
Summary
Combining a checkpoint inhibitor with an inhibitor of extracellular signal-regulated kinase (ERK) may result in synergistic antitumor activity. We evaluated MK-8353, an ERK1 and ERK2 inhibitor, plus pembrolizumab in a phase 1b study in patients with advanced solid tumors. This open-label, nonrandomized, dose-escalation study (NCT02972034) enrolled adults with advanced solid tumors previously treated with 1‒5 prior lines of therapy. MK-8353 was administered orally in combination with pembrolizumab 200 mg every 3 weeks as follows: twice daily (arm A; MK-8353 50‒350 mg), once daily (arm B; MK-8353 50‒600 mg), or once daily every other week (arm C; MK-8353 50‒300 mg). The primary objective was evaluation of safety via occurrence of dose-limiting toxicities (DLTs). A secondary objective was objective response by RECIST v1.1 per investigator assessment. Among 110 evaluable patients (arm A, n = 22; arm B, n = 50; arm C, n = 38), median age was 58.0 (range, 35‒79) years and 50% had received 1 or 2 prior lines of therapy. DLTs occurred in 19 patients (n = 6 [27%], n = 8 [16%], and n = 5 [13%], respectively); the most frequent was grade 3 maculopapular rash (n = 15). Grade 3/4 treatment-related AEs occurred in 35% of patients; the most common were maculopapular rash (13%) and increased lipase (5%); none were grade 5. Eight patients (7%) attained an objective response (arm B, n = 7 [complete response, n = 1; partial response, n = 6]; arm C, n = 1 [complete response]). In conclusion, MK-8353 once daily plus pembrolizumab could be administered with a manageable toxicity profile but had modest antitumor activity in patients with advanced solid tumors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Extracellular signal‒regulated kinase (ERK) is a component of the mitogen-activated protein kinase (MAPK) pathway, which has a key role in cellular signaling including cell proliferation, differentiation, apoptosis, and survival [1,2,3]. Constitutive activation of the MAPK pathway is often observed in human cancers and is associated with high rates of cancer cell proliferation. For example, dysregulation of the pathway through genetic mutations is implicated in a number of tumor types including melanoma, thyroid, ovarian, colorectal, and lung cancer [1, 2]. Furthermore, elevated ERK expression has been reported in ovarian, colon, breast, and lung cancers [1].
MK-8353 is an orally available, adenosine triphosphate‒competitive, small-molecule inhibitor of ERK1 and ERK2. MK-8353 inhibits ERK1 and ERK2 activity and induces conformational change in these kinases that prevents their phosphorylation and activation by mitogen-activated protein/extracellular signal-regulated kinase (MEK) [4]. Monotherapy with MK-8353 was evaluated in a phase 1 study of patients with advanced solid tumors [5]. In that study, twice-daily doses of MK-8353 up to 400 mg were safe and well tolerated, and 3/15 patients who received a 300- to 400-mg dose achieved a partial response. Notably, all 3 responses occurred among the 8 patients who had BRAF V600‒mutant melanoma. The combination of MK-8353 with selumetinib was evaluated in a phase 1b study of patients with advanced or metastatic solid tumors [6]. The maximum tolerated dose was MK-8353 100 mg twice daily (BID) plus selumetinib 50 mg BID, but there were no objective responses [6].
Preclinical evidence has suggested that combining inhibitors of programmed cell death protein 1 (PD-1) or programmed cell death ligand 1 (PD-L1) with agents that target the MAPK pathway might have a synergistic effect and lead to durable tumor regression [7, 8]. The preclinical findings were supported by early studies in patients with advanced BRAF V600‒mutated melanoma, including preliminary findings from the phase 2 KEYNOTE-022 study of the PD-1 inhibitor pembrolizumab in combination with the MEK1 and MEK2 inhibitor trametinib and the BRAF inhibitor dabrafenib [9] and the IMspire150 study of the PD-L1 inhibitor atezolizumab in combination with the MEK1 and MEK2 inhibitor cobimetinib and the BRAF V600E inhibitor vemurafenib [10]. Both studies demonstrated extended progression-free survival (PFS) with a regimen incorporating a checkpoint inhibitor and a MEK1 and MEK2 inhibitor. Additionally, the combination of atezolizumab and cobimetinib demonstrated clinical benefit in a phase 1/1b study in patients with metastatic colorectal cancer, melanoma, or non-small-cell lung cancer [11].
Based on the preclinical and preliminary clinical findings for MK-8353 and early studies of MEK inhibitors in combination with checkpoint inhibitors, we conducted a phase 1b study of MK-8353 in combination with pembrolizumab in patients with previously treated advanced solid tumors (ClinicalTrials.gov, NCT02972034).
Methods
Patients
Adults (≥ 18 years old) were eligible if they had histologic or cytologic documentation of locally advanced or metastatic solid tumors and had received between 1 and 5 prior lines of anticancer treatment, excluding neoadjuvant, adjuvant, and maintenance treatment; ≥ 1 measurable lesion per Response Evaluation Criteria in Solid Tumors (RECIST) v1.1; an Eastern Cooperative Oncology Group (ECOG) performance status ≤ 1; and adequate organ function.
Key exclusion criteria included solid tumors suitable for local treatment with curative intent, Gilbert syndrome, immunodeficiency, additional malignancy that was progressing or required active treatment (exceptions: basal or squamous cell carcinoma of the skin or in situ cervical cancer), active central nervous system metastases and/or carcinomatous meningitis, active autoimmune disease requiring systemic treatment within 2 years, pneumonitis, history of interstitial lung disease, active infection requiring systemic therapy, or any other condition that might confound the trial results or interfere with the patient’s participation. Patients were excluded from parts 1 and 2 if they had received prior therapy with cancer vaccines, or compounds targeting PD-1 (including pembrolizumab), PD-L1, PD-L2, CTLA-4, or MEK (including, but not limited to, trametinib and cobimetinib) and were also excluded from part 2 if they had previously received compounds targeting LAG-3, CD-137, OX-40, CD-40, GITR, BRAF, MEK or other molecules in the MAPK pathway; treatment that interfered with CYP3A4 (including certain strong CYP3A4 inducers, inhibitors, moderate inhibitors, and substrates with narrow therapeutic range) or CYP2C8 (including certain substrates with narrow therapeutic range) within 14 days of enrollment; received chronic systemic steroids or other immunosuppressive therapy within 7 days of study treatment; received an anticancer monoclonal antibody within ≤ 4 weeks of study treatment; received chemotherapy, targeted small-molecule therapy, or radiation therapy within 14 days before study day 1 or did not recover from adverse events (AEs) caused by a previously administered agent; or had transfusion of blood products or colony-stimulating factors within ≤ 4 weeks of study day 1.
Study design and treatment
This was a two-part, multicenter, open-label, nonrandomized study. Part 1 was a dose-escalation study with an objective of identifying the recommended phase 2 dose of MK-8353 in combination with pembrolizumab. Part 2 was an expansion study with an objective of assessing the efficacy and safety of the recommended phase 2 dose. Herein, we report the methodology and results of part 1.
In the original protocol, there was a single dose-finding arm (later became arm A) with a starting dose of MK-8353 350 mg BID plus pembrolizumab 200 mg every 3 weeks. The protocol was amended based on the occurrence of dose-limiting toxicities (DLTs) at that dose level to restart MK-8353 at a dose of 100 mg BID and potentially de-escalate to 50 mg BID. Following the development of grade 3 toxicities at the MK-8353 100 mg BID dose level, the protocol was amended further to lower the starting dose for arm A to MK-8353 50 mg BID and added arm B, which used once-daily (QD) dosing of MK-8353 50 mg in combination with pembrolizumab 200 mg every 3 weeks, as well as the option to enroll arm C (administration of MK-8353 QD for 1 week on/1 week off in combination with pembrolizumab 200 mg every 3 weeks) and arm D (2-week run-in with MK-8353 monotherapy followed by continuous MK-8353 plus pembrolizumab 200 mg every 3 weeks).
During dose escalation, a minimum of 3 patients were required at each dose. At the starting dose in arm A, 1 patient was enrolled and then subsequent patients were enrolled once the first patient had reached cycle 1, day 15. All dose escalation and de-escalation decisions were based on the occurrence of DLTs from a given dose during the DLT-evaluable period and were made jointly by the investigators and the sponsor.
Study treatment continued for up to 35 cycles in arms A, B and C, and for up to 36 cycles in arm D, or until disease progression, unacceptable toxicity, intercurrent illness, investigator’s decision, or patient withdrawal. Patients who discontinued one study drug could continue the other study drug. Enrollment was planned to alternate between arms A and B.
Dose escalation for each treatment arm followed a modified toxicity probability interval (mTPI) design as described by Ji et al. (Supplemental Fig. 1) [12], with a target DLT rate of approximately 30% during treatment cycle 1 (21 days; ie, the DLT window) to confirm the maximum tolerated dose of combination therapy. DLTs were defined as any of the following toxicities determined by the investigator to be possibly, probably, or definitely related to study drug administration that occurred during the DLT window: grade 4 nonhematologic (nonlaboratory) toxicity; grade 4 hematologic toxicity (except thrombocytopenia) lasting ≥ 7 days; grade 4 thrombocytopenia of any duration or grade 3 thrombocytopenia associated with bleeding; grade 3 nonhematologic (nonlaboratory) toxicity lasting more than 3 days despite optimal supportive care; grade 3 or 4 nonhematologic laboratory finding that required medical intervention or persisted for more than 1 week, except for clinically nonsignificant, treatable, or reversible abnormalities (eg, liver function tests, uric acid); grade 3 or 4 febrile neutropenia (grade 3: absolute neutrophil count < 1000/mm3 with single temperature of 38.3 °C or with temperature ≥ 38 °C sustained for more than 1 h; grade 4: similar criteria as for grade 3 febrile neutropenia with life-threatening consequences and urgent intervention indicated); more than 2-week delay in initiating treatment cycle 2 due to treatment-related toxicity; treatment-related toxicity that leads to treatment discontinuation during cycle 1; missing > 25% of the MK-8353 doses due to treatment-related AEs during cycle 1; and death.

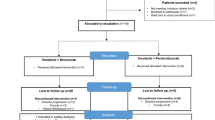
Patient disposition. BID, twice daily; Q3W, every 3 weeks; QD, once daily. aOne patient did not receive treatment due to screen failure
The protocol was approved by an independent institutional review board or institutional ethics committee at each study site. Patients provided written informed consent.
Endpoints
The primary endpoint was the occurrence of DLTs. Other safety endpoints included AEs, serious AEs, laboratory assessments, ECGs, vital signs, and physical examinations. The secondary endpoint was objective response rate (ORR) per RECIST v1.1 assessed by the investigator; objective response was defined as a confirmed complete response (CR) or partial response (PR).
Assessments
Adverse event monitoring began during screening and continued through 90 days after the last dose of study drug. AEs were graded based on investigator assessment using the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0 or later. Tumor imaging was done at baseline, every 9 weeks during treatment, and at treatment discontinuation. Response was evaluated by investigators per RECIST v1.1.
Statistical analysis
The safety population included all patients who received at least 1 dose of study treatment (ie, all patients as-treated [APaT] population). The DLT-evaluable population included all patients in the APaT population who were observed for ≥ 21 days after the first dose of assigned treatment or experienced a DLT prior to 21 days after the first dose of assigned treatment. The efficacy population included all patients with RECIST-measurable disease at baseline who received ≥ 1 dose of study treatment.
Dose-limiting toxicities were summarized by dose level and DLT rates estimated using the pool adjacent-violators algorithm, which forces rate estimates to be nondecreasing with increasing dose levels and pools adjacent violators for weighted samples by sample size [12]. Estimates were provided for DLT rates among patients treated at the maximum tolerated dose, and 80% CIs were provided based on Bayesian credible intervals with a prior distribution of beta (1, 1). For safety assessments, AEs were summarized as counts and frequencies at each dose level. ORR was estimated using an exact method based on the binomial distribution (Clopper-Pearson interval).
Results
Patients
The study was conducted between January 13, 2017, and December 2, 2022. 111 patients were enrolled, one of whom did not receive study treatment. 110 patients across arms A (n = 22), B (n = 50), and C (n = 38) were evaluable for DLTs, safety, and efficacy; although planned, arm D was not initiated (Fig. 1). Across treatment arms, median age was 58.0 (range, 35‒79) years, 61 patients (55%) were men, 69 (63%) had a baseline ECOG performance status of 1, and 55 (50%) had received either 1 or 2 prior lines of anticancer therapy (Table 1). Colorectal cancer was the most common tumor type (n = 37 [34%]).
As of the data cutoff date (December 2, 2022), 3 patients (3%) had completed study treatment and 107 patients (97%) had discontinued study treatment. The most common reasons for treatment discontinuation were disease progression (n = 84 [76%]) and AEs (n = 16 [15%]). Median time on therapy across all arms was 2.0 (range, 0.1‒25.8) months; patients received a median of 3 (range, 1‒35) cycles of pembrolizumab treatment. Part 2 of this study did not open.
Dose finding
Overall, 19 of 110 patients experienced at least 1 DLT (arm A, n = 6; arm B, n = 8; arm C, n = 5) with MK-8353 plus pembrolizumab. In arm A, 3 of 4 patients (75% [80% CI, 48%‒93%]) enrolled at the MK-8353 350 mg BID starting dose experienced a DLT (all grade 3 maculopapular rash; Supplemental Table 1). Following the first protocol amendment, the single patient that was enrolled at the MK-8353 100 mg BID dose experienced a DLT (grade 3 maculopapular rash). The dose was de-escalated to MK-8353 50 mg in the morning plus 100 mg in the evening, and 2 of 3 patients (67% [80% CI, 37%‒89%]) experienced a DLT (grade 3 maculopapular rash). None of the 13 patients enrolled when the MK-8353 dose was further de-escalated to 50 mg BID experienced a DLT.
In arm B, no DLTs were observed at increasing MK-8353 dose levels of 50 mg QD (n = 4), 100 mg QD (n = 3), and 150 mg QD (n = 4) each given with pembrolizumab (Supplemental Table 2). One of 7 patients at the MK-8353 200 mg QD plus pembrolizumab dose level (14% [80% CI, 3%‒35%]) experienced a DLT (grade 3 maculopapular rash). No DLTs occurred at the MK-8353 300 mg QD plus pembrolizumab dose level. At the MK-8353 400 mg QD dose level, 4 of 13 enrolled patients (31% [80% CI, 17%‒47%]) experienced DLTs (grade 2 maculopapular rash, n = 1; grade 3 maculopapular rash, n = 3). At the MK-8353 600 mg QD dose level, 3 of 7 enrolled patients (43% [80% CI, 23%‒65%]) experienced 4 total DLTs (grade 4 hypotension, n = 1; grade 3 fatigue, n = 1; and grade 3 maculopapular rash, n = 2).
In arm C, in which MK-8353 was dosed QD every other week, no DLTs occurred in the 3 patients enrolled at the MK-8353 starting dose of 100 mg (Supplemental Table 3). At the MK-8353 150 mg dose level, 2 of 12 enrolled patients (17% [80% CI, 6%‒32%]) experienced DLTs (grade 3 colitis, n = 1; grade 3 rash, n = 1). At the MK-8353 200 mg dose level, 1 of 9 patients (11% [80% CI, 2%‒28%]) experienced a DLT (grade 3 maculopapular rash). At the MK-8353 300 mg dose level, 2 of 10 patients (20% [80% CI, 8%‒38%]) experienced a DLT (grade 4 immune-mediated hepatitis, n = 1; grade 3 maculopapular rash, n = 1).
Safety
Eighty of 110 patients (73%) experienced treatment-related AEs (Table 2 and Supplemental Tables 4–6). Across treatment arms, the most common treatment-related AEs were diarrhea (22%), maculopapular rash (21%), fatigue (17%), and nausea (13%). Grade 3/4 treatment-related AEs occurred in 38 patients (35%). The most common grade 3/4 treatment-related AEs were maculopapular rash (13%), increased lipase (4%), diarrhea (4%), and increased gamma-glutamyltransferase (3%). No deaths were attributed to treatment-related AEs. Fifteen of 110 patients (14%) discontinued 1 or more components of study treatment because of a treatment-related AE. Serious treatment-related AEs occurred in 23 patients (21%). The most common serious treatment-related AEs were maculopapular rash (7%), abdominal pain, diarrhea, immune-mediated hepatitis (all 2%).
Immune-mediated AEs and infusion reactions, which were based on a list of preferred terms intended to capture known risks of pembrolizumab and were considered regardless of attribution to study treatment by the investigator, were reported for 28 patients (25%). Across treatment arms, the most common immune-mediated AEs were severe skin reactions (14%), colitis (5%), pneumonitis (4%), and hypothyroidism (3%). Twenty-two patients (20%) experienced grade 3/4 immune-mediated AEs.
Antitumor activity
Eight of 110 evaluable patients (7% [95% CI, 3%‒14%]) achieved an objective response (Table 3). An additional 20 patients (18%) had stable disease. None of the 22 patients in arm A achieved a CR or PR (Supplemental Table 7). In arm B, 7 of 50 patients (14% [95% CI, 6%‒27%]) had an objective response, including 1 CR and 1 PR at the MK-8353 300 mg QD dose level (n = 10), 3 PRs at the MK-8353 400 mg QD dose level (n = 14) and 2 PRs at the MK-8353 600 mg QD dose level (n = 8) (Supplemental Table 8). Median duration of response in arm B was not reached (range, 4.0‒37.8 + months). The patient with a CR had a tumor type of alveolar soft part sarcoma. The patients with PR had tumor types of pancreatic cancer (excluding islets; n = 2), skin cancer (not otherwise specified, n = 1), salivary duct squamous cell carcinoma (n = 1), cervical cancer (n = 1), and colorectal cancer (n = 1). Only the patient with salivary duct squamous cell carcinoma had received prior immune checkpoint inhibitor therapy (urabrelimab, an anti-CD47 monoclonal antibody). In arm C (n = 38), 1 patient at the 150 mg QD every other week dose level achieved a CR, for an ORR of 3% (95% CI, 0%‒14%; Supplemental Table 9). Duration of response for this patient was 35.3 months (ongoing). This patient had a tumor type of colorectal cancer and had not received prior immune checkpoint inhibitor therapy.
Discussion
In this phase 1b study of patients with previously treated advanced solid tumors, BID dosing of MK-8353 in combination with pembrolizumab led to DLTs above the 30% threshold and required de-escalation of the MK-8353 dose to 50 mg QD. Tolerability was improved with both continuous QD dosing and with QD dosing every other week, permitting the evaluation of MK-8353 at doses up to 600 mg QD in combination with pembrolizumab. Based on the totality of the safety data in the study, MK-8353 400 mg QD plus pembrolizumab 200 mg Q3W was identified as a potential recommended phase 2 dose. However, the antitumor activity of the combination was modest, with an ORR of 7% in the overall population. Of the 8 patients who experienced a response, 7 were in arm B (QD dosing), in which the ORR was 14% across all dose levels, and 21% with MK-8353 400 mg QD (the recommended phase 2 dose). Across all dose levels, the ORR was 0% in arm A, 14% in arm B, and 3% in arm C. Although the number of patients in each treatment arm was sufficient to determine clinical viability, these responses were not considered clinically meaningful given the heterogeneity of the population. Although tumor PD-L1 status was assessed in this study, the sample sizes for this and other biomarker measurements were too small after the initial treatment cycles to justify analyses according to biomarker status.
ORRs were modest across all 3 arms, with little evidence of an association between dose and antitumor activity. The optional arm D (run-in dosing of MK-8353 followed by continuous treatment with MK-8353 plus pembrolizumab) was not enrolled. Although pharmacokinetic data were not collected in the current analysis, a prior study of MK-8353 monotherapy in patients with advanced cancer found that MK-8353 was well tolerated at doses up to 400 mg BID [5]. Findings from the MK-8353 monotherapy study showed that doses between 300 and 400 mg led to pharmacokinetic exposure that resulted in tumor growth inhibition or regression in preclinical models [5]. Based on these pharmacokinetic data, the MK-8353 400 mg QD dose in the current study should have yielded exposure to inhibit tumor growth or regression. However, we cannot exclude the possibility that the inability to dose BID may have meant that sufficient steady-state exposure could not be achieved and that this may explain, at least in part, the limited activity observed.
While this study was ongoing, results were published from several studies of MEK pathway inhibitors in combination with PD-1 or PD-L1 inhibition for the treatment of advanced solid tumors, all of which demonstrated high toxicity and/or lack of clinical benefit. For example, the combination of the MEK inhibitor cobimetinib plus the PD-L1 inhibitor atezolizumab had high toxicity and limited antitumor activity in patients with previously treated solid tumors [11] and had a less tolerable safety profile without PFS benefit as first-line therapy in patients with BRAF V600 wild-type advanced melanoma [13]. Furthermore, trametinib plus pembrolizumab had limited antitumor activity in patients with BRAF wild-type melanoma and advanced solid tumors [14], and atezolizumab plus cobimetinib had limited antitumor activity in patients with (predominantly microsatellite-stable) metastatic colorectal cancer [15]. Together, the results from the previously mentioned studies suggest that combinations of a MEK inhibitor with a PD-1/PD-L1 inhibitor have limited clinical benefit in patients whose tumors lack a BRAF mutation or that are not microsatellite-stable. Additionally, response can vary by tumor type and prior PD-1/PD-L1 inhibitor treatment [16]. Furthermore, the combination of spartalizumab plus dabrafenib and trametinib was associated with higher AE rates than dabrafenib and trametinib alone, with ORRs of 69% versus 64%, and without significantly improving the median PFS (16.2 [95% CI, 12.7‒23.9] months vs 12.0 [95% CI, 10.2‒15.4] months, respectively) in patients with BRAF V600‒mutant advanced melanoma [17]. Secondly, in a phase 1 study, the combination of pembrolizumab and vemurafenib with or without cobimetinib demonstrated an ORR of 78% and median overall survival of 35.3 months [18]; however, DLTs were reported for 8 of 9 enrolled patients, and the study was closed early due to unacceptable toxicity. Finally, the combination of atezolizumab plus vemurafenib and cobimetinib improved investigator-assessed PFS versus vemurafenib and cobimetinib (hazard ratio [HR], 0.79 [95% CI, 0.64‒0.97]) but did not improve median overall survival (HR, 0.84 [95% CI, 0.66‒1.06]) in patients with BRAF V600-mutation‒positive advanced melanoma [19]. Together with the findings from the current analyses, these results suggest the combination of agents targeting MAPK signaling and PD-1/PD-L1 has limited clinical utility in this setting.
In conclusion, the combination of MK-8353 plus pembrolizumab had a generally manageable safety profile and had modest antitumor activity in patients with previously treated advanced solid tumors. Since the initiation of this study, several studies have reported high toxicity with limited antitumor activity with PD-1 and PD-L1 inhibitors in combination with inhibitors of the MAPK pathway in patients with advanced tumors. MK-8353 plus pembrolizumab did not elicit sufficient antitumor activity to warrant additional study of this combination.
Data availability
Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA (MSD), is committed to providing qualified scientific researchers access to anonymized data and clinical study reports from the company’s clinical trials for the purpose of conducting legitimate scientific research. MSD is also obligated to protect the rights and privacy of trial participants and, as such, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The MSD data sharing website (available at: http://engagezone.msd.com/ds_documentation.php) outlines the process and requirements for submitting a data request. Applications will be promptly assessed for completeness and policy compliance. Feasible requests will be reviewed by a committee of MSD subject matter experts to assess the scientific validity of the request and the qualifications of the requestors. In line with data privacy legislation, submitters of approved requests must enter into a standard data-sharing agreement with MSD before data access is granted. Data will be made available for request after product approval in the US and EU or after product development is discontinued. There are circumstances that may prevent MSD from sharing requested data, including country or region-specific regulations. If the request is declined, it will be communicated to the investigator. Access to genetic or exploratory biomarker data requires a detailed, hypothesis-driven statistical analysis plan that is collaboratively developed by the requestor and MSD subject matter experts; after approval of the statistical analysis plan and execution of a data-sharing agreement, MSD will either perform the proposed analyses and share the results with the requestor or will construct biomarker covariates and add them to a file with clinical data that is uploaded to an analysis portal so that the requestor can perform the proposed analyses.
References
Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL (2020) ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med 19:1997–2007. https://doi.org/10.3892/etm.2020.8454
Inamdar GS, Madhunapantula SV, Robertson GP (2010) Targeting the MAPK pathway in melanoma: why some approaches succeed and other fail. Biochem Pharmacol 80:624–637. https://doi.org/10.1016/j.bcp.2010.04.029
Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22:153–183. https://doi.org/10.1210/edrv.22.2.0428
Boga SB, Deng Y, Zhu L, Nan Y, Cooper AB, Shipps GW Jr, Doll R, Shih NY, Zhu H et al (2018) MK-8353: discovery of an orally bioavailable dual mechanism ERK inhibitor for oncology. ACS Med Chem Lett 9:761–767. https://doi.org/10.1021/acsmedchemlett.8b00220
Moschos SJ, Sullivan RJ, Hwu WJ, Ramanathan RK, Adjei AA, Fong PC, Shapira-Frommer R, Tawbi HA, Rubino J et al (2018) Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight 3:e92352. https://doi.org/10.1172/jci.insight.92352
Stathis A, Tolcher AW, Wang JS, Renouf DJ, Chen LC, Suttner LH, Freshwater T, Webber AL, Nayak T et al (2023) Results of an open-label phase 1b study of the ERK inhibitor MK-8353 plus the MEK inhibitor selumetinib in patients with advanced or metastatic solid tumors. Invest New Drugs 41:380–390. https://doi.org/10.1007/s10637-022-01326-3
Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, Gould SE, Maecker H, Irving BA et al (2016) MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 44:609–621. https://doi.org/10.1016/j.immuni.2016.01.024
Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, Yang J, Seestaller-Wehr L, Zhang SY et al (2015) The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res 21:1639–1651. https://doi.org/10.1158/1078-0432.CCR-14-2339
Ferrucci PF, Di Giacomo AM, Del Vecchio M, Atkinson V, Schmidt H, Schachter J, Queirolo P, Long GV, Stephens R et al (2020) KEYNOTE-022 part 3: a randomized, double-blind, phase 2 study of pembrolizumab, dabrafenib, and trametinib in BRAF-mutant melanoma. J Immunother Cancer 8:e001806. https://doi.org/10.1136/jitc-2020-001806
Gutzmer R, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, Pereira RP, Eigentler T, Rutkowski P et al (2020) Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 395:1835–1844. https://doi.org/10.1016/S0140-6736(20)30934-X
Hellmann MD, Kim TW, Lee CB, Goh BC, Miller WH Jr, Oh DY, Jamal R, Chee CE, Chow LQM et al (2019) Phase Ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann Oncol 30:1134–1142. https://doi.org/10.1093/annonc/mdz113
Ji Y, Wang SJ (2013) Modified toxicity probability interval design: a safer and more reliable method than the 3 + 3 design for practical phase I trials. J Clin Oncol 31:1785–1791. https://doi.org/10.1200/JCO.2012.45.7903
Gogas H, Dreno B, Larkin J, Demidov L, Stroyakovskiy D, Eroglu Z, Francesco Ferrucci P, Pigozzo J, Rutkowski P et al (2021) Cobimetinib plus atezolizumab in BRAF(V600) wild-type melanoma: primary results from the randomized phase III IMspire170 study. Ann Oncol 32:384–394. https://doi.org/10.1016/j.annonc.2020.12.004
Maio M, Carlino MS, Joshua AM, McWhirter E, Ribas A, Ascierto PA, Miller WH Jr, Butler MO, Ferrucci PF et al (2022) KEYNOTE-022: pembrolizumab with trametinib in patients with BRAF wild-type melanoma or advanced solid tumours irrespective of BRAF mutation. Eur J Cancer 160:1–11. https://doi.org/10.1016/j.ejca.2021.09.024
Eng C, Kim TW, Bendell J, Argiles G, Tebbutt NC, Di Bartolomeo M, Falcone A, Fakih M, Kozloff M et al (2019) Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol 20:849–861. https://doi.org/10.1016/S1470-2045(19)30027-0
Sherman E, Lee JL, Debruyne PR, Keam B, Shin SJ, Gramza A, Caro I, Amin R, Shah K et al (2023) Safety and efficacy of cobimetinib plus atezolizumab in patients with solid tumors: a phase II, open-label, multicenter, multicohort study. ESMO Open 8:100877. https://doi.org/10.1016/j.esmoop.2023.100877
Dummer R, Long GV, Robert C, Tawbi HA, Flaherty KT, Ascierto PA, Nathan PD, Rutkowski P, Leonov O et al (2022) Randomized phase III trial evaluating spartalizumab plus dabrafenib and trametinib for BRAF V600-mutant unresectable or metastatic melanoma. J Clin Oncol 40:1428–1438. https://doi.org/10.1200/JCO.21.01601
Shaikh SS, Zang Y, Hanmer J, Wang H, Lin Y, Davar D, Zarour HM, Kirkwood JM, Najjar YG (2022) Phase I trial of pembrolizumab plus vemurafenib and cobimetinib in patients with metastatic melanoma. Front Oncol 12:1022496. https://doi.org/10.3389/fonc.2022.1022496
Ascierto PA, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, Pereira RP, Eigentler T, Rutkowski P et al (2023) Overall survival with first-line atezolizumab in combination with vemurafenib and cobimetinib in BRAF(V600) mutation-positive advanced melanoma (IMspire150): second interim analysis of a multicentre, randomised, phase 3 study. Lancet Oncol 24:33–44. https://doi.org/10.1016/S1470-2045(22)00687-8
Acknowledgements
This study was funded by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA (MSD). We thank the patients and their families and caregivers for participating in this study, along with all investigators and site personnel. Medical writing assistance was provided by Autumn Kelly, MA, of ICON plc (Blue Bell, PA, USA). This assistance was funded by MSD.
Funding
This work was supported by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA. The sponsor was involved in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, and approval of the manuscript; and decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Contributions
Conception, design, or planning of the study: Howard Burris III, Lillian L. Siu. Acquisition, analysis, or interpretation of the data: Nehal J. Lakhani, Howard Burris III, Wilson H. Miller Jr, Mo Huang, Lin-Chi Chen, Lillian L. Siu. Provision of study materials/patients: Nehal J. Lakhani, Howard Burris III, Wilson H. Miller Jr, Lillian L. Siu. Review of the manuscript for important intellectual content: Nehal J. Lakhani, Howard Burris III, Wilson H. Miller Jr, Mo Huang, Lin-Chi Chen, Lillian L. Siu. Decision to submit the manuscript for publication: Nehal J. Lakhani, Howard Burris III, Wilson H. Miller Jr, Mo Huang, Lin-Chi Chen, Lillian L. Siu.
Corresponding author
Ethics declarations
Ethics approval
The study was compliant with International Council for Harmonisation Good Clinical Practice guidelines and the Declaration of Helsinki. The protocol (MK-8353–013) was approved by an institutional review board or independent ethics committee at each site.
Consent to participate
Written informed consent was obtained from all participants before enrollment.
Competing interest
Nehal J. Lakhani: Research funding for clinical trial costs from Merck, during the conduct of the study; research funding for clinical trial costs from ALX Therapeutics, Ascentage, Asana, Beigene, Constellation Pharma/Morphosys, Alexion, Cerulean, Forty Seven, Alpine, Merck, Pfizer, InhibRx, Regeneron, Apexian, Formation Biologics (forbius), Symphogen, CytomX, Sapience Therapeutics, Incyte, Jounce, Livzon, Northern Biologics, Innovent Biologics, Ikena, Odonate, Loxo/Lilly, Ikena, Mersana, Macrogenics, Helsinn, Seagen/Pfizer, Shattuck Labs, Samumed, Astellas, Alkermes/Mural Oncology, SK Life Sciences, KSQ Therapeutics/Roche, Tizona, Servier, Sapience, Repare Therapeutics, Gilead, Bristol Myers/Celgene, GSK, Janssen, Artios, Arcus Bio, Volastra, Incyte, Revolution Medicines, outside the submitted work; and honoraria for Advisory Board from Innovent Biologics and Ikena. Howard Burris III: Research funding to institution from AbbVie, Agios, Arch Oncology, ARMO Biosciences, Array BioPharma, Arvinas, AstraZeneca, Bayer, BeiGene, BioAtla, Boehringer Ingelheim, Bristol-Myers Squibb, CALGB, Celgene, Coordination Pharmaceuticals, CytomX, Lilly, EMD Serono, Roche/Genentech, Gossamer Bio, Harpoon Therapeutics, Hengrui Therapeutics, Incyte, Jounce Therapeutics, Kymab, MacroGenics, MedImmune, Merck, Millennium/Takeda, Moderna, NGM Biopharmaceuticals, Novartis, Pfizer, Revolution Medicines, Ryvu Therapeutics, Foundation Medicine, SeaGen, Tesaro, TG Therapeutics, Verastem, Vertex Pharmaceuticals, XBiotech, Zymeworks. Consulting fees (non-compensated) from Bristol-Myers Squibb and Novartis. Consulting fees paid to institution from AstraZeneca, GRAIL, Incyte, Roche, Vincerx Pharma. Stock ownership (self) HCA Healthcare. Wilson H. Miller Jr: Personal fees for consultant or Honoraria for BMS, Merck & Co., Inc., Rahway, NJ, USA, Roche, Novartis, Amgen, GSK, Sanofi, Mylan and EMD Serono. Research grant to institution from CIHR; CRS; SWCRF, Research Funding to Institution from Merck, BMS, Novartis, GSK, Roche, AstraZeneca, Methlgene, MedImmune, Sanofi, Array, MiMic, Ocellaris, Astellas, Pfizer, Genentech and Seagen. Mo Huang: Employee of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA, and stockholder in Merck & Co., Inc., Rahway, NJ, USA. Lin-Chi Chen: Employee of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA and stockholder in Merck & Co., Inc., Rahway, NJ, USA. Lillian L. Siu: Advisory Role/Consultation: Merck, Pfizer, AstraZeneca, Roche, GSK, Voronoi, Arvinas, Navire, Relay, Marengo, Daiichi Sankyo, Amgen, Medicenna, LTZ Therapeutics, Tubulis, Nerviano, Pangea, Incyte, Gilead. Research Support (institution): Merck, Novartis, Bristol-Myers Squibb, Pfizer/SeaGen, Boehringer-Ingelheim, GlaxoSmithKline, Roche/Genentech, AstraZeneca/Medimmune, Bayer, Abbvie, Amgen, Symphogen, EMD Serono, 23Me, Daiichi Sankyo, Gilead, Marengo, Incyte, LegoChem, Loxo/Eli Lilly, Medicenna, Takara. Leadership position: Treadwell Therapeutics (founder). Stock ownership: Agios.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lakhani, N.J., Burris, H., Miller, W.H. et al. A phase 1b study of the ERK inhibitor MK-8353 plus pembrolizumab in patients with advanced solid tumors. Invest New Drugs (2024). https://doi.org/10.1007/s10637-024-01461-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10637-024-01461-z