
Abstract
Despite increasing therapeutic options to treat rheumatoid arthritis (RA), many patients fail to reach treatment targets. The use of antidiabetic drugs like thiazolidinediones has been associated with lower RA risk. We aimed to explore the repurposing potential of antidiabetic drugs in RA prevention by assessing associations between genetic variation in antidiabetic drug target genes and RA using Mendelian randomization (MR). A two-sample MR design was used to estimate the association between the antidiabetic drug and RA risk using summary statistics from genome-wide association studies (GWAS). We selected independent genetic variants from the gene(s) that encode the target protein(s) of the investigated antidiabetic drug as instruments. We extracted the associations of instruments with blood glucose concentration and RA from the UK Biobank and a GWAS meta-analysis of clinically diagnosed RA, respectively. The effect of genetic variation in the drug target(s) on RA risk was estimated by the Wald ratio test or inverse-variance weighted method. Insulin and its analogues, thiazolidinediones, and sulfonylureas had valid genetic instruments (n = 1, 1, and 2, respectively). Genetic variation in thiazolidinedione target (gene: PPARG) was inversely associated with RA risk (odds ratio [OR] 0.38 per 0.1mmol/L glucose lowering, 95% confidence interval [CI] 0.20–0.73). Corresponding ORs (95%CIs) were 0.83 (0.44–1.55) for genetic variation in the targets of insulin and its analogues (gene: INSR), and 1.12 (0.83, 1.49) 1.25 (0.78-2.00) for genetic variation in the sulfonylurea targets (gene: ABCC8 and KCNJ11). In conclusion, genetic variation in the thiazolidinedione target is associated with a lower RA risk. The underlying mechanisms warrant further exploration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory joint disease affecting around 0.5 to 1.0% of the adult population [1]. Therapeutic drugs against RA include synthetic and biological disease-modifying anti-rheumatic drugs (DMARDs), and glucocorticoids [2]. Despite available treatment options, a significant proportion of patients with RA fail to reach low disease activity or remission [2]. Because of lower accessibility to biological DMARDs in low-income countries, the true failure rate may be higher [3].
There are unmet preventive and therapeutic needs for RA. De novo drug discovery can be time- and resource-consuming. Drug repurposing is an alternative, which aims to apply licensed drugs, with well-studied safety and pharmacokinetic profiles, to new indications [4]. Established RA has been associated with impaired glucose metabolism, especially insulin resistance [5]. Major drugs to improve glucose metabolism include metformin as the first-line treatment, and glucagon-like peptide-1 (GLP-1) receptor agonists, sodium-glucose cotransporter-2 (SGLT2) inhibitors, dipeptidyl peptidase 4 (DPP-4) inhibitors, insulin and its analogues, thiazolidinediones and sulfonylureas as second-line options [6]. Metformin use was reported to be associated with a decreased risk of developing RA [7]. Likewise, the use of thiazolidinediones, a drug class activating peroxisome proliferator-activated receptor-γ (PPARγ) to reduce glucose concentration and increase insulin sensitivity, was associated with a reduced RA risk [8]. Glucagon-like peptide-1 (GLP-1) is an incretin hormone inducing insulin secretion and suppressing glucagon production [9]. Dipeptidyl peptidase 4 (DPP-4) inhibitors can prevent GLP-1 from degradation and GLP-1 receptor agonists can mimic the physiological function of GLP-1 [9]. A meta-analysis of four retrospective cohorts of patients with type 2 diabetes mellitus (T2DM) found that DPP4 inhibitor users had a 28% lower risk of incident RA compared with those who did not receive DPP4 inhibitors [10]. Likewise, in vitro studies found anti-inflammatory effects of GLP-1 receptor agonists in fibroblast-like synoviocytes (FLS) from patients with RA [11, 12]. Evidence is scarce concerning the impact of using SGLT2 inhibitors, insulin and sulfonylureas on RA risk.
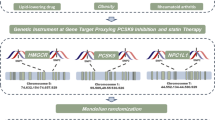
Because of their observational designs, these studies have demonstrated associations but not necessarily causality, for instance, reverse causation may have been at play. Mendelian randomization (MR) can be a viable approach to gauge the repurposing potential of a drug [13]. Briefly, MR uses genetic variants, usually single nucleotide polymorphisms (SNPs), as instruments of an exposure to provide the ideally unconfounded effect of the exposure on the outcome [14]. Genetic variants from the gene locus that encodes the drug target protein are likely to influence the expression or function of the protein. Leveraging these variants as genetic instruments can mimic how the drug modulates its target protein, allowing us to estimate the effect of genetic variation in the drug target on a new indication like a randomized controlled trial (Fig. 1) [13, 15]. This study aimed to evaluate the repurposing potential of antidiabetic agents to prevent RA in an MR framework, using genetic variants in the encoding gene loci of the targets of different antidiabetic drugs.

Overview of the Mendelian randomization study design. The Mendelian randomization (MR) design uses alleles randomized at germ cell formation and conception as instruments to estimate unconfounded associations between an exposure and an outcome, and can be a viable method to gauge the potential of drug repurposing. This study selects genetic variants from the gene(s) encoding the target protein(s) of the antidiabetic drug as instruments. The main downstream effect of antidiabetic drugs is decreased glucose concentration. Hence, this study leverages associations of the selected genetic instruments with blood glucose concentration to proxy the pharmacological modulation of the drug target protein of interest (relevance assumption). Genetic variants are expected to not be associated with confounders (independence assumption) and affect RA through other pathways (exclusion restriction assumption). The two samples used in the analyses were summary data from genome-wide association studies on blood glucose (UK Biobank) and clinically diagnosed RA.
Methods
Study overview
We utilized a two-sample MR design to provide evidence for the potential usefulness of antidiabetic drug repurposing on RA prevention by deriving summary statistics of instrument-exposure and instrument-outcome associations from large-scale genome-wide association studies (GWAS) in separate populations (Fig. 1). We first identified the gene(s) that encode the target protein(s) of each antidiabetic drug class from Drugbank and ChEMBL databases, and chose independent variants from the gene locus as instruments to proxy the pharmacological modulation of the drug target protein. We extracted associations between genetic instruments and blood glucose concentration from the glucose GWAS in the UK Biobank (UKB)[16], and associations between genetic instruments and RA risk from the so far largest GWAS of clinically diagnosed RA [17]. Then we combined two datasets to estimate the effects of genetic variation in the antidiabetic drug targets on the risk of RA using MR analysis. Details about all the GWAS used in our study are listed in Table 1.
GWAS of blood glucose (instrument-exposure associations)
We performed a GWAS analysis of blood glucose concentration in the UKB. Briefly, the UKB recruited about half a million participants aged 40 to 69 years across the UK between 2006 and 2010 [18]. UKB collected a random blood sample for glucose concentration. We excluded participants with self-reported diabetes or diabetes treatment at baseline, and those diagnosed with diabetes (International Classification of Diseases [ICD] 10th code: E10-E14; ICD9 code: 250) before the enrollment, as identified through the linkage to multiple databases (Supplementary Fig. 1). Genotype data after imputation were available in the UKB [16]. We excluded individuals having a high proportion of heterozygous genotypes (the indication of low sample quality), sex chromosome aneuploidy, or > 5% missing rate of variants. To correct for relatedness between individuals, one relative was randomly retained within pairs with a kinship threshold of 0.0884. We removed variants with a missing rate > 0.1 or a minor allele frequency < 0.01. We restricted the analysis to White British participants without missing information on glucose concentration (N = 309,895) and adjusted for age at baseline, sex, array and ten principal genetic components.
GWAS of rheumatoid arthritis (instrument-outcome associations)
We obtained information on genetic association data of clinically diagnosed RA from a GWAS meta-analysis of 22,350 cases and 74,823 controls from 25 European RA cohorts (downloaded on 2021/12/07; Table 1) [17]. Genetic associations specifically for seropositive RA were also available in this GWAS that encompassed 17,221 seropositive cases. Seropositive RA case was defined as RA positive for either rheumatoid factor or anti-citrullinated peptide antibodies. Covariates included age, sex and principal components, as appropriate.
Genetic instruments for anti-diabetic drugs
Major antidiabetic drugs included metformin, GLP-1 receptor agonists, SGLT2 inhibitors, DPP-4 inhibitors, insulin and its analogues, thiazolidinediones and sulfonylureas [6]. Genes encoding the target proteins of these antidiabetic drugs were identified from Drugbank (v5.0) and ChEMBL (v29.0) databases (Table 2) [19, 20]. Metformin was removed because two databases listed different target proteins and its drug mechanisms have not been fully clarified.
We extracted genetic variants from the target gene region and its neighboring 2.5 kb window of each drug class, retained those having associations with the glucose concentration with a false discovery rate less than 0.05, and then applied a linkage disequilibrium (LD) clumping (r2 < 0.001) to select nearly independent variants as genetic instruments (Supplementary Table 1). Palindromic variants were allowed with a minor allele frequency below 0.3. For variants absent in the RA GWAS, we used the European panel from the 1000 Genome Project Phase 3 as the reference panel and chose the variant in high LD (r2 > 0.8) with the drug-target-associated variant as a proxy [21]. To examine whether the genetic instruments would affect RA risk through other pathways other than the drug target of interest, we also searched for traits associated with these instruments in the GWAS Catalog [22].
In addition, we selected genetic variants of biological relevance as instruments for thiazolidinediones and sulfonylureas. A common variant (rs1801282) within the PPARG gene is a missense variant and results in the substitution of Ala for Pro at position 12 in the PPARγ2-specific exon B. The rs1801282 variant can regulate binding affinity to PPARγ response element and ability to activate transcription [23]. Its minor allele (G) was associated with a lower T2DM risk [24]. The rs757110 variant within the ABCC8 gene encodes the subunit of sulfonylurea target protein, ATP-sensitive potassium (KATP) channel. The KATP channel locates on pancreatic β-cell membranes and its inhibition promotes insulin release [25]. The minor allele (A) of rs757110 could facilitate closing the KATP channel and was associated with decreased 2-hour glucose concentration and T2DM risk [25, 26]. Genetic instruments of biological relevance were independently leveraged in MR analysis.
Positive control analysis
A positive control MR analysis serves to justify the genetic instruments of the drug by demonstrating the expected effect on the outcome which has an established causal relationship with the drug of interest [27]. The intended indication for antidiabetic drugs is T2DM. Besides their glucose-lowering properties, GLP-1 receptor agonists and SGLT2 inhibitors have weight loss effects and DDP4 inhibitors are neutral to weight change, whereas insulin and its analogues, thiazolidinediones and sulfonylureas could confer weight gain [28]. We leveraged body mass index (BMI), hip circumference and waist circumference and waist-to-hip ratio as additional positive control outcomes [29, 30]. Thiazolidinediones can ameliorate insulin sensitivity, so we used insulin resistance (IR) as a positive control outcome for thiazolidinediones [31]. This positive control analysis excluded the functional variant (rs1801282) as the instrument for thiazolidinediones because its minor allele had a high allele frequency (G = 0.903) in the IR GWAS. Meanwhile, GLP-1 receptor agonists and sulfonylureas can stimulate insulin secretion, so we used fasting proinsulin concentration to reflect the insulin secretion ability of pancreatic β-cells [32]. The primary MR analysis was only performed for antidiabetic drugs that demonstrated expected associations with these positive control outcomes. Details about genetic association data for these positive control outcomes, e.g., sample size, inclusion/exclusion criteria, covariates, are listed in Table 1 [29, 30, 33,34,35,36].
Sensitivity analyses
We conducted a sensitivity analysis for thiazolidinediones. Fasting insulin concentration significantly decreases in obese or type 2 diabetes patients receiving thiazolidinediones in clinical studies [37, 38]. Hence we additionally used the fasting insulin concentration as a downstream biomarker of thiazolidinediones. We obtained effect sizes of associations between the selected genetic instruments for thiazolidinedione and fasting insulin concentration from the largest GWAS meta-analysis of fasting insulin, which involved 105,056 white European individuals from 38 studies [35]. The GWAS analysis was performed in each sex and then pooled together using meta-analysis. Covariates included age, study site and principal components, as appropriate. Then we repeated MR analysis to estimate the effect of genetic variation in the thiazolidinedione target, which was predicted by insulin decrement, on RA risk. The minor allele frequency of the functional variant (rs1801282) was high (MAFG=0.874) in the fasting insulin GWAS so it was excluded from this sensitivity analysis. Additionally, a relaxed clumping threshold (r2 < 0.1) was applied to include more genetic instruments for the primary MR analyses.
Statistical methods
The effect size of the genetic association between each genetic instrument and the blood glucose concentration was estimated by the linear regression using PLINK v1.9 [39]. The mean F statistic was calculated to test the genetic instrument strength for each drug class [40]. An F statistic above ten typically indicates a strong instrument [40]. If the effects of the genetic instrument on the exposure and the outcome did not correspond to the same allele on the same DNA strand, we aligned the allele in the outcome dataset to that in the exposure dataset and flipped its genetic effect size accordingly. If a single genetic instrument was obtained, the causal effect was estimated by the ratio of genetic associations with RA and blood glucose concentration (Wald ratio test). If multiple genetic instruments were selected, inverse-variance weighted (IVW) method was used by combining ratio estimates for all instruments in a fixed effect meta-analysis [41]. Furthermore, MR Egger regression, weighted median and weighted mode methods were used to relax the IVW assumption that the average pleiotropic effect was zero [27]. We set the significance level at 0.008 (0.05/6 drug classes) after Bonferroni correction. The MR estimate was scaled to the odds ratio (OR) of RA per 0.1mmol/L lower blood glucose concentration. MR analyses were completed using the R package ‘TwoSampleMR’ [42].
For drugs that emerged as causally related to RA risk in the MR analysis, we checked whether blood glucose concentration and RA colocalized within the gene region of the drug target protein in question using a Bayesian framework [43]. We set the prior probability of the association between each variant with either trait to be 1 × 10− 4 and the prior probability of a shared causal variant between two traits to be 1 × 10− 5 (R package ‘coloc’) [43].
Standard protocol approvals, registrations, and patient consents
This research has been conducted using the UK Biobank Resource under Application Number ‘22224’. The UK Biobank has approval from the North West Multi-center Research Ethics Committee (MREC) in the UK and the aim of this study has the approval from the Regional Ethics Review Board in Stockholm. All cohort data included in the GWAS used in the present study have individual approvals from relevant ethical review boards.
Results
Genetic instruments for antidiabetic drugs
The blood glucose GWAS was performed in 309,895 UKB participants (Manhattan plot and Q-Q plot in Supplementary Fig. 2). We identified one variant for each of GLP-1 receptor agonists, insulin and thiazolidinediones, and two variants for sulfonylureas, respectively, as instruments for the pharmacological modulation of their corresponding drug target proteins (Supplementary Table 1). No valid genetic instruments were found for SGLT2 and DDP4 inhibitors. Mean F statistics of instruments were 19.3 for GLP-1 receptor agonists, 17.4 for insulin, 14.2 for thiazolidinediones and 31.4 for sulfonylureas, respectively. Corresponding F statistics for the two instruments of biological relevance were 11.0 for thiazolidinediones and 48.5 for sulfonylureas, respectively. These indicated that strong genetic instruments were chosen for each drug type.
We searched for traits associated with these instruments in the GWAS Catalog to examine if the exclusion restriction assumption was violated. The rs35240997 variant (the instrument for thiazolidinediones) was linked to red blood cell count and high-density lipoprotein (HDL) cholesterol and the functional variant rs1801282 additionally showed associations with triglycerides, systolic blood pressure, sex hormone-binding globulin and serum albumin. The rs5219 variant (the instrument for sulfonylureas) was additionally associated with cortical surface area and blood pressure (Supplementary Table 2).
Positive control analysis
Genetic variations in the targets of insulin and its analogues, thiazolidinedione and sulfonylurea were associated with a risk reduction in T2DM, with ORs (95%CI) per 0.1mmol/L glucose lowering being 0.40 (0.25–0.65), 0.30 (0.18–0.50) and 0.48 (0.37–0.63), respectively (Supplementary Fig. 3, panel A). Corresponding associations for thiazolidinediones and sulfonylureas were similar when using their functional variants as instruments. In contrast, genetic variation in the GLP-1 receptor agonist target did not predict a deceased T2DM risk (OR 1.25, 95%CI 0.81–1.92). Overall, genetic variation in target(s) of thiazolidinediones, of sulfonylureas, and of insulin and its analogues demonstrated associations with higher risk of obesity-related phenotypes (Supplementary Fig. 3, panel B-D). However, the positive associations between genetic variation in the target of GLP-1 receptor agonists and all obesity-related phenotypes were inconsistent with existing evidence that GLP-1 receptor agonists have a weight loss effect. We particularly treated IR as the positive control outcome for the genetic instrument selected for thiazolidinediones and observed an inverse association between the two (Supplementary Fig. 3, panel E). Genetic variation in the sulfonylureas target was associated with increased insulin secretion (Supplementary Fig. 3, panel F). In summary, positive control analyses justified the selected genetic instruments of insulin and its analogues, thiazolidinediones and sulfonylureas, but not the instrument of GLP-1 receptor agonists.
Mendelian randomization and sensitivity analysis
Genetic variation in the thiazolidinedione target (gene: PPARG) was significantly associated with a RA risk after multiple testing correction (OR 0.38 per 0.1mmol/L glucose lowering, 95%CI 0.20–0.73, P = 0.004; Fig. 2). The association was consistent when using the functional variant rs1801282 as the genetic instrument (OR 0.30, 95%CI 0.14–0.61, P = 0.001). By contrast, genetic variation in the targets of insulin and its analogues, and of sulfonylurea, were not associated with RA risk (OR [95%CI]: 0.83 [0.44–1.55] and 1.25 [0.78-2.00], respectively).

Estimated effects of genetic variations in antidiabetic drug targets on rheumatoid arthritis. OR, odds ratio; CI, confidence interval; RA, rheumatoid arthritis. * The rs1801282 variant is a functional variant within the PPARG gene region and can regulate binding affinity to PPARγ (encoded by the PPARG gene) response element and ability to activate transcription. † The rs757110 variant is a functional variant within the ABCC8 gene. It can promote insulin release by inhibiting ATP-sensitive potassium channel, of which the subunit is encoded by the ABCC8 gene. Genetic instruments from the gene region encoding the drug target protein can proxy the antidiabetic drug of interest. Combining variant-glucose and variant-RA associations, the effect of the antidiabetic drug on RA is estimated by the Wald ratio test or inverse-variance weighted method. MR estimates are scaled to RA risk per 0.1 mmol/L glucose lowering
When the MR analyses were restricted to seropositive RA, genetic variation in thiazolidinedione target was associated with an OR of 0.27 per 0.1mmol/L glucose decrement for seropositive RA risk (95%CI 0.13–0.57, P = 0.001; Fig. 3). Consistent association was observed using the functional genetic instrument (OR 0.23, 95% 0.10–0.51, P = 3.08 × 10− 4). Corresponding ORs (95%CIs) were 0.77 (0.37–1.58) for insulin and its analogues and 1.33 (0.81–2.18) for sulfonylureas.

Estimated effects of genetic variation(s) in antidiabetic drug targets on seropositive rheumatoid arthritis. OR, odds ratio; CI, confidence interval; RA, rheumatoid arthritis. * The rs1801282 variant is a functional variant within the PPARG gene region and can regulate binding affinity to PPARγ (encoded by the PPARG gene) response element and ability to activate transcription. † The rs757110 variant is a functional variant within the ABCC8 gene. It can promote insulin release by inhibiting ATP-sensitive potassium channel, of which the subunit is encoded by the ABCC8 gene. Genetic instruments from the gene region encoding the drug target protein can proxy the antidiabetic drug of interest. Combining variant-glucose and variant-seropositive RA associations, the effect of the antidiabetic drug on RA risk is estimated by the Wald ratio test or the inverse-variance weighted method. MR estimates are scaled to RA risk per 0.1 mmol/L glucose lowering
We also utilized the fasting insulin concentration as the downstream biomarker of thiazolidinediones and repeated MR analysis using genetic associations of the thiazolidinedione instrument with fasting insulin and RA. Here, genetic variation in the thiazolidinedione target, which was predicted by the decrement of fasting insulin concentration, was associated with lower risks for RA (OR 0.76 per log-transformed unit of lower fasting insulin, 95%CI 0.63–0.91) and seropositive RA 0.69 (95%CI 0.56–0.85). To avoid chance finding due to few instruments, we used a relaxed clumping threshold (r2 < 0.1) to select more genetic instruments and MR analyses showed robust results (Supplementary Table 3).
Colocalization analysis
The probability of a causal variant associated with glucose concentration, or RA risk, within the PPARG gene was 0.7%, and 60.6%, respectively (Supplementary Table 4). The probability of the existence of distinct causal variants between two traits was 1.1% and the probability of a shared causal variant was only 1.9%. The regional association plots of both traits showed similar pattern around the genetic instrument of thiazolidinediones (Supplementary Fig. 4).
Discussion
This is the first MR study to investigate whether genetic variations in antidiabetic drug targets were associated with RA risk. Leveraging existing large-scale genetic association data on RA risk, our genetic investigation suggests that thiazolidinediones might exert protective effects on RA risk, at least seropositive RA. By contrast, no such association was observed with other antidiabetic drugs.
Our MR findings suggested a causal relationship between genetic variation in the target of thiazolidinediones and RA, but colocalization results did not find a shared causal variant between the glucose concentration and RA within the PPARG gene. These seemingly discrepant results may stem from the different methodological theories of two methods: MR selects variants associated with the exposure whereas colocalization is more conservative by requiring significant associations of the causal variant with both traits [44]. The regional association plots of both traits exhibited weak genetic associations of the instrument for thiazolindinediones, which could lead to a weak observed colocalization. Other study designs have reported that exposure to thiazolidinediones was related to a lower RA risk. A population-based case-control study compared the use of thiazolidinediones between T2DM patients with and without an RA diagnosis, and observed an RA risk reduction, although without reaching statistical significance, associated with thiazolidinedione use compared with non-use (OR 0.91, 95%CI 0.81–1.02) [8]. After further excluding participants who took thiazolidinediones 90 days before the diagnosis of RA from the case group, there was a dose-response relationship, with a 25% (95%CI 11-39%) lower RA risk among patients exposing to the highest cumulative defined daily doses of thiazolidinediones [8]. Their finding about lower RA risk in thiazolidinedione users is concordant with our results that genetic variation in the thiazolidinedione target is preventive against RA.
The mechanism behind any preventive effect of thiazolidinediones on RA risk is unclear, but experimental data regarding the therapeutic effects of thiazolidiendiones could provide some insights. PPARγ was remarkably downregulated in FLS from patients with RA compared with FLS from healthy controls [45]. FLS are a dominant cell population in synovium. They can secrete pro-inflammatory cytokines like interleukin-6, interact with macrophage-like synoviocytes and other immune-related cells, and subsequently cause inflamed joints [46]. Murine models have observed that upregulating PPARγ could inhibit the proliferation and migration of FLS [45]. Patients with RA also upregulated PPARγ in peripheral macrophages in comparison with healthy controls, and the extent of PPARγ expression was negatively related to RA disease activity.[47] In the joint pathology of RA, activated macrophages can produce various pro-inflammatory factors and interact with other immune cells [48]. Thiazolidinediones can activate PPARγ and may repress gene transcription by negatively interfering with other transcription-factor pathways to achieve anti-inflammatory effects [31]. Crossover trials also reported that thiazolidinediones could decrease the disease activity in non-diabetic patients who received stable DMARDs treatment for RA [49, 50]. Yet in these studies it was uncertain whether PPARγ dysregulation was driven by the natural pathological course of the RA disease itself, by DMARD treatment, or by other factors [46, 47]. Our results thus highlight the potential for further mechanistically oriented research on the role of PPARγ in RA and other states of chronic inflammation.
To the best of our knowledge, there is no prior research on the impact of insulin, its analogues and sulfonylureas on RA risk. Irrespective, our study did not support a causal effect of insulin and sulfonylureas on RA risk, but on the other hand suggest that whatever mechanism that drives the association between the genetic variation in the thiazolidinedione target and RA risk is not shared with any glucose-lowering drugs, or mediated via other mechanisms shared by the genetic variation in targets of these drugs.
This study has several strengths. Firstly, we applied an MR design to make a causal inference without confounding bias and reverse causation. Secondly, we restricted analyses within the European ancestry to avoid spurious associations due to population stratification. Thirdly, we specifically chose genetic variants from within a narrow window (2.5 kb) of the encoding gene as instruments, which may be related to the gene function or expression. Fourthly, we performed positive control analyses that predicted the effects of antidiabetic drugs on the intended indication and other established outcomes to justify the validity of selected genetic instruments. Finally, the large F statistic indicated the small chance of a weak instrument bias.
Yet a few limitations should be acknowledged. Firstly, this study could only predict the drug effect on RA risk via perturbing documented proteins (on-target effects). We could not rule out the possibility of drugs modifying RA risk through other proteins (off-target effects). Secondly, few genetic instruments could lead to a chance finding. However, our study selected genetic instruments of biological relevance for thiazolidinediones and sulfonylureas and validated them using various positive control outcomes. We relaxed the clumping threshold to obtain more genetic instruments and observed robust MR estimates. For insulin and its analogues, GLP-1 receptor agonists, DDP4 inhibitors and SGLT2 inhibitors, MR study could use genetic instruments that are associated with the drug target protein or other downstream biomarkers to estimate the drug effect on RA risk when more relevant data emerge. Thirdly, horizontal pleiotropy could bias MR estimates, but we selected variants from the vicinity of gene locus that are unlikely to have pleiotropic effects through other genes. Regional association plots also showed that variants in high LD with the instrument fell within the PPARG gene locus. We found that instruments for thiazolidinediones were related to traits like red blood cell count, HDL cholesterol, triglycerides, blood pressure, sex hormone-binding globulin and serum albumin. But these traits could be vertical pleiotropic effects because thiazolidinediones have been reported to decrease red blood cell count [51], increase HDL concentration and triglycerides [31], lower blood pressure [52], increase sex hormone-binding globulin [53], and reduce albuminuria [54]. Fourthly, the MR estimate reflects the life-long exposure to a drug, while the drug usually exerts their impact in a short window. This means effect sizes in our study may not be comparable to those reported in trials or observational studies. Fifthly, we could not explore whether genetic variation in the thiazolidinedione target has a beneficial effect on seronegative RA because the GWAS of seronegative RA is lacking. Sixthly, we excluded T2DM patients and individuals on antidiabetic agents in the glucose GWAS to avoid the confounding bias from antidiabetic treatments. But this could inflate the ‘healthy volunteer’ bias in the UKB population and therefore lead to null colocalization findings. Finally, analyses within the European ancestry limited the generalizability of our findings to other ethnicities.
Conclusion
The present study provides MR evidence about thiazolidinediones as a potential drug class for RA prevention. Genetic evidence offers valuable insights into therapeutic development because the genetic link between a drug target and an indication substantially expedites prioritizing drugs and lowers failure rates in clinical trials [55, 56]. Hence, thiazolidinediones and its target protein should be prioritized as a potential therapeutic target to prevent RA. Further studies could incorporate large-scale GWAS of RA progression phenotypes to assess the disease-modifying potential of thiazolidinediones or even the combined effect of DMARDs and thiazolidinediones on RA.
References
Safiri S, Kolahi AA, Hoy D, et al. Global, regional and national burden of rheumatoid arthritis 1990–2017: a systematic analysis of the Global Burden of Disease study 2017. Ann Rheum Dis. 2019;78(11):1463–71. https://doi.org/10.1136/annrheumdis-2019-215920.
Smolen JS, Landewe RBM, Bijlsma JWJ, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. 2020;79(6):685–99. https://doi.org/10.1136/annrheumdis-2019-216655.
Putrik P, Ramiro S, Kvien TK, et al. Inequities in access to biologic and synthetic DMARDs across 46 european countries. Ann Rheum Dis. 2014;73(1):198–206. https://doi.org/10.1136/annrheumdis-2012-202603.
Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3(8):673–83. https://doi.org/10.1038/nrd1468.
Ristic GG, Subota V, Stanisavljevic D, et al. Impact of disease activity on impaired glucose metabolism in patients with rheumatoid arthritis. Arthritis Res Ther. 2021;23(1):95. https://doi.org/10.1186/s13075-021-02476-0.
American Diabetes Association Professional Practice Committee. 9. Pharmacologic approaches to Glycemic Treatment: Standards of Medical Care in Diabetes—2022. Diabetes Care. 2021;45 Suppl 1:125–S43. https://doi.org/10.2337/dc22-S009.
Naffaa ME, Rosenberg V, Watad A, et al. Adherence to metformin and the onset of rheumatoid arthritis: a population-based cohort study. Scand J Rheumatol. 2020;49(3):173–80. https://doi.org/10.1080/03009742.2019.1695928.
Hsieh MS, Hung PS, Hsieh VC, Liao SH, How CK. Association between thiazolidinedione use and rheumatoid arthritis risk in patients with type II diabetes, a population-based, case-control study. Int J Clin Pract. 2021;75(3):e13804. https://doi.org/10.1111/ijcp.13804.
Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368(9548):1696–705. https://doi.org/10.1016/S0140-6736(06)69705-5.
Charoenngam N, Rittiphairoj T, Ponvilawan B, Ungprasert P. Use of dipeptidyl peptidase-4 inhibitors is associated with a lower risk of rheumatoid arthritis in patients with type 2 diabetes mellitus: a systematic review and meta-analysis of cohort studies. Diabetes Metab Syndr. 2021;15(1):249–55. https://doi.org/10.1016/j.dsx.2020.12.042.
Du X, Zhang H, Zhang W, et al. The protective effects of lixisenatide against inflammatory response in human rheumatoid arthritis fibroblast-like synoviocytes. Int Immunopharmacol. 2019;75:105732. https://doi.org/10.1016/j.intimp.2019.105732.
Tao Y, Ge G, Wang Q, et al. Exenatide ameliorates inflammatory response in human rheumatoid arthritis fibroblast-like synoviocytes. IUBMB Life. 2019;71(7):969–77. https://doi.org/10.1002/iub.2031.
Walker VM, Davey Smith G, Davies NM, Martin RM. Mendelian randomization: a novel approach for the prediction of adverse drug events and drug repurposing opportunities. Int J Epidemiol. 2017;46(6):2078–89. https://doi.org/10.1093/ije/dyx207.
Smith GD, Ebrahim S. Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22. https://doi.org/10.1093/ije/dyg070.
Gill D, Georgakis MK, Walker VM, et al. Mendelian randomization for studying the effects of perturbing drug targets. Wellcome open research. 2021;6:16. https://doi.org/10.12688/wellcomeopenres.16544.2.
Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–9. https://doi.org/10.1038/s41586-018-0579-z.
Ishigaki K, Sakaue S, Terao C et al. Trans-ancestry genome-wide association study identifies novel genetic mechanisms in rheumatoid arthritis. medRxiv. 2021:2021.12.01.21267132. https://doi.org/10.1101/2021.12.01.21267132
Allen NE, Sudlow C, Peakman T, Collins R. UK biobank data: come and get it. Sci Transl Med. 2014;6(224):224ed4. https://doi.org/10.1126/scitranslmed.3008601.
Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–D82. https://doi.org/10.1093/nar/gkx1037.
Gaulton A, Hersey A, Nowotka M, et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017;45(D1):D945–D54. https://doi.org/10.1093/nar/gkw1074.
Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31(21):3555–7. https://doi.org/10.1093/bioinformatics/btv402.
Buniello A, MacArthur JAL, Cerezo M, et al. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47(D1):D1005–D12. https://doi.org/10.1093/nar/gky1120.
Deeb SS, Fajas L, Nemoto M, et al. A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat Genet. 1998;20(3):284–7. https://doi.org/10.1038/3099.
Altshuler D, Hirschhorn JN, Klannemark M, et al. The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000;26(1):76–80. https://doi.org/10.1038/79216.
Hamming KS, Soliman D, Matemisz LC, et al. Coexpression of the type 2 diabetes susceptibility gene variants KCNJ11 E23K and ABCC8 S1369A alter the ATP and sulfonylurea sensitivities of the ATP-sensitive K(+) channel. Diabetes. 2009;58(10):2419–24. https://doi.org/10.2337/db09-0143.
Emdin CA, Klarin D, Natarajan P, et al. Genetic variation at the Sulfonylurea receptor, type 2 diabetes, and Coronary Heart Disease. Diabetes. 2017;66(8):2310–5. https://doi.org/10.2337/db17-0149.
Burgess S, Davey Smith G, Davies NM, et al. Guidelines for performing mendelian randomization investigations. Wellcome Open Res. 2019;4:186. https://doi.org/10.12688/wellcomeopenres.15555.2.
Tsapas A, Karagiannis T, Kakotrichi P, et al. Comparative efficacy of glucose-lowering medications on body weight and blood pressure in patients with type 2 diabetes: a systematic review and network meta-analysis. Diabetes Obes Metab. 2021;23(9):2116–24. https://doi.org/10.1111/dom.14451.
Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518(7538):197–206. https://doi.org/10.1038/nature14177.
Shungin D, Winkler TW, Croteau-Chonka DC, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518(7538):187–96. https://doi.org/10.1038/nature14132.
Yki-Jarvinen H, Thiazolidinediones. N Engl J Med. 2004;351(11):1106–18. https://doi.org/10.1056/NEJMra041001.
Chaudhury A, Duvoor C, Reddy Dendi VS, et al. Clinical review of antidiabetic drugs: implications for type 2 diabetes Mellitus Management. Front Endocrinol (Lausanne). 2017;8:6. https://doi.org/10.3389/fendo.2017.00006.
Scott RA, Scott LJ, Magi R, et al. An expanded genome-wide Association study of type 2 diabetes in Europeans. Diabetes. 2017;66(11):2888–902. https://doi.org/10.2337/db16-1253.
Dupuis J, Langenberg C, Prokopenko I, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42(2):105–16. https://doi.org/10.1038/ng.520.
Lagou V, Magi R, Hottenga JJ, et al. Sex-dimorphic genetic effects and novel loci for fasting glucose and insulin variability. Nat Commun. 2021;12(1):24. https://doi.org/10.1038/s41467-020-19366-9.
Broadaway KA, Yin X, Williamson A, et al. Loci for insulin processing and secretion provide insight into type 2 diabetes risk. Am J Hum Genet. 2023;110(2):284–99. https://doi.org/10.1016/j.ajhg.2023.01.002.
Nolan JJ, Ludvik B, Beerdsen P, Joyce M, Olefsky J. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N Engl J Med. 1994;331(18):1188–93. https://doi.org/10.1056/NEJM199411033311803.
Lebovitz HE, Dole JF, Patwardhan R, Rappaport EB, Freed MI. Rosiglitazone Monotherapy is effective in patients with type 2 diabetes. J Clin Endocrinol Metabolism. 2001;86(1):280–8. https://doi.org/10.1210/jcem.86.1.7157.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. https://doi.org/10.1186/s13742-015-0047-8.
Burgess S, Thompson SG, Collaboration CCG. Avoiding bias from weak instruments in mendelian randomization studies. Int J Epidemiol. 2011;40(3):755–64. https://doi.org/10.1093/ije/dyr036.
Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658–65. https://doi.org/10.1002/gepi.21758.
Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7. https://doi.org/10.7554/eLife.34408.
Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383. https://doi.org/10.1371/journal.pgen.1004383.
Zuber V, Grinberg NF, Gill D, et al. Combining evidence from mendelian randomization and colocalization: review and comparison of approaches. Am J Hum Genet. 2022. https://doi.org/10.1016/j.ajhg.2022.04.001.
Li XF, Sun YY, Bao J, et al. Functional role of PPAR-gamma on the proliferation and migration of fibroblast-like synoviocytes in rheumatoid arthritis. Sci Rep. 2017;7(1):12671. https://doi.org/10.1038/s41598-017-12570-6.
Tu J, Hong W, Zhang P, Wang X, Korner H, Wei W. Ontology and function of Fibroblast-Like and Macrophage-Like Synoviocytes: how do they talk to each other and can they be targeted for rheumatoid arthritis therapy? Front Immunol. 2018;9:1467. https://doi.org/10.3389/fimmu.2018.01467.
Palma A, Sainaghi PP, Amoruso A, et al. Peroxisome proliferator-activated receptor-gamma expression in monocytes/macrophages from rheumatoid arthritis patients: relation to disease activity and therapy efficacy–a pilot study. Rheumatology (Oxford). 2012;51(11):1942–52. https://doi.org/10.1093/rheumatology/kes177.
Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376(9746):1094–108. https://doi.org/10.1016/s0140-6736(10)60826-4.
Marder W, Khalatbari S, Myles JD, et al. The peroxisome proliferator activated receptor-γ pioglitazone improves vascular function and decreases disease activity in patients with rheumatoid arthritis. J Am Heart Assoc. 2013;2(6):e000441. https://doi.org/10.1161/jaha.113.000441.
Ormseth MJ, Oeser AM, Cunningham A, et al. Peroxisome proliferator-activated receptor gamma agonist effect on rheumatoid arthritis: a randomized controlled trial. Arthritis Res Ther. 2013;15(5):R110. https://doi.org/10.1186/ar4290.
Lin KD, Lee MY, Feng CC, Chen BK, Yu ML, Shin SJ. Residual effect of reductions in red blood cell count and haematocrit and haemoglobin levels after 10-month withdrawal of pioglitazone in patients with type 2 diabetes. Diabet Med. 2014;31(11):1341–9. https://doi.org/10.1111/dme.12481.
Lebovitz HE. Thiazolidinediones: the Forgotten Diabetes Medications. Curr Diab Rep. 2019;19(12):151. https://doi.org/10.1007/s11892-019-1270-y.
Azziz R, Ehrmann D, Legro RS, et al. Troglitazone improves ovulation and hirsutism in the polycystic ovary syndrome: a multicenter, double blind, placebo-controlled trial. J Clin Endocrinol Metab. 2001;86(4):1626–32. https://doi.org/10.1210/jcem.86.4.7375.
Sarafidis PA, Bakris GL. Protection of the kidney by thiazolidinediones: an assessment from bench to bedside. Kidney Int. 2006;70(7):1223–33. https://doi.org/10.1038/sj.ki.5001620.
Reay WR, Cairns MJ. Advancing the use of genome-wide association studies for drug repurposing. Nat Rev Genet. 2021;22(10):658–71. https://doi.org/10.1038/s41576-021-00387-z.
Nelson MR, Tipney H, Painter JL, et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47(8):856–60. https://doi.org/10.1038/ng.3314.
Acknowledgements
The analyses on UK biobank genotypes were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) at Uppmax and partially funded by the Swedish Research Council through grant agreement no. 2018–05973. We thank the participants of the UK Biobank and the investigators who contributed to the data collection. Summary statistics for the genetic associations with RA, T2DM, fasting insulin, fasting proinsulin, HOMI-IR, BMI, waist circumference, and hip circumference were obtained from the GWAS previously published. We thank all investigators for sharing the genome-wide summary statistics. This research has been conducted using the UK Biobank Resource under Application Number ‘22224’.
Funding
Open access funding provided by Karolinska Institute. The work was conducted with support from the Swedish Research Council (2015-03255; 2019-01272, 2020-06101), Karolinska Institutet joint NIH-grant, Karolinska Institutet Foundation, Karolinska Institutet Strategic Research Program in Epidemiology, King Gustaf V and Queen Victoria?s Foundation of Freemasons, Swedish Heart Lung Foundation, and by the National Institutes of Health R01AG067996 to Sara Hägg.
Author information
Authors and Affiliations
Contributions
CQ and SH conceptualized the study design and interpreted the results. CQ analyzed the data and drafted the manuscript. SH and BT provided the methodological suggestions. YW managed the UKB data. YW, TN, AH and YL offered statistical support for the glucose GWAS analysis. BT, YW, LMDG, TN, AH, YL, LP, JA and SH revised the manuscript.
Corresponding author
Ethics declarations
Conflicting interests
All the authors declare no conflicting interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qin, C., Diaz-Gallo, LM., Tang, B. et al. Repurposing antidiabetic drugs for rheumatoid arthritis: results from a two-sample Mendelian randomization study. Eur J Epidemiol 38, 809–819 (2023). https://doi.org/10.1007/s10654-023-01000-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10654-023-01000-9