
Abstract
Sitravatinib (MGCD516), a spectrum-selective receptor tyrosine kinase inhibitor targeting TAM (TYRO3, AXL, MERTK) and split kinase family receptors, has demonstrated preclinical anti-tumor activity and modulation of tumor microenvironment. This first-in-human phase 1/1b study included sitravatinib dose exploration and anti-tumor activity evaluation in selected patients with advanced solid tumors. Primary objectives included assessment of safety, pharmacokinetics and clinical activity of sitravatinib. Secondary objectives included identifying doses for further investigation and exploring molecular markers for patient selection. In phase 1, 32 patients received 10–200 mg, while phase 1b dose expansion comprised 161 patients (150 mg n = 99, 120 mg n = 62). Maximum tolerated dose was determined as 150 mg daily. Dose-limiting toxicity was reported in 4/28 evaluable phase 1 patients (three at 200 mg, one at 80 mg). In phase 1b, 120 mg was defined as the recommended dose due to tolerability. Treatment-related adverse events (TRAEs) were experienced by 174/193 patients (90.2%); grade ≥ 3 TRAEs in 103 patients (53.4%). Most common TRAEs were diarrhea, fatigue, hypertension and nausea; TRAEs led to treatment discontinuation in 26 patients (13.5%) and death in one patient. Sitravatinib was steadily absorbed and declined from plasma with a terminal elimination half-life of 42.1–51.5 h following oral administration. Overall objective response rate was 11.8% in phase 1b, 13.2% in patients with non-small cell lung cancer (NSCLC) and 4.2% in patients with NSCLC with prior checkpoint inhibitor experience. Sitravatinib demonstrated manageable safety and modest clinical activity in solid tumors. NCT02219711 (first posted August 14, 2014).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Receptor tyrosine kinases (RTKs) regulate numerous cellular processes including cell proliferation, apoptosis and migration. Aberrant RTK activation is very common in cancer and represents an important therapeutic target for cancer treatment [1]. The utility of RTK-targeted therapies is well-documented in cancers with appropriate genetic alterations, with many targeted therapies now approved worldwide [1].
Sitravatinib (MGCD516) is an orally available small molecule inhibitor targeting closely related spectrum of RTKs, including TAM family receptors (TYRO3, AXL, MERTK) and split kinase family receptors (vascular endothelial growth factor receptor 2 [VEGF-R2], MET, RET and KIT). Several sitravatinib targets, such as TAM receptors, MET, RET and KIT, are dysregulated in many types of cancer through overexpression or genetic alteration, and contribute to tumor development [2]. Additionally, it is well-known that VEGF and its receptors can drive tumor angiogenesis in cancer [3]. Therefore, by targeting this collection of RTKs, sitravatinib may have meaningful anti-tumor effects.
The potent inhibitory activity of sitravatinib was demonstrated with biochemical half-maximal inhibitory concentration values ranging from 1.5–20 nM against target RTKs, including AXL, MERTK, VEGF-R, KIT and MET [4]. Additionally, sitravatinib has demonstrated anti-proliferative effects against solid tumor cells with a variety of phenotypes in vitro, as well as potent anti-tumor activity in xenograft tumor models of lung cancer and sarcoma with RTK dysfunction [4, 5].
Here, we report results for the first-in-human phase 1/1b study of sitravatinib, in patients with advanced solid tumors, including non-small cell lung cancer (NSCLC; clinicaltrials.gov identifier: NCT02219711) [6].
Materials and methods
Study design
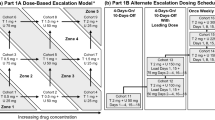
This study was a multicenter, phase 1/1b clinical trial evaluating the safety, pharmacokinetics (PK) and clinical activity of sitravatinib (free base formulation) in patients with advanced solid tumors. The study comprised two main parts: (1) dose escalation (phase 1); and (2) evaluation of clinical activity in patients selected based on histological diagnoses and/or the presence of defined molecular markers (phase 1b).
In the PK lead-in period, patients received a single oral dose of sitravatinib (10–200 mg) under fasted conditions with at least 200 mL of water, followed by PK sample collection for 3–7 days, depending on emerging PK information. After the PK lead-in period, patients commenced the daily regimen planned for their cohort. Blood samples were collected pre-dose and 0.5 (for 10 mg only), 1, 2, 4, 6, 8, 12, 24, 36, 48, 72 and 168 (for 20–200 mg dose levels) h post-dose from patients following a single oral dose, and at pre-dose, 0.5, 1, 2, 4, 6, 8, 12 and 24 h post-dose following multiple oral doses for the 10–200 mg levels.
The starting dose for the phase 1 dose escalation study was 10 mg once daily (QD). Dose escalation was carried out using the modified toxicity probability interval (mTPI) method [7] with the maximum tolerated dose (MTD) defined as the dose associated with a risk of dose-limiting toxicity (DLT) in 30 ± 5% of patients during the first treatment cycle.
Phase 1b cohorts were organized by diagnosis (renal cell carcinoma [RCC] or castrate-resistant prostate cancer [CRPC]) or by diagnosis of a solid tumor malignancy with a molecular alteration of interest for sitravatinib (such as gene amplification, mutation or rearrangement in MET, AXL, RET, NTRK, DDR2, KDR, PDGFRA, KIT or CBL).
This study was approved by an institutional review board at each participating site and was conducted in accordance with Good Clinical Practice guidelines, defined by the International Conference on Harmonisation. All patients provided written informed consent.
Choice of starting dose
The starting dose of 10 mg QD was selected based on non-clinical, 4-week toxicology studies conducted in rats and dogs. In rat studies, 10 mg/kg was the highest dose that did not exceed the severely toxic dose in 10% of the animals (STD10). The proposed human dose was based on one-tenth of the STD10 in rats corrected for body surface area (mg/m2).
Patient eligibility
Eligible patients were ≥ 18 years old with a histologically confirmed advanced, unresectable or metastatic solid tumor for which standard treatment was not available. Eligible patients had discontinued their most recent prior therapy ≥ 2 weeks before their first dose of study treatment and had recovered from any adverse events (AEs) of their prior therapy back to baseline or grade 1 (excluding alopecia); they also had an Eastern Cooperative Oncology Group performance score (ECOG PS) of 0–2.
Patients included in phase 1b cohorts had a selected diagnosis or tested positive for a designated target tumor molecular marker. The following populations were included: patients with NSCLC with a qualifying molecular alteration in MET, AXL, RET, NTRK, DDR2, KDR, PDGFRA, KIT or CBL; patients with other solid tumor types with a qualifying molecular alteration; patients with clear cell RCC (ccRCC) refractory to angiogenesis inhibitors; and patients with metastatic CRPC (mCRPC) with bone metastases.
Patients with symptomatic or uncontrolled brain metastases and/or with a second active cancer (excluding basal-cell carcinoma or cervical intraepithelial neoplasia) were excluded. For the phase 1b part, patients who had received prior treatment targeting the molecular marker of interest or patients with ccRCC or mCRPC previously treated with cabozantinib were excluded. Further eligibility and discontinuation criteria can be found in the Supplementary Information (Sects. 1.1 and 1.2, respectively).
Study objectives and assessments
The primary objectives were to characterize the safety profile, PK and clinical activity of sitravatinib. The secondary objectives included exploration of potential pharmacodynamic (PD) markers in blood plasma, identification of doses and regimens of sitravatinib for investigation of clinical activity and exploration of the use of molecular markers for the selection of patients with increased potential for response to sitravatinib.
Safety assessments included evaluation of DLTs, AEs, physical examinations, vital sign measurements, electrocardiogram recordings and laboratory tests. AEs, including laboratory abnormalities, were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03 from the day of the first dose of study treatment until ≥ 28 days after the last dose.
PK samples were collected after a single dose during the PK lead-in period and following multiple oral doses during the study. Plasma PK samples were assayed for quantification of sitravatinib. The lower limit of quantification was 0.05 ng/mL. PK parameters were determined using a noncompartmental analysis approach including maximum (peak) concentration (Cmax), time to reach Cmax following drug administration (tmax), area under the plasma concentration–time curves (AUCs), apparent total clearance of the drug from plasma after oral administration, apparent volume of distribution during the terminal phase after administration and terminal elimination half-life (t1/2). PD effects were examined by analyzing VEGF-A ligand and soluble (s)-VEGF-R2 levels in patients’ plasma samples collected before and after sitravatinib administration.
Clinical activity was assessed by objective response rate (ORR) according to Response Evaluation Criteria In Solid Tumors (RECIST) v1.1. Additional endpoints included duration of response (DoR), progression-free survival (PFS) and overall survival (OS) in phase 1b cohorts of patients based on diagnosis and tumor molecular alterations. An exploratory post-hoc analysis of patients with non-squamous NSCLC who experienced disease progression on prior checkpoint inhibitor (CPI) therapy was performed.
Disease status was evaluated according to RECIST v1.1 at baseline and every three cycles in phase 1, and every two cycles (6-week intervals) in phase 1b. Assessments were performed until objective disease progression was documented or until subsequent anti-cancer therapy started (see Supplementary Information [Sect. 1.3] for details).
Statistical analyses
The mTPI method [7] was applied for dose escalation. Assumptions applied in establishing the mTPI method included the involvement of up to 30 patients in each regimen explored, a 0.3 probability of DLT at the MTD and an acceptable variance around the MTD of ± 0.05. At least three patients were planned for each cohort, safety permitting.
A DLT was defined as a grade ≥ 4 hematologic abnormality lasting ≥ 4 days; grade 3 thrombocytopenia with clinically significant bleeding; febrile neutropenia; clinically significant grade ≥ 3 non-hematologic AEs not related to underlying malignancy; intolerable grade 2 AEs; or toxicity resulting in an inability to deliver 80% of the dose during the first treatment cycle.
The safety population included all patients who received ≥ 1 dose of sitravatinib. The DLT evaluable population included all phase 1 patients who had taken ≥ 80% of the assigned doses of treatment and were evaluated for toxicity 21 days in the first cycle or had experienced a DLT in cycle 1. The PK evaluable population included all patients with sufficient concentration-time data for PK parameter evaluation. The modified intent-to-treat (mITT) population included all phase 1b patients who received ≥ 1 dose of study drug.
Cohorts of patients defined by tumor molecular markers were evaluated using an optimal Simon 2-stage design. Additionally, an exploratory analysis to describe the ORR in patients with NSCLC was performed.
DoR, PFS and OS were reported descriptively and summarized using the Kaplan–Meier method. DoR was defined as the time from first documentation of objective tumor response (complete response [CR] or partial response [PR]) until first documentation of disease progression per RECIST 1.1 or death (any cause). PFS was defined as the time from first dose of study treatment until progressive disease as defined by RECIST 1.1 or death (any cause). OS was defined as the time from first dose of study treatment until death (any cause).
Results
Baseline characteristics
Overall, 193 patients received ≥ 1 dose of sitravatinib (safety population). The phase 1 dose escalation cohort comprised 32 patients treated with 10–200 mg, while 161 patients comprised the phase 1b dose expansion cohorts (Fig. 1). In the overall population (n = 193), median age was 65.0 years; 51.8% were male; most patients had ECOG PS 1 (61.7%), had received prior systemic therapy (93.3%) and had mainly NSCLC (29.0%) or RCC (21.2%) (Table 1). Other primary diagnoses are summarized in Supplementary Table S1. For the 53 patients with NSCLC in phase 1b, the histology was adenocarcinoma (n = 45), squamous carcinoma (n = 5) and ‘other’ (n = 3); median age was 66.0 years; 39.6% were male; 60.4% were white and 26.4% were Asian; and 60.4% had ECOG PS 1. In these patients, the median number of prior therapies was two (range, 1–8); 24 patients had received prior immunotherapy, with 20 also having received prior platinum-based chemotherapy. Among the 29 patients who did not receive prior immunotherapy, 24 had received prior platinum-based chemotherapy.

Flow diagram of patients included in this study (N = 193)
DLTs in Phase 1
Dose levels evaluated among the 32 patients in phase 1 were 10 mg (n = 4), 20 mg (n = 4), 40 mg (n = 5), 80 mg (n = 7), 110 mg (n = 4), 150 mg (n = 4) and 200 mg (n = 4). In phase 1, 4/28 (14.3%) DLT-evaluable patients experienced one DLT each (three at 200 mg and one at 80 mg). Reported DLTs (n = 1 [3.6% of the overall phase 1 population] for each) were intolerable grade 2 fatigue, mucosal inflammation and peripheral sensory neuropathy (all at 200 mg), and grade 3 palmar-plantar erythrodysesthesia (PPE) syndrome (at 80 mg). Thus, 150 mg QD was determined to be the MTD. During phase 1b, the starting dose was decreased to 120 mg QD based on tolerability. Overall, 99 patients in phase 1b received 150 mg sitravatinib as the starting dose; 62 patients received 120 mg sitravatinib as the starting dose.
Safety
In the safety population (N = 193), the median number of cycles was six and four for patients receiving 150 mg sitravatinib and 120 mg sitravatinib, respectively. In total, 174 patients (90.2%) experienced treatment-related AEs (TRAEs), including 103 (53.4%) who experienced grade ≥ 3 TRAEs (Table 2). The most common TRAEs were diarrhea (50.8%), fatigue (43.0%), hypertension (40.4%) and nausea (30.1%). The most common grade ≥ 3 TRAEs were hypertension (20.7%), diarrhea (10.4%) and fatigue (7.3%). Overall, 26 patients (13.5%) discontinued sitravatinib due to TRAEs; the most common reasons were diarrhea, nausea and fatigue (all in 2.1% of patients). Notably, more patients receiving 150 mg sitravatinib (17.2%) discontinued treatment due to TRAEs compared with patients receiving 120 mg sitravatinib (11.3%). Furthermore, the proportion of patients experiencing serious TRAEs and grade ≥ 3 TRAEs was higher in the 150 mg arm (22.2% and 61.6%, respectively) than in the 120 mg arm (8.1% and 51.6%, respectively; Table 2). Evaluation of TRAEs in patients treated with 120 or 150 mg suggested that 120 mg should be the recommended dose for further exploration. Overall, TRAEs led to treatment modification (dose reduction or treatment interruption) in 120 patients (62.2%), with the most common being diarrhea (17.6%), fatigue (15.0%), hypertension (15.0%) and PPE syndrome (11.9%). Cardiac arrest was the only TRAE leading to death (n = 1, 0.5% of the overall population). This patient was a past smoker with a medical history that included hypothyroidism, mesenteric vein thrombus and hyperlipidemia. Additional safety data are in Supplementary Table S2.
PK and PD analyses
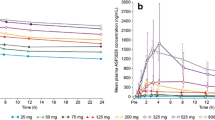
The PK evaluable population comprised 53 patients from the phase 1 and phase 1b cohorts; 40 patients participated in both the PK lead-in and cycle 1 portions, while seven patients participated only in the PK lead-in period and six patients participated only in the cycle 1 PK portion. A few patients in phase 1b receiving 120 mg sitravatinib also participated in the PK lead-in. After single oral administration of 10–200 mg under fasting conditions, sitravatinib was steadily absorbed with a median tmax ranging from 3.02–8.87 h and arithmetic mean t1/2 ranging from 42.1–51.5 h. After multiple oral administrations of 10–150 mg sitravatinib QD under fasting conditions, median tmax,ss ranged between 2.00–8.13 h. At the proposed clinical dose (120 mg QD), the interpatient variability for Cmax and AUCτ,ss was ~60%.
Steady-state appeared to have been reached by cycle 1 day 8, and Cmax,ss and AUCτ,ss accumulation ratios ranged from 1.82–6.89 and 2.13–8.34, respectively. Peak to trough ratios (PTR) in plasma for sitravatinib concentrations at steady state ranged from approximately 1.5–2.1-fold. Sitravatinib exposure (Cmax and AUCs) appeared to increase in an approximately dose-proportional manner following single- and multiple-dose administration from 10–200 mg, based on a statistical power model where the 95% confidence interval [CI] of the slope estimate for these PK parameters included the value of 1. Figure 2 shows the change in plasma concentration of sitravatinib over time after single and multiple doses. Key PK parameters are in Supplementary Table S3.

Plasma concentrations of sitravatinib following A a single dose and B multiple doses over time
PD analysis demonstrated a concentration-dependent modulation of each PD marker with a percent change from baseline for VEGF-A determined as a 200% increase (Supplementary Fig. S1). Based on the EC50 (30.9 ng/mL) from an exposure-response analysis, 120 mg sitravatinib QD is expected to achieve an approximately near maximal effect on the drug target VEGF-R2.
Clinical activity
In the overall phase 1b mITT population, the ORR was 11.8% (19/161), with all responses being PRs (Table 3). Additionally, phase 1b cohorts were analyzed by diagnosis (RCC or CRPC) or by identification of a tumor molecular alteration of interest (gene amplification, mutation or rearrangement involving MET, AXL, RET, NTRK, DDR2, KDR, PDGFRA, KIT or CBL gene loci). Responses were observed in patients with RCC and NSCLC, and included patients with tumor RET rearrangements, and MET, CBL and AXL alterations (Supplementary Table S4). For patients with NSCLC, the following molecular alterations were reported: RET alterations in 24 patients, MET alterations in 12 patients, CBL alterations in ten patients, Chr4q12 amplification in four patients and AXL, KDR and NTRK alterations in one patient each. The ORR for patients with NSCLC with a molecular alteration of interest was 13.2% (7/53), while that for patients with non-squamous NSCLC and prior CPI experience (exploratory analysis) was 4.2% (1/24) (Table 3).
In the overall phase 1b mITT population, at the time of data cut-off (median follow-up, 27.6 months), 6-month DoR was 71.3% (95% CI: 44.0, 87.0), with median DoR being 8.2 months (95% CI: 4.3, 16.6) (Supplementary Fig. S2A). In this population, 6-month PFS was 37.5% (95% CI: 29.2, 45.9), with median PFS being 4.3 months (95% CI: 3.1, 5.6) (Supplementary Fig. S2B); 12-month OS was 41.3% (95% CI: 32.7, 49.6), with median OS being 10.7 months (95% CI: 9.9, 11.9) (Supplementary Fig. S2C). Respective clinical activity data stratified by diagnosis and molecular sub-class are in Supplementary Table S4.
Discussion
Sitravatinib is a potent inhibitor of several RTKs that act as oncogenic drivers, including RET, TAM receptors and split kinase family receptors. This first-in-human phase 1/1b study demonstrated that sitravatinib had a manageable safety profile with AEs consistent with on-target inhibition and clinical activity was observed in selected populations.
Evaluation of sitravatinib in the phase 1 dose escalation stage resulted in a recommended phase 1b dose of 150 mg daily based on first cycle observations. However, after sequential evaluations of both 150 and 120 mg sitravatinib in phase 1b, 120 mg emerged as the recommended dose for further exploration based on a lower number of discontinuations, serious TRAEs and grade ≥ 3 TRAEs, compared with 150 mg.
Here, the PK profile of sitravatinib was characterized in patients with advanced solid tumor malignancies following single and multiple daily oral administrations from 10–200 mg. Under fasting conditions, sitravatinib was steadily absorbed with a median tmax ranging from 3.02–8.87 h and arithmetic mean t1/2 ranging from 42.1–51.5 h. At 120 mg QD, the between-patient variability for Cmax and AUCτ,ss was ~60%. Steady-state appeared to have been reached by cycle 1 day 8 and exposure (Cmax and AUCs) appeared to increase in a dose-proportional manner. PTR in plasma for sitravatinib concentrations at steady state ranged from approximately 1.5- to 2.1-fold, demonstrating a relatively small difference in steady-state Cmax and Cmin. The long t1/2 and low PTR strongly support a once-daily dosing regimen for sitravatinib. Regarding PD effects, the magnitude of increase in VEGF-A and decrease in s-VEGF-R2 following sitravatinib treatment is consistent with effectively targeting the VEGF-R family and with the effects observed for other agents targeting VEGF-R, including sunitinib, axitinib and cabozantinib [8,9,10].
Modest clinical activity of sitravatinib was demonstrated in the overall phase 1b population (ORR 11.8%), where almost 60% of patients had received ≥ 3 prior systemic therapies. The ORR for patients with NSCLC with a molecular alteration of interest was 13.2%, which is lower than that reported for next-generation therapies selectively targeting a single kinase, such as MET or RET [11, 12]. A post-hoc exploratory analysis of patients with NSCLC who experienced disease progression on prior CPI therapy showed that these patients did not gain a clinically meaningful benefit from sitravatinib monotherapy alone (ORR of 4.2%). Overall, these results suggested that sitravatinib, as a monotherapy, did not have significant anti-tumor activity in the analyzed cohorts, including NSCLC. However, sitravatinib is being investigated in combination with CPIs, based on its immunomodulatory role of the tumor microenvironment (TME).
CPI therapy is now established as a breakthrough treatment for various solid tumors, including NSCLC. Although many patients benefit from this treatment, some patients experience disease progression and develop resistance to CPIs through various mechanisms, such as the establishment of an immunosuppressive TME. Previous studies have revealed that targeting TAM receptors has an immunomodulatory effect on the TME, particularly involving polarization of tumor-associated macrophage populations [13]. Additionally, it has been demonstrated that targeting VEGF or VEGF-R decreases the number of immunosuppressive cells, such as regulatory T cells and myeloid-derived suppressor cells (MDSCs), in tumor models and patients with cancer [14]. Therefore, the role of sitravatinib in the modulation of the TME has been further explored. Preclinical data demonstrated that sitravatinib could modulate the TME by affecting macrophage polarization through inhibition of the expression of IL-4-stimulated arginase 1 (a marker of M2 polarization) [5]. Additionally, sitravatinib inhibited expression of the M2 markers arginase 1, YM-1 and Fizz-1 upon stimulation with conditioned media from murine cancer cells – a source of TAM receptor ligands – and reduced immunosuppressive cell populations, such as MDSCs and M2 macrophages, in vivo [5]. Notably, these changes facilitated a T effector cell response and augmented the effects of anti-programmed death (PD)-1/PD-ligand-1 (anti-PD-1) therapy in these xenograft models [5], and it was therefore hypothesized that the combination of sitravatinib with an anti-PD-1 agent, such as nivolumab, may have a synergistic clinical effect. This hypothesis was tested in a phase 1 window-of-opportunity trial evaluating sitravatinib monotherapy followed by sitravatinib combined with nivolumab in oral cavity cancer [15]. Sitravatinib monotherapy resulted in a less immunosuppressive TME with a reduction in MDSCs and repolarization of macrophages from the M2 to the M1 phenotype [15, 16]. Additionally, sitravatinib followed by the combination with nivolumab for one cycle prior to surgery resulted in tumor reduction for all patients, including one CR [15].
Based on these preliminary data, the anti-tumor efficacy of sitravatinib with CPI therapy has been explored in the MRTX-500 phase 2 study, which evaluated sitravatinib plus nivolumab in advanced NSCLC and indicated encouraging results in patients who had progressed on, or after, prior CPI therapy [17]. These promising data have led to the evaluation of sitravatinib plus nivolumab compared with docetaxel in patients with non-squamous NSCLC in the ongoing phase 3 SAPPHIRE study (NCT03906071) [18]. Additionally, another phase 3 study (NCT04921358) [19] is evaluating sitravatinib plus tislelizumab (a PD-1 inhibitor) compared with docetaxel in patients with locally advanced or metastatic NSCLC.
Conclusion
In this study, the PK profile of sitravatinib was well characterized, indicating a steady absorption following oral administration and an appropriate t1/2 for a once-daily dosing regimen. Sitravatinib had a manageable safety profile and demonstrated modest clinical activity in patients with heavily pretreated advanced solid tumors. Ongoing studies are evaluating sitravatinib in combination with other agents, such as anti-PD-1 inhibitors, in multiple tumor types, including NSCLC.
Data availability
Mirati will honor legitimate requests for clinical trial data from qualified researchers, upon request, as necessary for conducting methodologically sound research. Mirati will provide access to data and clinical study reports (CSRs) for clinical trials for which results are posted on the clinicaltrials.gov registry for products or indications that have been approved by regulators in the US and EU. In general, data will be made available for request approximately 12 months after clinical trial completion. Relevant components of the protocol and statistical analysis plan for this study will also be made available upon request.
References
Yamaoka T, Kusumoto S, Ando K, Ohba M, Ohmori T (2018) Receptor tyrosine kinase-targeted cancer therapy. Int J Mol Sci 19:3491. https://doi.org/10.3390/ijms19113491
Akalu YT, Rothlin CV, Ghosh S (2017) TAM receptor tyrosine kinases as emerging targets of innate immune checkpoint blockade for cancer therapy. Immunol Rev 276:165–177. https://doi.org/10.1111/imr.12522
Goel HL, Mercurio AM (2013) VEGF targets the tumour cell. Nat Rev Cancer 13:871–882. https://doi.org/10.1038/nrc3627
Patwardhan PP, Ivy KS, Musi E, de Stanchina E, Schwartz GK (2016) Significant blockade of multiple receptor tyrosine kinases by MGCD516 (sitravatinib), a novel small molecule inhibitor, shows potent anti-tumor activity in preclinical models of sarcoma. Oncotarget 7:4093–4109. https://doi.org/10.18632/oncotarget.6547
Du W, Huang H, Sorrelle N, Brekken RA (2018) Sitravatinib potentiates immune checkpoint blockade in refractory cancer models. JCI Insight 3:e124184. https://doi.org/10.1172/jci.insight.124184
Clinicaltrials.gov (2020) Phase 1/1b Study of MGCD516 in Patients With Advanced Cancer. https://clinicaltrials.gov/ct2/show/NCT02219711. Updated January 30, 2020. Accessed October 21, 2021
Ji Y, Wang S-J (2013) Modified toxicity probability interval design: a safer and more reliable method than the 3 + 3 design for practical phase I trials. J Clin Oncol 31:1785–1791. https://doi.org/10.1200/jco.2012.45.7903
DePrimo SE, Bello CL, Smeraglia J, Baum CM, Spinella D, Rini BI et al (2007) Circulating protein biomarkers of pharmacodynamic activity of sunitinib in patients with metastatic renal cell carcinoma: modulation of VEGF and VEGF-related proteins. J Transl Med 5:32. https://doi.org/10.1186/1479-5876-5-32
Bruce JY, Scully PC, Carmichael LL, Eickhoff JC, Perlman SB, Kolesar JM et al (2015) Pharmacodynamic study of axitinib in patients with advanced malignancies assessed with (18)F-3’deoxy-3’fluoro-L-thymidine positron emission tomography/computed tomography. Cancer Chemother Pharmacol 76:187–195. https://doi.org/10.1007/s00280-015-2779-7
Leibowitz-Amit R, Pintilie M, Khoja L, Azad AA, Berger R, Laird AD et al (2016) Changes in plasma biomarkers following treatment with cabozantinib in metastatic castration-resistant prostate cancer: a post hoc analysis of an extension cohort of a phase II trial. J Transl Med 14:12. https://doi.org/10.1186/s12967-015-0747-y
Drilon A, Oxnard GR, Tan DSW, Loong HHF, Johnson M, Gainor J et al (2020) Efficacy of selpercatinib in RET fusion–positive non–small-cell lung cancer. N Engl J Med 383:813–824. https://doi.org/10.1056/NEJMoa2005653
Paik PK, Felip E, Veillon R, Sakai H, Cortot AB, Garassino MC et al (2020) Tepotinib in non–small-cell lung cancer with MET exon 14 skipping mutations. N Engl J Med 383:931–943. https://doi.org/10.1056/NEJMoa2004407
Aehnlich P, Powell RM, Peeters MJW, Rahbech A, thor Straten P (2021) TAM receptor inhibition–implications for cancer and the immune system. Cancers (Basel) 13:1195. https://doi.org/10.3390/cancers13061195
Yang J, Yan J, Liu B (2018) Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol 9:978. https://doi.org/10.3389/fimmu.2018.00978
Oliva M, Chepeha D, Araujo DV, Diaz-Mejia JJ, Olson P, Prawira A et al (2021) Antitumor immune effects of preoperative sitravatinib and nivolumab in oral cavity cancer: SNOW window-of-opportunity study. J Immunother Cancer 9:e003476. https://doi.org/10.1136/jitc-2021-003476
Oliva M, Araujo DV, Chepeha DB, Prawira A, Spreafico A, Bratman SV et al (2020) SNOW: sitravatinib and nivolumab in oral cavity cancer (OCC) window of opportunity study. J Immunother Cancer 38:6569–6569. https://doi.org/10.1200/JCO.2020.38.15_suppl.6569
Leal TA, Berz D, Rybkin I, Iams WT, Bruno D, Blakely C et al (2021) 11910 – MRTX-500: phase II trial of sitravatinib (sitra) + nivolumab (nivo) in patients (pts) with non-squamous (NSQ) non-small cell lung cancer (NSCLC) progressing on or after prior checkpoint inhibitor (CPI) therapy. Ann Oncol 32(suppl_5):S949–S1039
Clinicaltrials.gov (2021) Phase 3 Study of Sitravatinib Plus Nivolumab vs Docetaxel in Patients With Advanced Non-Squamous Non-Small Cell Lung Cancer (SAPPHIRE). https://clinicaltrials.gov/ct2/show/NCT03906071. Updated September 30, 2021. Accessed October 21, 2021
Clinicaltrials.gov (2021) Tislelizumab in Combination With Sitravatinib in Patients With Locally Advanced or Metastatic Non-Small Cell Lung Cancer. https://clinicaltrials.gov/ct2/show/NCT04921358. Updated September 21, 2021. Accessed October 21, 2021.
Acknowledgements
Medical writing support under the direction of the authors was provided by Flaminia Fenoaltea, MSc, and Charlotte Kennerley, PhD, of Ashfield MedComms, an Ashfield Health company, and funded by Mirati Therapeutics, Inc.
Funding
This work was supported by Mirati Therapeutics, Inc.
Author information
Authors and Affiliations
Contributions
Conception and design: SG, JGC, STCN, RCC, SP. Collection and assembly of data: TMB, RH, LB, SG, DA, RDC, AA, KDE, JSW, JGC, SP. Data analysis and interpretation: TMB, BCC, LB, SG, DWK, DA, RDC, KDE, YL, XY, JGC, STCN, RCC, SP. Manuscript writing: TMB, BCC, LB, TLW, SG, DWK, DA, KDE, JSW, YL, XY, JGC, RCC, SP. Final approval of manuscript: All authors. Accountable for all aspects of the work: All authors.
Corresponding author
Ethics declarations
Ethics statement
This study was approved by an institutional review board at each participating site and was conducted in accordance with Good Clinical Practice guidelines, defined by the International Conference on Harmonisation. All patients provided written informed consent.
Competing interests
TMB declares Consulting or advisory role: Guardant Health, Loxo, Pfizer, Exelixis, Blueprint Medicines, Foundation Medicine, Bayer, AstraZeneca; Consulting or advisory role to institution: Ignyta, Moderna Therapeutics, Pfizer; Speakers’ bureau: Bayer, Bristol-Myers Squibb, Eli Lilly; Research funding to institution: Daiichi Sankyo, Medpacto, Inc., Incyte, Mirati Therapeutics, Inc., MedImmune, Abbvie, AstraZeneca, Leap Therapeutics, MabVax, Stemline Therapeutics, Merck, Eli Lilly, GlaxoSmithKline, Novartis, Pfizer, Genentech/Roche, Deciphera, Merrimack, Immunogen, Millennium, Ignyta, Calithera Biosciences, Kolltan Pharmaceuticals, Principa Biopharma, Peleton, Immunocore, Roche, Aileron Therapeutics, Bristol-Myers Squibb, Amgen, Moderna Therapeutics, Sanofi, Boehringer Ingelheim, Astellas Pharma, Five Prime Therapeutics, Jacobio, Top Alliance BioScience, Loxo, Janssen, Clovis Oncology, Takeda, Karyopharm Therapeutics, Onyx, Phosplatin Therapeutics, Foundation Medicine, ARMO BioSciences; Travel, accommodation, expenses: Astellas Pharma, AstraZeneca, Celgene, Clovis Oncology, EMD Serono, Genentech, Eli Lilly, Merck, Novartis, Pharmacyclics, Sysmex, Pfizer BCC declares Research funding: Novartis, Bayer, AstraZeneca, MOGAM Institute, Dong-A ST, Champions Oncology, Janssen, Yuhan, Ono, Dizal Pharma, MSD, Abbvie, Medpacto, GIInnovation, Eli Lilly, Blueprint Medicines, Interpark Bio Convergence Corp.; Consulting role: Novartis, AstraZeneca, Boehringer Ingelheim, Roche, Bristol Myers Squibb, Ono, Yuhan, Pfizer, Eli Lilly, Janssen, Takeda, MSD, Janssen, Medpacto, Blueprint Medicines; Stock ownership: TheraCanVac Inc., Gencurix Inc., Bridgebio Therapeutics, KANAPH Therapeutic Inc., Cyrus Therapeutics, Interpark Bio Convergence Corp.; Scientific Advisory Board: KANAPH Therapeutic Inc., Brigebio Therapeutics, Cyrus Therapeutics, Guardant Health, Joseah BIO; Board of director: Gencurix Inc., Interpark Bio Convergence Corp.; Royalty: Champions Oncology; Founder: DAAN Biotherapeutics. RH declares Consulting: Abbvie, Novartis, EMD Serono, Daichii Sankyo; Research funding to institution, not to self: Abbvie, Agios, Corvus, Daichii Sankyo, Exelixis, Mirati Therapeutics, Inc., Novartis, Eli Lilly, Turning Point. LB declares Data Monitoring Committee: ORIC; Advisory board: Turning Point Therapeutics, Daichi, Bristol Myers Squibb, Janssen, Merck, Regeneron, Bayer, Takeda, Boehringer Ingleheim, Novartis, Genentech and Sanofi; Scientific committee: Neuvogen; Research funding to institution: Beyondspring. TLW declares research funding to institution from: AbbVie, AstraZeneca, Clovis Oncology, Genmab, GlaxoSmithKline-Tesaro, Mersana, Repare, Roche-Genentech. SG was funded by Mirati Therapeutics, Inc. to conduct this trial (to the institution). DWK declares research funding to institution from: Alpha Biopharma, Amgen, AstraZeneca/MedImmune, Boehringer Ingelheim, Bridge BioTherapeutics, Chong Keun Dang, Daiichi-Sankyo, GlaxoSmithKline, Hanmi, Janssen, Merus, Mirati Therapeutics, Inc., MSD, Novartis, ONO Pharmaceutical, Pfizer, Roche/Genentech, Takeda, Turning Point Therapeutics, Xcovery, Yuhan; Travel and accommodation support for advisory board meeting attendance from: Amgen, Daiichi-Sankyo. DA declares Consulting or scientific advisory board support: Vaccinex, Merck, Blueprint Medicines, Boehringer Ingelheim, Cue Biopharma, Kura Oncology, Eisai, Exelixus, twoXAR, Immunitas, Natco Pharma, TargImmune Therapeutics, and Xilio; Institutional research support: Vaccinex, Pfizer, Eli Lilly, Merck, Celgene/Bristol Myers Squibb, Novartis, AstraZeneca, Atara Bio, Blueprint Medicine, Celldex, Enzychem, Kura, Exelixis, Innate, Sensei, Debiopharm International, ISA Therapeutics, Gilead Sciences, BeiGene, Roche, Hookpia Biotech, Adlai Nortye USA, Rubius Therapeutics, Epizyme, and Matrix Biomed. RDC declares Consulting: Alkermes, Bristol Myers Squibb, Castle Biosciences, Ideaya, Immunocore, InxMed, Iovance, Merck, Novartis, Oncosec, Pierre Fabre, PureTech Health, Regeneron, Sanofi Genzyme, Sorrento Therapeutics, Trisalus; Clinical/Scientific Advisory Boards: Aura Biosciences, Chimeron, Rgenix; Research Funding to Columbia University: Amgen, Astellis, AstraZeneca, Bristol Myers Squibb, Corvus, Ideaya, Immunocore, Iovance, Merck, Mirati Therapeutics, Inc., Novartis, Pfizer, Plexxikon, Regeneron, Roche/Genentech. AA declares to be an advisory board member/consultant for: Merck, Sharp & Dohme Corp, Bristol Myers Squibb, AstraZeneca, Genentech, Roche, Pfizer, Progenics, Prometheus; Travel/Accommodations/Expenses: Merck Sharp & Dohme Corp, Bristol Myers Squibb; Research funding through institution: Merck Sharp & Dohme Corp, AstraZeneca, Bristol Myers Squibb, Astellas, Seattle Genetics, Genentech, Pfizer, Progenics, Prometheus, Eli Lilly, ASCO, Celgene, Harpoon Therapeutics. KDE declares research support for clinical trials from Mirati Therapeutics, Inc. JSW declares research funding to institution from: Agenus, AstraZeneca, AstraZeneca/MedImmune, Bicycle therapeutics, BioNTech, Boehringer Ingelheim, Celgene, CicloMed, Clovis Oncology, Cyteir, Daiichi Sankyo, Genentech/Roche, GlaxoSmithKline, H3 Biomedicine, Hutchison MediPharma, Ignyta, Jacobio, Janssen Research & Development, Klus Pharma, Kymab, Loxo, LSK BioPharma, Macrogenics, Merck, Moderna Therapeutics, Phoenix Pharmaceuticals, Prelude Therapeutics, QiLu Pharmaceutical, Revolution Medicines, Ribon Therapeutics, Syndax, Stemline Therapeutics, Taiho Pharmaceutical, Tesaro, TopAlliance BioSciences Inc, Xencor, Artios, Erasca, Inc., immuno-Gen, Cullinan Oncology, Immuno-Onc, Bayer Health, Biosplice, Zymeworks, BioTheryX, TeneoBio, Nurix, IgM Biosciences, Aevi Genomic Medicine, PureTech, StingThera, Forty Seven, Treadwell Therapeutics, MabSpace Biosciences, Novartis, Olema Oncology, Seven and Eight Biopharmaceuticals, ORIC Pharmaceuticals, Relay Therapeutics, Black Diamond Therapeutics. YL is a full-time employee of Mirati Therapeutics, Inc. XY is a full-time employee of Mirati Therapeutics, Inc. and owns Mirati stocks. JGC is an employee, shareholder and executive officer at Mirati Therapeutics, Inc. STCN is a former employee of Mirati Therapeutics, Inc. and owns Mirati stocks. RCC is a full-time employee of Mirati Therapeutics, Inc. SP declares Clinical Trials Research (paid to institution) from: Arcus, Bristol Myers Squibb, Eli Lilly, GlaxoSmithKline, Holy Stone Healthcare Co., Tyme, Ipsen, Mirati Therapeutics, Inc., Novartis, Xencor, Astellas, Janssen; Advisory Board/Consultant: 4D Pharma, Xencor, Ipsen, Zymeworks, Amal Therapeutics, Novartis.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Previous presentations: Presented in part at the American Society of Clinical Oncology annual meeting 2016 and 2018.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bauer, T., Cho, B.C., Heist, R. et al. First-in-human phase 1/1b study to evaluate sitravatinib in patients with advanced solid tumors. Invest New Drugs 40, 990–1000 (2022). https://doi.org/10.1007/s10637-022-01274-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-022-01274-y