation during adult gland maturation and alveologenesis of the breast terminal duct-lobular units, the site of origin for most breast cancers, a crosstalk between progesterone, prolactin and receptor signaling pathways may not only be relevant in normal, but also malignant breast cells [46].
MAPK8 (JNK, c-Jun N-terminal kinase) in our study has been associated with breast-cancer risk at rs10857561, both in the individual main and hormone-receptor positive (ER + and PR +) stratified analyses, as well as in the SNP-SNP interaction analyses. While all three canonical MAPK pathways (ERK MAPK, p38 MAPK, and JNK MAPK) serve as input to the circadian clock in distinct ways [9], JNK in particular has been shown to be essential for the normal oscillation of the mammalian circadian clock and its photic regulation. The JNK-imparted transmission of light signals to the BMAL1-CLOCK complex controls oscillation speed and phase response of the master clock [9].
Aside from their role as master clock regulators via SCN or peripheral tissue, MAPK genes play a role in cellular processes like proliferation and cell death [47]. It is therefore not surprising that they were associated with the development of cancer at many sites [48]. While rs10857561 in MAPK8 showed noteworthy risk estimates for breast cancer in general as well as ER-positive and PR-positive subgroups in our analyses, this SNP has been previously shown to be associated with rectal cancer once more highlighting the role in carcinogenesis [49].
A major strength of this study is the large number of study participants (22,992 cases and 21,413 controls) retrieved from population-based case–control studies with defined reference age (age at diagnosis for cases, age at interview for controls) and availability of comprehensive epidemiological and tumor immunohistochemistry as well as genotype data. This allowed us to calculate precise SNP-specific OR main effect and interaction estimates. Our hypothesis-driven approach of a putative role of polymorphic regulators or signaling mediators of melatonin in breast-cancer risk limited the number of potentially detectable false-positive associations. Moreover, we used the BFDP to measure the noteworthiness of our effect estimates and to account for false-positive results via multiple testing. Furthermore, our interaction analysis based on logic regression models enabled us to model the effects of complex interaction scenarios considering the multivariate structure of SNP interplay.
In spite of the large study size, limitations of the study include a high number of missing values in included confounders. We used the category ‘unknown’ for categorical variables in these participants to maintain the remaining information. Moreover, the sample size in subgroup analyses was reduced, for example, ER status was missing for 21% of samples and PR status for 36% of samples. However, missingness is likely to be random with respect to genotypes. There was also minor heterogeneity in definition of stage, grade, and cut-off levels for ER and PR across studies. The tumor subtypes were strictly defined by immunohistochemical markers as other data, such as intrinsic subtypes from expression profiles nowadays used for subtype definition, are not available in large-scale epidemiological studies. Finally, our interpretations strictly rely on functional and physiological data reported in the literature and include in vitro, animal in vivo as well as post mortem findings.
Our newly identified breast-cancer risk associations justify continued research into the relationship between breast-cancer risk and putative modulators of the intricate network of rhythmic circadian regulators such as melatonin and serotonin upon photic stimulation at night. The observed interactions between genetic variants of TPH2 and MAPK8 highlight their cooperation as putative breast-cancer risk modulators and call upon the comprehensive scrutiny of the circadian clock system and its input and output effectors in large breast-cancer cohorts. This research holds the potential to reveal new insights into the breast-cancer risk of women exposed to light-at-night which is particularly relevant for female night shift workers.
Data availability
The datasets analyzed in the current study are available via the BCAC Data Access Coordinating Committee (bcac@medschl.cam.ac.uk) upon reasonable request. Summary-level genotype data are available via http://bcac.ccge.medschl.cam.ac.uk and in supplementary table S2. Individual-level data are available via the BCAC Data Access Coordinating Committee (bcac@medschl.cam.ac.uk).
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49. https://doi.org/10.3322/caac.21660.
Momenimovahed Z, Salehiniya H. Epidemiological characteristics of and risk factors for breast cancer in the world. Breast Cancer (Dove Med Press). 2019;11:151–64. https://doi.org/10.2147/BCTT.S176070.
Stevens RG, Brainard GC, Blask DE, Lockley SW, Motta ME. Breast cancer and circadian disruption from electric lighting in the modern world. CA Cancer J Clin. 2014;64:207–18. https://doi.org/10.3322/caac.21218.
Straif K, Baan R, Grosse Y, Secretan B, Ghissassi FE, Bouvard V, et al. Carcinogenicity of shift-work, painting, and fire-fighting. Lancet Oncol. 2007;8:1065–6. https://doi.org/10.1016/S1470-2045(07)70373-X.
Ward EM, Germolec D, Kogevinas M, McCormick D, Vermeulen R, Anisimov VN, et al. Carcinogenicity of night shift work. Lancet Oncol. 2019;20:1058–9. https://doi.org/10.1016/S1470-2045(19)30455-3.
Cordina-Duverger E, Menegaux F, Popa A, Rabstein S, Harth V, Pesch B, et al. Night shift work and breast cancer: a pooled analysis of population-based case-control studies with complete work history. Eur J Epidemiol. 2018;33:369–79. https://doi.org/10.1007/s10654-018-0368-x.
Buijs FN, León-Mercado L, Guzmán-Ruiz M, Guerrero-Vargas NN, Romo-Nava F, Buijs RM. The circadian system: a regulatory feedback network of periphery and brain. Physiology (Bethesda). 2016;31:170–81. https://doi.org/10.1152/physiol.00037.2015.
Ostrin LA. Ocular and systemic melatonin and the influence of light exposure. Clin Exp Optom. 2019;102:99–108. https://doi.org/10.1111/cxo.12824.
Goldsmith CS, Bell-Pedersen D. Diverse roles for MAPK signaling in circadian clocks. Adv Genet. 2013;84:1–39. https://doi.org/10.1016/B978-0-12-407703-4.00001-3.
Reszka E, Przybek M, Muurlink O, Pepłonska B. Circadian gene variants and breast cancer. Cancer Lett. 2017;390:137–45. https://doi.org/10.1016/j.canlet.2017.01.012.
Rabstein S, Harth V, Justenhoven C, Pesch B, Plöttner S, Heinze E, et al. Polymorphisms in circadian genes, night work and breast cancer: results from the GENICA study. Chronobiol Int. 2014;31:1115–22. https://doi.org/10.3109/07420528.2014.957301.
Lv Y, Li Y, Li J, Bian C, Qin C, Shi Q. A comparative genomics study on the molecular evolution of serotonin/melatonin biosynthesizing enzymes in vertebrates. Front Mol Biosci. 2020;7:11. https://doi.org/10.3389/fmolb.2020.00011.
Claustrat B, Leston J. Melatonin: physiological effects in humans. Neurochirurgie. 2015;61:77–84. https://doi.org/10.1016/j.neuchi.2015.03.002.
Acuña-Castroviejo D, Escames G, Venegas C, Díaz-Casado ME, Lima-Cabello E, López LC, et al. Extrapineal melatonin: sources, regulation, and potential functions. Cell Mol Life Sci. 2014;71:2997–3025. https://doi.org/10.1007/s00018-014-1579-2.
Lucas RJ, Peirson SN, Berson DM, Brown TM, Cooper HM, Czeisler CA, et al. Measuring and using light in the melanopsin age. Trends Neurosci. 2014;37:1–9. https://doi.org/10.1016/j.tins.2013.10.004.
Cipolla-Neto J, Amaral FG, Soares JM, Gallo CC, Furtado A, Cavaco JE, et al. The crosstalk between melatonin and sex steroid hormones. Neuroendocrinology. 2022;112:115–29. https://doi.org/10.1159/000516148.
Hill SM, Belancio VP, Dauchy RT, Xiang S, Brimer S, Mao L, et al. Melatonin: an inhibitor of breast cancer. Endocr Relat Cancer. 2015;22:R183-204. https://doi.org/10.1530/ERC-15-0030.
Cos S, González A, Martínez-Campa C, Mediavilla MD, Alonso-González C, Sánchez-Barceló EJ. Estrogen-signaling pathway: a link between breast cancer and melatonin oncostatic actions. Cancer Detect Prev. 2006;30:118–28. https://doi.org/10.1016/j.cdp.2006.03.002.
Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, Milne RL, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. 2013;45:353–61. https://doi.org/10.1038/ng.2563.
Michailidou K, Lindström S, Dennis J, Beesley J, Hui S, Kar S, et al. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017;551:92–4. https://doi.org/10.1038/nature24284.
Schwender H, Ickstadt K. Identification of SNP interactions using logic regression. Biostatistics. 2008;9:187–98. https://doi.org/10.1093/biostatistics/kxm024.
Wakefield J. A Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am J Hum Genet. 2007;81:208–27. https://doi.org/10.1086/519024.
R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2016.
Schwender H. logicFS: Identification of SNP Interactions. 2013. https://bioconductor.org/packages/release/bioc/manuals/logicFS/man/logicFS.pdf. Accessed 18 Jan 2023.
Kooperberg C, Ruczinski I. LogicReg: Logic Regression. 2016. https://CRAN.R-project.org/package=LogicReg. Accessed 18 Jan 2023.
Zhang H, Ahearn TU, Lecarpentier J, Barnes D, Beesley J, Qi G, et al. Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. Nat Genet. 2020;52:572–81. https://doi.org/10.1038/s41588-020-0609-2.
Milne RL, Kuchenbaecker KB, Michailidou K, Beesley J, Kar S, Lindström S, et al. Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat Genet. 2017;49:1767–78. https://doi.org/10.1038/ng.3785.
Bose M, Benada J, Thatte JV, Kinalis S, Ejlertsen B, Nielsen FC, et al. A catalog of curated breast cancer genes. Breast Cancer Res Treat. 2022;191:431–41. https://doi.org/10.1007/s10549-021-06441-y.
Peters EJ, Slager SL, McGrath PJ, Knowles JA, Hamilton SP. Investigation of serotonin-related genes in antidepressant response. Mol Psychiatry. 2004;9:879–89. https://doi.org/10.1038/sj.mp.4001502.
Preuss N, Salehi B, van der Veen JW, Shen J, Drevets WC, Hodgkinson C, et al. Associations between prefrontal γ-aminobutyric acid concentration and the tryptophan hydroxylase isoform 2 gene, a panic disorder risk allele in women. Int J Neuropsychopharmacol. 2013;16:1707–17. https://doi.org/10.1017/S1461145713000254.
Walther DJ, Peter J-U, Bashammakh S, Hörtnagl H, Voits M, Fink H, Bader M. Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science. 2003;299:76. https://doi.org/10.1126/science.1078197.
Zill P, Büttner A, Eisenmenger W, Bondy B, Ackenheil M. Regional mRNA expression of a second tryptophan hydroxylase isoform in postmortem tissue samples of two human brains. Eur Neuropsychopharmacol. 2004;14:282–4. https://doi.org/10.1016/j.euroneuro.2003.10.002.
Zill P, Büttner A, Eisenmenger W, Müller J, Möller H-J, Bondy B. Predominant expression of tryptophan hydroxylase 1 mRNA in the pituitary: a postmortem study in human brain. Neuroscience. 2009;159:1274–82. https://doi.org/10.1016/j.neuroscience.2009.01.006.
Chen G-L, Miller GM. Advances in tryptophan hydroxylase-2 gene expression regulation: new insights into serotonin-stress interaction and clinical implications. Am J Med Genet B Neuropsychiatr Genet. 2012;159B:152–71. https://doi.org/10.1002/ajmg.b.32023.
Nagy AD, Iwamoto A, Kawai M, Goda R, Matsuo H, Otsuka T, et al. Melatonin adjusts the expression pattern of clock genes in the suprachiasmatic nucleus and induces antidepressant-like effect in a mouse model of seasonal affective disorder. Chronobiol Int. 2015;32:447–57. https://doi.org/10.3109/07420528.2014.992525.
Morin LP. Serotonin and the regulation of mammalian circadian rhythmicity. Ann Med. 1999;31:12–33. https://doi.org/10.3109/07853899909019259.
Moyer RW, Kennaway DJ. Serotonin depletion decreases light induced c-fos in the rat suprachiasmatic nucleus. NeuroReport. 2000;11:1021–4. https://doi.org/10.1097/00001756-200004070-00025.
Liang J, Wessel JH, Iuvone PM, Tosini G, Fukuhara C. Diurnal rhythms of tryptophan hydroxylase 1 and 2 mRNA expression in the rat retina. NeuroReport. 2004;15:1497–500. https://doi.org/10.1097/01.wnr.0000131007.59315.66.
Sengupta A, Baba K, Mazzoni F, Pozdeyev NV, Strettoi E, Iuvone PM, Tosini G. Localization of melatonin receptor 1 in mouse retina and its role in the circadian regulation of the electroretinogram and dopamine levels. PLoS ONE. 2011;6:e24483. https://doi.org/10.1371/journal.pone.0024483.
Malek ZS, Labban LM. Photoperiod regulates the daily profiles of tryptophan hydroxylase-2 gene expression the raphe nuclei of rats. Int J Neurosci. 2021;131:1155–61. https://doi.org/10.1080/00207454.2020.1782903.
Debeljuk L, Lasaga M. Tachykinins and the control of prolactin secretion. Peptides. 2006;27:3007–19. https://doi.org/10.1016/j.peptides.2006.07.010.
Waldstreicher J, Duffy JF, Brown EN, Rogacz S, Allan JS, Czeisler CA. Gender differences in the temporal organization of proclactin (PRL) secretion: evidence for a sleep-independent circadian rhythm of circulating PRL levels- a clinical research center study. J Clin Endocrinol Metab. 1996;81:1483–7. https://doi.org/10.1210/jcem.81.4.8636355.
Tworoger SS, Eliassen AH, Sluss P, Hankinson SE. A prospective study of plasma prolactin concentrations and risk of premenopausal and postmenopausal breast cancer. J Clin Oncol. 2007;25:1482–8. https://doi.org/10.1200/JCO.2006.07.6356.
Tikk K, Sookthai D, Johnson T, Rinaldi S, Romieu I, Tjønneland A, et al. Circulating prolactin and breast cancer risk among pre- and postmenopausal women in the EPIC cohort. Ann Oncol. 2014;25:1422–8. https://doi.org/10.1093/annonc/mdu150.
Macias H, Hinck L. Mammary gland development. Wiley Interdiscip Rev Dev Biol. 2012;1:533–57. https://doi.org/10.1002/wdev.35.
Lee W, Bergen AW, Swan GE, Li D, Liu J, Thomas P, et al. Gender-stratified gene and gene-treatment interactions in smoking cessation. Pharmacogenomics J. 2012;12:521–32. https://doi.org/10.1038/tpj.2011.30.
Santen RJ, Song RX, McPherson R, Kumar R, Adam L, Jeng M-H, Yue W. The role of mitogen-activated protein (MAP) kinase in breast cancer. J Steroid Biochem Mol Biol. 2002;80:239–56. https://doi.org/10.1016/s0960-0760(01)00189-3.
Burotto M, Chiou VL, Lee J-M, Kohn EC. The MAPK pathway across different malignancies: a new perspective. Cancer. 2014;120:3446–56. https://doi.org/10.1002/cncr.28864.
Slattery ML, Lundgreen A, Wolff RK. MAP kinase genes and colon and rectal cancer. Carcinogenesis. 2012;33:2398–408. https://doi.org/10.1093/carcin/bgs305.
Acknowledgements
We thank all the individuals who took part in these studies and all the researchers, clinicians, technicians and administrative staff who have enabled this work to be carried out. ABCFS thank Maggie Angelakos, Judi Maskiell, Gillian Dite. ESTHER thanks Hartwig Ziegler, Sonja Wolf, Volker Hermann, Christa Stegmaier, Katja Butterbach. GENICA thanks Christian Baisch, Hans-Peter Fischer, Anne Lotz, and Beate Pesch. KARMA and SASBAC thank the Swedish Medical Research Counsel. MARIE thanks Petra Seibold, Sabine Behrens, Ursula Eilber and Muhabbet Celik. MTLGEBCS would like to thank Martine Tranchant (CHU de Québec – Université Laval Research Center), Marie-France Valois, Annie Turgeon and Lea Heguy (McGill University Health Center, Royal Victoria Hospital; McGill University) for DNA extraction, sample management and skilful technical assistance. J.S. is Chair holder of the Canada Research Chair in Oncogenetics. NBHS thanks study participants and research staff for their contributions and commitment to the study. The OFBCR thanks Teresa Selander, Nayana Weerasooriya and Steve Gallinger. PBCS thanks Louise Brinton, Mark Sherman, Neonila Szeszenia-Dabrowska, Beata Peplonska, Witold Zatonski, Pei Chao, Michael Stagner. SBCS thanks Sue Higham, Helen Cramp, Dan Connley, Ian Brock, Sabapathy Balasubramanian and Malcolm W.R. Reed. We thank the SEARCH team.
Funding
Open Access funding enabled and organized by Projekt DEAL. BCAC is funded by Cancer Research UK grant: PPRPGM-Nov20\100002, the European Union's Horizon 2020 Research and Innovation Programme (grant numbers 634935 and 633784 for BRIDGES and B-CAST respectively), and the PERSPECTIVE I&I project, funded by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research, the Ministère de l’Économie et de l'Innovation du Québec through Genome Québec, the Quebec Breast Cancer Foundation. The EU Horizon 2020 Research and Innovation Programme funding source had no role in study design, data collection, data analysis, data interpretation or writing of the report. Additional funding for BCAC is provided via the Confluence project which is funded with intramural funds from the National Cancer Institute Intramural Research Program, National Institutes of Health. Genotyping of the OncoArray was funded by the NIH Grant U19 CA148065, and Cancer Research UK Grant C1287/A16563 and the PERSPECTIVE project supported by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research (grant GPH-129344) and, the Ministère de l’Économie, Science et Innovation du Québec through Genome Québec and the PSRSIIRI-701 grant, and the Quebec Breast Cancer Foundation. Funding for iCOGS came from: the European Community's Seventh Framework Programme under grant agreement n° 223175 (HEALTH-F2-2009-223175) (COGS), Cancer Research UK (C1287/A10118, C1287/A10710, C12292/A11174, C1281/A12014, C5047/A8384, C5047/A15007, C5047/A10692, C8197/A16565), the National Institutes of Health (CA128978) and Post-Cancer GWAS initiative (1U19 CA148537, 1U19 CA148065 and 1U19 CA148112—the GAME-ON initiative), the Department of Defence (W81XWH-10-1-0341), the Canadian Institutes of Health Research (CIHR) for the CIHR Team in Familial Risks of Breast Cancer, and Komen Foundation for the Cure, the Breast Cancer Research Foundation, and the Ovarian Cancer Research Fund. The Australian Breast Cancer Family Study (ABCFS) was supported by grant UM1 CA164920 from the National Cancer Institute (USA). The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the USA Government or the BCFR. The ABCFS was also supported by the National Health and Medical Research Council of Australia, the New South Wales Cancer Council, the Victorian Health Promotion Foundation (Australia) and the Victorian Breast Cancer Research Consortium. John L. Hopper is a National Health and Medical Research Council (NHMRC) Senior Principal Research Fellow. Melissa C. Southey is a NHMRC Senior Research Fellow. The CECILE study was supported by Fondation de France, Institut National du Cancer (INCa), Ligue Nationale contre le Cancer, Agence Nationale de Sécurité Sanitaire, de l'Alimentation, de l'Environnement et du Travail (ANSES), Agence Nationale de la Recherche (ANR). The ESTHER study was supported by a grant from the Baden Württemberg Ministry of Science, Research and Arts. Additional cases were recruited in the context of the VERDI study, which was supported by a grant from the German Cancer Aid (Deutsche Krebshilfe). The GENICA was funded by the Federal Ministry of Education and Research (BMBF) Germany grants 01KW9975/5, 01KW9976/8, 01KW9977/0 and 01KW0114, the Robert Bosch Foundation, Stuttgart, Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, the Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, as well as the Department of Internal Medicine, Johanniter GmbH Bonn, Johanniter Krankenhaus, Bonn, Germany. The KARMA study was supported by Märit and Hans Rausings Initiative Against Breast Cancer. The MARIE study was supported by the Deutsche Krebshilfe e.V. [70-2892-BR I, 106332, 108253, 108419, 110826, 110828], the Hamburg Cancer Society, the German Cancer Research Center (DKFZ) and the Federal Ministry of Education and Research (BMBF) Germany [01KH0402]. The work of MTLGEBCS was supported by the Quebec Breast Cancer Foundation, the Canadian Institutes of Health Research for the “CIHR Team in Familial Risks of Breast Cancer” program – grant # CRN-87521 and the Ministry of Economic Development, Innovation and Export Trade – grant # PSR-SIIRI-701. The NBHS was supported by NIH grant R01CA100374. Biological sample preparation was conducted the Survey and Biospecimen Shared Resource, which is supported by P30 CA68485. The Ontario Familial Breast Cancer Registry (OFBCR) was supported by grant U01CA164920 from the USA National Cancer Institute of the National Institutes of Health. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the USA Government or the BCFR. The PBCS was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. The SASBAC study was supported by funding from the Agency for Science, Technology and Research of Singapore (A*STAR), the US National Institute of Health (NIH) and the Susan G. Komen Breast Cancer Foundation. The SBCS was supported by Sheffield Experimental Cancer Medicine Centre and Breast Cancer Now Tissue Bank. SEARCH is funded by Cancer Research UK [C490/A10124, C490/A16561] and supported by the UK National Institute for Health Research Biomedical Research Centre at the University of Cambridge. The University of Cambridge has received salary support for Paul D.P. Pharoah from the NHS in the East of England through the Clinical Academic Reserve. The SZBCS was supported by Grant PBZ_KBN_122/P05/2004 and the program of the Minister of Science and Higher Education under the name "Regional Initiative of Excellence" in 2019–2022 project number 002/RID/2018/19 amount of financing 12 000 000 PLN.
Author information
Authors and Affiliations
Contributions
Conception and design: VH, HB, TB, SR. Provision of data: RH, WYL, PG, TT, ILA, HB, JCC, AC, SSC, KC, ME, JDF, MGC, MSG, UH, WH, BH, JLH, AJ, YDK, JL, AMM, NO, VR, MS, XOS, JS, MCS, WZ, PH, HB, TB. Data management: MKB, QW, JD, KM, ML, AMD, PDPP, DFE. Statistical analyses: KW, KI, SW, RH, CT, SR. Manuscript writing: KW, RH, TB, SW, TB, VH, HB, SR. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Hermann Brenner is a member of the Editorial Board. Otherwise, the authors report no conflicts of interest.
Ethics approval and consent to participate
All studies were approved by local ethics committees and all participants gave informed consent.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Katharina Wichert, Reiner Hoppe, Thomas Behrens, Stefan Winter, Thomas Brüning, Hiltrud Brauch, Volker Harth, Sylvia Rabstein are the members of the writing group.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wichert, K., Hoppe, R., Ickstadt, K. et al. Polymorphisms in genes of melatonin biosynthesis and signaling support the light-at-night hypothesis for breast cancer. Eur J Epidemiol 38, 1053–1068 (2023). https://doi.org/10.1007/s10654-023-01048-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10654-023-01048-7