
Summary
Introduction. Erwinase® (native Erwinia chrysanthemi L-Asparaginase (nErA)) is an approved second-line treatment for acute lymphoblastic leukaemia (ALL) in children and adolescents, who develop hypersensitivity or neutralising antibodies to E.coli derived L-Asparaginases (ASNases). However, nErA has a short in vivo half-life requiring frequent dosing schedules in patients. In this study, nErA was covalently conjugated to PEG molecules with the aim of extending its half-life in vivo. Methods. Firstly, efficacy of this novel product PEG-nErA was investigated on human ALL cell lines (Jurkat, CCRF-CEM and CCRF-HSB2), in vitro. Secondly, its pharmacokinetic (PK) and pharmacodynamic (PD) characteristics were determined, in vivo (12 rats in each group). Results. It was found that the specific activity (U/mg of enzyme) and the kinetic constant (KM) of nErA remained unaltered post PEGylation. PEG-nErA was shown to have similar cytotoxicity to nErA (IC50: 0.06–0.17 U/mL) on human ALL cell lines, in vitro. Further, when compared to nErA, PEG-nErA showed a significantly improved half-life in vivo, which meant that L-Asparagine (Asn) levels in plasma remained depleted for up to 25 days with a four-fold lower dose (100 U/kg) compared with 72 h for nErA at 400 U/kg dose. Conclusion. Overall, this next generation product PEG-nErA (with improved PK and PD characteristics compared to nErA) would bring a significant advantage to the therapeutic needs of ALL patients and should be further explored in clinical trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
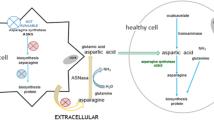
L-asparaginase (ASNase) is an integral part of the treatment of acute lymphoblastic leukaemia (ALL) in children and adolescents. ASNase catalyses the production of L-Aspartic acid (Asp) from L-Asparagine (Asn). ALL cells lack Asn synthase activity [1, 2], which means that ALL cells cannot generate their own supply of Asn and depend on circulating endogenous Asn. The mechanism of action of ASNase treatment is to deplete circulating Asn in blood, starving ALL cells of the essential amino acid Asn leading to cell death. There are now four ASNase preparations that have been approved for ALL treatment. For years, options for ASNase treatment have included native Escherichia coli ASNase (nEcA), native Erwinia chrysanthemi ASNase (nErA) and PEGylated Escherichia coli ASNase (PEG-nEcA). Recently, a recombinant version of Erwinia chrysanthemi ASNase has been approved by the US FDA [3]. However, to date no PEGylated Erwinia-based ASNase has been approved for clinical use in ALL treatment.
Most ALL patients receive PEG-nEcA as a first-line treatment, however around 30–40% of those patients develop hypersensitivity reactions [4, 5]. This hypersensitivity can either be due to an adverse immune reaction to the enzyme or due to the generation of neutralising antibodies (often referred to as silent inactivation), resulting in reduced clinical function of the enzyme [6]. nErA is immunologically distinct from PEG-nEcA and does not present cross-reactivity to antibodies generated from PEG-nEcA [7]. Hence, nErA (or, as of July 2021, rErA) is offered as a second-line treatment to those ALL patients who develop hypersensitivity to PEG-nEcA. Erwinase® or Erwinaze® are the proprietary names for nErA. Erwinase® was first developed in the late 1960s and early 1970s at Porton Down, UK [8, 9] and is marketed in many countries throughout the world.
Both nEcA and nErA consist of a homotetramer of 35 kDa subunits and although they are structurally similar proteins, their sequences have only 46% identity [10]. Both Asn and L-Glutamine (Gln) act as substates for nEcA and nErA enzymes, however they have significantly different affinities, as determined by the Michaelis constants (KM) [11, 12]. Similar to that of nEcA, the in vivo half-life of Erwinase® is low in comparison to PEG-nEcA, resulting in the requirement for frequent dosing of the native enzymes (several times per week) compared with the PEGylated version of nEcA.
It follows that one potential way to increase nErA half-life and reduce the dosing frequency is to PEGylate the enzyme, in a manner analogous to that of PEG-nEcA. Understanding the impact of PEGylation on the in vivo half-life of the drug is a critical step in the development of this form of nErA. Thus, in the research described here, PEGylation of nErA was investigated both in vitro on human ALL cell lines and in vivo in terms of its pharmacokinetic (PK) and pharmacodynamic (PD) profile.
Methods and materials
All reagents were purchased from Sigma Aldrich, UK, unless specified otherwise.
PEGylation of nErA
nErA drug substance (DS) was obtained from Porton Biopharma Limited (PBL), Porton Down, UK, manufactured as described previously [13]. nErA DS was then conjugated to NHS-activated monomethoxy poly (ethylene glycol) (PEG or mPEG) with a mean molecular weight of 5 kDa with succinimidyl carbonate (SC) linkers (Laysan Bio, USA), using standard protocols [17].
Specific activity and KM measurement
The specific activity (U per mg of enzyme) and KM of nErA and PEG-nErA were determined using methods described previously [14].
In vitro Study—Cytotoxicity assay
The in vitro efficacy of PEG-nErA vs nErA was assessed using cytotoxicity assay at an external contract research organisation (CRO) (Cyprotex, UK). The enzymes (± PEG) were supplied by PBL and the cell lines were acquired from ATCC, UK. The following acute lymphoblastic leukaemia (ALL) cell lines were used: Jurkat, CCRF-CEM and CCRF-HSB2. Cell lines used for negative control included non-ALL cell lines 1301 and THP1. Briefly, the cell lines were cultured in RPMI 1640 medium supplemented with 200 mM Glu, 10% Foetal bovine serum and 1% Penicillin–Streptomycin. nErA and PEG-nErA samples diluted in water were added to wells of a 96-well plate and serially diluted to arrive at final concentrations between 50U/mL and 5 × 10–5 U/mL, with one set of control wells (water only) in triplicates. To each well, 100µL of cells was added at either 1 × 105 or 2 × 105 cells per 100µL. The plates were incubated at 37 °C for 72 h. Then, 20µL of MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) (5 mg/mL) reagent was added to each well and the plate incubated for a further 2 h at 37 °C. Finally, 100µL of solubilisation reagent (0.37% hydrochloric acid in isopropanol) was added and mixed thoroughly with a pipette before reading the plate absorbance at 570 nm with background at 750 nm using a plate reader (Molecular Devices, UK). The cell viability data was normalised and IC50 data were measured at 72 h using Graph-Pad Prism 7.
In vivo study
The in vivo efficacy of PEG-nErA versus an nErA control was assessed at an external CRO (Envigo, UK). Briefly, male CD Sprague–Dawley rats (Charles River, UK) received a single intravenous administration of control PBS (0 U/kg), nErA (400 U/kg), or PEG-nErA (1, 5, 25, 100 or 400 U/kg) (Table 1). There were 12 animals in each group. Venous blood samples were taken from each animal at defined timepoints (pre-dose and up to 1032 h) for measuring ASNase activity (PK parameter) and Asn and Gln concentration (PD parameter). Blood samples were taken in K2EDTA tubes and separated by centrifugation (2000 g at 4 °C for 10 min) before aliquoting the resulting plasma into 2 separate vials and freezing at -70 °C. Samples were shipped to PBL on dry ice. The animal experiment protocol was approved by the local ethics committee at Envigo.
Measurement of plasma ASNase activity
ASNase activity in plasma samples was measured using the method described in Lanvers et al., [15]. Briefly, AHA solution (10 mM L-Aspartic acid Beta-Hydroxamate (AHA) was prepared in 50 mM MOPS, 0.015% (w/v) BSA, pH 7.2) was incubated at 37 °C for at least 10 min. In a 96-well microplate, 10µL of plasma and 90µL of pre-warmed AHA solution was added. As a reference standard, 10µL of nErA reconstituted in water with known activity was substituted for plasma. The microplate was incubated at 37 °C for 40 min and 125µL of 24.5% (v/v) trichloroacetic acid was added. The plate was centrifuged at 2500 rpm for 5 min. In a separate microplate, 150µL of 1 M disodium carbonate and 50µL of 2% 8-hydroxyquinoline in ethanol were added to each well. Then, 50µL of the supernatant from the original plate was added, and the plate was heated at 55 °C for 40 min, allowed to cool to room temperature before reading in a plate reader (Molecular Devices, UK) at 710 nm.
Measurement of plasma Asn and Gln levels
Plasma Asn and Glu concentrations were determined using LC–MS/MS at Q3 Analytical Ltd. Briefly, Asn and Glu were weighed, dissolved in deionised water to a concentration of 1 mg/mL. Midodrine was dissolved in dimethyl sulphoxide (DMSO) as a 1 mg/mL stock solution and diluted to 1 μg/mL in acetonitrile to make the internal standard (IS) solution. Samples were prepared by mixing 50μL of plasma (standard or sample) with 150μL of IS solution, centrifuged and the supernatant transferred to a 1.5 mL Eppendorf. The solution was reduced to 50µL using a centrifugal evaporator (Genevac, UK) for 45 min at 40 °C and added to 70μL AccQ Tag borate buffer (Waters, UK) and 20μL Waters AccQ Tag reagent solution (Waters, UK) (1 vial dissolved in 4 mL reconstitution solution). The tube was sealed and incubated at 60 °C for 10 min. After cooling to room temperature, 5μL was injected onto the LC–MS system. Amino acids were separated on a reverse-phase symmetry C18 column (Waters, UK), using gradient of mobile phase A (0.1% formic acid, 5 mM heptafluorobutyric acid) and mobile phase B (0.1% formic acid in acetonitrile, 5 mM heptafluorobutyric acid) for 7 min. The peak area ratios of the amino acids to the IS were used as a surrogate measure of the concentrations of each amino acid.
Statistics
For cytotoxicity assays, IC50 values were obtained using GraphPad Prism 7 and are presented with the standard error from four-parameter curve fitting. The difference in efficacy of nErA vs PEG-nErA in vitro was calculated using t-test, p value of < 0.05 was considered statistically significant. For PK/PD studies, results were presented as ± 1 standard deviation around the mean.
Results
PEGylation of nErA
PEGylation of nErA (up to 350 mol PEG per mole 35kD monomer) did not alter the specific activity and KM values of nErA (Table 2). Using a capillary isoelectric focussing technique, it was previously found that PEGylation of nErA incorporated approximately 7 mPEG molecules per 35 kDa nErA monomer for reaction molar ratios on the order of 40 PEG molecules to one enzyme molecule [16]. PEG-nErA was formulated using trehalose and lyophilised, which was found to offer extended stability at 5 °C ± 3 °C for up to 3 years (data not shown).
Cytotoxicity of PEG-nErA vs nErA, in vitro
The viability of ALL cell lines treated with serial dilutions of nErA and PEG-nErA was measured at 72 h using the MTT assay. The data showed similar ALL cell viability when cell lines were treated with nErA or PEG-nErA (Fig. 1). The absolute viability data differed from cell line to cell line and this may be due to simple variation in the way the various cells reacted with the MTT reagent. IC50 values, representing the interpolated enzyme dose at which 50% of the ALL cells were killed were calculated. Overall, both nErA and PEG-nErA showed equivalent cytotoxicity on human ALL cell lines after 72 h (Fig. 1, Table 3). Note that cytotoxicity data in Fig. 1 were normalised using Graph Pad Prism 7 so that all data sets were on a 0 – 100% scale (0% representing the lower horizontal asymptote and 100% representing the upper horizontal asymptote), in order to allow a direct comparison between all data sets. In the negative control cell lines, there was no cytotoxic effect from both nErA and PEG-nErA (data not shown).

Cytotoxic effect of PEG-nErA and nErA on human ALL cell lines after 72 h, in vitro using MTT assay. Cells were treated with increasing dose ± PEG Erwinase (5 × 10–5 to 50 U/mL), in triplicates. The data shows that PEGylation of nErA did not alter the overall cytotoxicity of nErA against human ALL cells. IC50 values were obtained using GraphPad Prism and are presented with the standard error from 4-parameter curve fitting
In vivo studies
Effect of treatment on plasma Asn and Gln concentration in vivo
The effect of PEGylation of nErA on plasma Asn and Gln was clearly observed during in vivo investigation. With respect to Asn concentration in plasma, animals receiving 400 U/kg nErA (without PEG) showed a typical response in terms of the timing and duration of the Asn trough, where Asn is depleted after dosing (time ‘zero’) to a non-detectable level for approximately 72 h before rebound to endogenous pre-dose levels. However, for PEG-nErA, dependent on dose, animals maintained a much longer Asn trough period. In the groups receiving 1, 5, 25, 100 and 400 U/kg of PEG-nErA, Asn depletion followed a dose-dependent relationship (Fig. 2A). Plasma Asn levels remained at undetectable levels for at least 25 and 31 days at 100 and 400U/kg PEG-nErA dose levels, respectively. For all dose levels except the highest (400U/kg PEG-nErA), complete rebound in Asn levels was observed over the duration of the study; for the highest dose level, only a partial rebound was observed.

For plasma Gln, a reduction from endogenous levels was only observed for PEG-nErA. For the non-PEGylated nErA control, there was no detectable reduction in plasma Gln level after dosing. At PEG-nErA doses ≥ 25U/kg, plasma Gln levels were depleted in a dose-dependent manner (Fig. 2B). There was no detectable reduction in circulating Gln levels for the lowest two PEG-nErA groups (1 and 5U/kg).
Plasma ASNase activity
nErA PEGylation was found to prolong the presence of ASNase in vivo in a dose-dependent manner. The plasma ASNase activity levels remained high in the PEG-nErA groups for longer duration as can be observed by the decreasing slope of the activity data points with increasing PEG-nErA dose (Fig. 3). Plasma activity was measured at 0.05 U/mL in the nErA group at 48 h, and a similar activity was measured at 32 days in the 400 U/kg PEG-nErA group. Data in Fig. 3 was plotted as the geometric mean of each activity from the 12 animals in a group. In lieu of a direct PK measurement, the plasma activity data in Fig. 3 were used as a surrogate marker to calculate PK parameters for this study.

Pharmacokinetic study performed in rats (n = 12 per dose, per group). Figure 3A: L-Asparaginase activity measured in rat plasma, determined using enzymatic activity method described in Lanvers et al.. [15]. Each data point represents the geometric mean of results from 12 animals. Error bars refer to ± 1 standard deviation around the geometric mean. Figure 3B: Correlation between Cmax from Regression Analysis and Theoretical Cmax. Error bars refer to ± 1 standard error around the fitted Cmax data
The Cmax from the activity data was estimated using a curve fitting technique (Table 4). As the animals in the study received only one dose of drug, by definition the Cmax is the concentration of drug in the animal’s blood at time zero. However, no plasma samples were taken from the animals in the PEG-nErA groups until 72 h, so a regression was performed and the Cmax back-calculated as the y-intercept. The geometric mean activity data were plotted per Fig. 3 and fitted using a logarithmic curve fit in Graph Pad Prism 7. Graph Pad was also used to calculate the area under the curve (AUC). In addition to Cmax and AUC, a number of additional PK parameters including clearance and half-life were calculated using standard equations for a single intravenous bolus [13]. Note that the calculated PK parameters were not meaningful at the lower dose levels (1 and 5 U/kg), as the enzyme activity levels were low for these dose levels resulting in poor curve fitting. Moreover, this study was geared towards observing the effects of higher PEG-nErA doses, and hence the time spacing between observations and samples was insufficient to observe effects at the lowest PEG doses. Hence, PK parameters for the 1 and 5 U/kg dose levels are not reported in Table 4.
Observing the AUC data, a dose-dependent increase was observed for the PEGylated enzyme, and the effect of PEGylation on AUC was evident (comparing nErA and PEG-nErA groups at 400 U/kg dosage). A predictable increase in half-life (t1/2) was also observed, at 9.7 vs 96.5 for nErA and PEG-nErA groups respectively at the 400U/kg dosing level. Further as expected, the clearance rate was significantly higher for animals in the nErA group vs those receiving PEG-nErA.
The calculated Cmax from logarithmic regression may also be compared to the theoretical Cmax. The theoretical Cmax (also shown in Table 4) is calculated based simply on the amount of drug dosed to an animal divided by the animal’s blood volume. The average weight of a rat at time zero in this study was 365 g. The blood volume of a 365 g rat was estimated at 28.5 mL and used to calculate the theoretical Cmax. A plot of this theoretical Cmax versus the Cmax obtained from linear regression of the activity data (Fig. 3B) showed that a linear relationship exists between the two sets of data. Based on this, it can be concluded that the PK data obtained in this study are valid and represent a reasonable representation (within the constraints of using a surrogate PK marker) of the animal metabolism of PEG-nErA.
Discussion
This study demonstrated that PEGylation of nErA had no impact on its specific activity and kinetic enzyme performance. PEGylation of biomolecules sometimes results in decreased drug-target binding affinity due to steric interference [18]. Similar studies have reported loss of 35–60% of the ASNase activity as a result of PEGylation [19, 20]. The apparent lack of impact of PEGylation may be due to the characteristics of the nErA molecule, but further investigation would be required to understand this relationship.
The in vitro cell viability data and IC50 values demonstrated that nErA and PEG-nErA appear identical with respect to cytotoxicity against human ALL cells. Furthermore, no cytotoxic effect was observed in the negative control cell lines (i.e., non-ALL cell lines) for both nErA and PEG-nErA. This further demonstrated that PEGylation was not the pivotal variable in cytotoxicity, and that the relationship between nErA (or ASNase more generally) and the ASNS negative characteristic of ALL cell lines is the important determinant for cytotoxicity [12].
The in vivo study showed that PEG-nErA had improved PK and PD characteristics in comparison to nErA. PEG-nErA exhibited a significantly improved half-life and depleted plasma Asn levels for longer duration in a dose-dependent manner in rats, in comparison to nErA. A single intravenous dose of 400 U/kg was found to be sufficient to deplete the plasma Asn levels for 31 days with PEG-nErA vs 72 h with nErA. In fact, PEG-nErA at ≥ 25U/kg dose outperformed nErA in terms of duration of Asn depletion, suggesting that 25–100 U/kg PEG-ErA dose could fit the typical two to three-week therapeutic window typically observed for currently used PEGylated ASNase products. Note that this final assertion does not take into account the potential effects of differences in treatment species with respect to dosing.
Further, there was no measurable depression in circulating Gln levels for the nErA and low-dose PEG-nErA groups (1 and 5U/kg dosage). However, plasma Gln levels remained depleted for a longer duration in PEG-nErA treated groups (≥ 25U/kg dosage) in a dose-dependent manner. There is significant debate in the scientific literature about the importance of anti-Gln activity in ASNase treatment of ALL. While glutaminase activity of ASNase is sometimes cited as being responsible for ALL side effects and toxicity in humans [21, 22], other literature reports claim that glutaminase side activity is critical to ALL blast apoptosis and overall efficacy of treatment [23,24,25]. Although the role of glutaminase activity in the clinical efficacy of Erwinia asparaginases and in particular PEG-nErA is not clear, the relationship between PEGylation and measurable, prolonged Gln depletion may be an important factor in any resulting clinical trials and thus, should continue to be monitored.
The plasma activity data compared well against the amino acid profile data. Plasma activity levels remained elevated for a longer duration in the PEG-nErA groups (48 h vs 32 days at 400 U/kg dose), and plasma activity also followed a dose-dependent relationship. Plasma ASNase activity level in the range of 0.05–0.4 U/mL is reported to be necessary for anti-leukemic therapeutic activity in humans [1, 4, 15, 26,27,28,29,30,31,32,33]. Plasma enzyme activity threshold of 0.1 U/mL was reached within only 24 h with nErA (400 U/kg), compared to 15 days with a lower dose of 100 U/kg PEG-nErA. This data compared well with other PEG-ASNase products (100 U/kg), reporting 0.1 U/mL plasma enzyme activity levels at 15 days in rats [34]. Furthermore, at lower plasma activity threshold of 0.01 U/mL, PEG-nErA appears to perform better at the same dose level (100 U/kg); this threshold was reached at 34 days with PEG-nErA vs 21 and 26 days with oncaspar and MC0609, respectively [34].
ALL patients who develop hypersensitivity or neutralising antibodies to E.coli based ASNase products rely on Erwinase® (nErA) as a second line treatment. However, Erwinase® has a limited supply issue, often leading to patient prioritisation [35]. Supposedly, recently approved JZP-458 (rErA produced in Pseudomonas fluorescens) potentially helps fulfil this unmet need by providing additional asparaginase preparation for those patients [3], however the hypersensitivity rate from this product is not yet understood or reported. Furthermore, PEG-nErA would also help resolve Erwinase® shortage problem, as clinically, it may be expected that PEG-nErA can be given at lower dosage and less frequently to patients compared with nErA or rErA, while achieving the same desired therapeutic outcome and improved patient experience.
PEGylation of biomolecules is already a well-established technology, shown to have significant net beneficial effects for patients [36,37,38,39]. Based on the results reported here, it is reasonable to expect that PEG-nErA would have an overall similar clinical safety and immunogenicity profile to nErA. However, further research will be necessary to advance this potential product variant to clinical use. It is envisaged that a pre-clinical study will be necessary to investigate the toxicity profile and PK/PD characteristics of PEG-nErA in a bigger animal model such as beagle dog. It is known that up to 33% of patients who switch from E.coli based ASNase products to nErA after developing clinical hypersensitivity suffer from allergic reactions to nErA [4]. It is expected that PEGylation of nErA would potentially reduce immunogenicity of nErA as well as the formation of neutralising anti-ASNase antibodies. Furthermore, repeated administration of PEGylated therapeutics is known to induce formation of anti-PEG antibodies associated with hypersensitivity reactions and rapid clearance [40, 41]. Hence, it would be useful to monitor anti-ASNase antibody and anti-PEG antibody levels during future pre-clinical studies. However, it has to be noted that measuring anti-drug antibody levels in animals may be of limited benefit in predicting clinical consequences in humans [42]. Subsequently, a phase I clinical trial in humans can be proposed to investigate safety and effective dose range in healthy volunteers.
Data availability
Yes.
Abbreviations
- AHA:
-
L-aspartic β-hydroxamate
- ALL:
-
Acute Lymphoblastic Leukaemia
- Asn:
-
L-Asparagine
- ASNase:
-
L-Asparaginase
- AUC:
-
Area Under the Curve
- Cmax :
-
Maximum Concentration
- CRO:
-
Contract Research Organisation
- DS:
-
Drug substance
- Gln:
-
L-Glutamine
- Glu:
-
Glutamic acid
- IC50 :
-
Interpolated Dose Representing 50% of Cells Killed
- IS:
-
Internal Standard
- KM :
-
Michaelis constant
- K2EDTA:
-
Potassium Ethylene Diamine Tetraacetic Acid
- LC–MS/MS:
-
Tandem Mass Spectrometry
- mPEG:
-
Monomethoxy poly(ethylene glycol)
- MTT:
-
(3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide
- nEcA:
-
Escherichia coli ASNase
- nErA:
-
Native Erwinia chrysanthemi ASNase
- PBL:
-
Porton Biopharma Limited
- PBS:
-
Phosphate Buffered Saline
- PD:
-
Pharmacodynamics
- PEG:
-
Poly(ethylene glycol)
- PEG-nEcA:
-
PEGylated Escherichia coli ASNase
- PK:
-
Pharmacokinetics
- SC-mPEG:
-
Succinimidyl Carbonate Monomethoxy Poly(ethylene glycol)
- SD:
-
Standard Deviation
- U:
-
Units (of Enzyme
References
Dubbers A, Wurthwein G, Muller HJ, Schulze-Westhoff P, Winkelhorst M, Kurzknabe E et al (2000) Asparagine synthetase activity in paediatric acute leukaemias: AML-M5 subtype shows lowest activity. Br J Haematol 109:427–429. https://doi.org/10.1046/j.1365-2141.2000.02015.x
Leslie M, Case MC, Hall AG, Coulthard SA (2006) Expression levels of asparagine synthetase in blasts from children and adults with acute lymphoblastic leukaemia. Br J Haematol 132:740–742. https://doi.org/10.1111/j.1365-2141.2005.05945.x
Maese L, Rizzari C, Coleman R, Power A, van der Sluis I, Rau RE (2021) Can recombinant technology address asparaginase Erwinia chrysanthemi shortages?. Pediatr Blood Cancer 2021:e29169. https://doi.org/10.1002/pbc.29169
Pieters R, Hunger SP, Boos J, Rizzari C, Silverman L, Baruchel A et al (2011) L-asparaginase treatment in acute lymphoblastic leukemia: a focus on Erwinia asparaginase. Cancer 117:238–249. https://doi.org/10.1002/cncr.25489
Vrooman LM, Supko JG, Neuberg DS, Asselin BL, Athale UH, Clavell L et al (2010) Erwinia asparaginase after allergy to E. coli asparaginase in children with acute lymphoblastic leukemia. Pediatr Blood Cancer 54:199–205. https://doi.org/10.1002/pbc.22225
Salzer W, Bostrom B, Messinger Y, Perissinotti AJ, Marini B (2018) Asparaginase activity levels and monitoring in patients with acute lymphoblastic leukemia. Leuk Lymphoma 59:1797–1806. https://doi.org/10.1080/10428194.2017.1386305
Zalewska-Szewczyk B, Gach A, Wyka K, Bodalski J, Mlynarski W (2009) The cross-reactivity of anti-asparaginase antibodies against different L-asparaginase preparations. Clin Exp Med 9:113–116. https://doi.org/10.1007/s10238-008-0026-9
Buck PW, Elsworth R, Miller GA, Sargeant K, Stanley JL, Wade HE (1971) The batch production of L-asparaginase from Erwinia carotovora. J Gen Microbiol 65:i
Cammack KA, Marlborough DI, Miller DS (1972) Physical properties and subunit structure of L-asparaginase isolated from Erwinia carotovora. Biochem J 126:361–379. https://doi.org/10.1042/bj1260361
Sudhir AP, Agarwaal VV, Dave BR, Patel DH, Subramanian RB (2016) Enhanced catalysis of L-asparaginase from Bacillus licheniformis by a rational redesign. Enzyme Microb Technol 86:1–6. https://doi.org/10.1016/j.enzmictec.2015.11.010
Nguyen HA, Su Y, Lavie A (2016) Design and Characterization of Erwinia Chrysanthemi l-Asparaginase Variants with Diminished l-Glutaminase Activity. J Biol Chem 291:17664–17676. https://doi.org/10.1074/jbc.M116.728485
Beckett A, Gervais D (2019) What makes a good new therapeutic L-asparaginase? World J Microbiol Biotechnol 35:152. https://doi.org/10.1007/s11274-019-2731-9
Gervais D, Allison N, Jennings A, Jones S, Marks T (2013) Validation of a 30-year-old process for the manufacture of L-asparaginase from Erwinia chrysanthemi. Bioprocess Biosyst Eng 36:453–460. https://doi.org/10.1007/s00449-012-0802-5
Gervais D, Foote N (2014) Recombinant deamidated mutants of Erwinia chrysanthemi L-asparaginase have similar or increased activity compared to wild-type enzyme. Mol Biotechnol 56:865–877. https://doi.org/10.1007/s12033-014-9766-9
Lanvers C, Vieira Pinheiro JP, Hempel G, Wuerthwein G, Boos J (2002) Analytical validation of a microplate reader-based method for the therapeutic drug monitoring of L-asparaginase in human serum. Anal Biochem 309:117–126. https://doi.org/10.1016/s0003-2697(02)00232-4
Minshull TC, Wood A, Roberts D, Hallam C, Lewis J, Orekoya A et al (2020) Determination of extent of PEGylation using denaturing capillary isoelectric focussing. Anal Biochem 611:113953. https://doi.org/10.1016/j.ab.2020.113953
Equations for PK/PD measurements. Acessed 2017
Fishburn CS (2008) The pharmacology of PEGylation: balancing PD with PK to generate novel therapeutics. J Pharm Sci 97:4167–4183. https://doi.org/10.1002/jps.21278
Chien WW, Allas S, Rachinel N, Sahakian P, Julien M, Le Beux C et al (2014) Pharmacology, immunogenicity, and efficacy of a novel pegylated recombinant Erwinia chrysanthemi-derived L-asparaginase. Invest New Drugs 32:795–805. https://doi.org/10.1007/s10637-014-0102-9
Soares AL, Guimaraes GM, Polakiewicz B, de Moraes Pitombo RN, Abrahao-Neto J (2002) Effects of polyethylene glycol attachment on physicochemical and biological stability of E. coli L-asparaginase. Int J Pharm 237:163–70. https://doi.org/10.1016/s0378-5173(02)00046-7
Mahajan RV, Kumar V, Rajendran V, Saran S, Ghosh PC, Saxena RK (2014) Purification and characterization of a novel and robust L-asparaginase having low-glutaminase activity from Bacillus licheniformis: in vitro evaluation of anti-cancerous properties. PLoS ONE 9:e99037. https://doi.org/10.1371/journal.pone.0099037
Chan WK, Lorenzi PL, Anishkin A, Purwaha P, Rogers DM, Sukharev S et al (2014) The glutaminase activity of L-asparaginase is not required for anticancer activity against ASNS-negative cells. Blood 123:3596–3606. https://doi.org/10.1182/blood-2013-10-535112
Panosyan EH, Grigoryan RS, Avramis IA, Seibel NL, Gaynon PS, Siegel SE et al (2004) Deamination of glutamine is a prerequisite for optimal asparagine deamination by asparaginases in vivo (CCG-1961). Anticancer Res 24:1121–1125
Offman MN, Krol M, Patel N, Krishnan S, Liu J, Saha V et al (2011) Rational engineering of L-asparaginase reveals importance of dual activity for cancer cell toxicity. Blood 117:1614–1621. https://doi.org/10.1182/blood-2010-07-298422
Parmentier JH, Maggi M, Tarasco E, Scotti C, Avramis VI, Mittelman SD (2015) Glutaminase activity determines cytotoxicity of L-asparaginases on most leukemia cell lines. Leuk Res 39:757–762. https://doi.org/10.1016/j.leukres.2015.04.008
Schore RJ, Devidas M, Bleyer A, Reaman GH, Winick N, Loh ML et al (2019) Plasma asparaginase activity and asparagine depletion in acute lymphoblastic leukemia patients treated with pegaspargase on Children's Oncology Group AALL07P4(.). Leuk Lymphoma. 60:1740–8. https://doi.org/10.1080/10428194.2018.1542146
Ahlke E, Nowak-Gottl U, Schulze-Westhoff P, Werber G, Borste H, Wurthwein G et al (1997) Dose reduction of asparaginase under pharmacokinetic and pharmacodynamic control during induction therapy in children with acute lymphoblastic leukaemia. Br J Haematol 96:675–681. https://doi.org/10.1046/j.1365-2141.1997.d01-2089.x
Boos J, Werber G, Ahlke E, Schulze-Westhoff P, Nowak-Gottl U, Wurthwein G et al (1996) Monitoring of asparaginase activity and asparagine levels in children on different asparaginase preparations. Eur J Cancer 32A:1544–1550. https://doi.org/10.1016/0959-8049(96)00131-1
Vieira Pinheiro JP, Ahlke E, Nowak-Gottl U, Hempel G, Muller HJ, Lumkemann K et al (1999) Pharmacokinetic dose adjustment of Erwinia asparaginase in protocol II of the paediatric ALL/NHL-BFM treatment protocols. Br J Haematol 104:313–320. https://doi.org/10.1046/j.1365-2141.1999.01192.x
Riccardi R, Holcenberg JS, Glaubiger DL, Wood JH, Poplack DG (1981) L-asparaginase pharmacokinetics and asparagine levels in cerebrospinal fluid of rhesus monkeys and humans. Cancer Res 41:4554–4558
Avramis VI, Sencer S, Periclou AP, Sather H, Bostrom BC, Cohen LJ et al (2002) A randomized comparison of native Escherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-risk acute lymphoblastic leukemia: a Children’s Cancer Group study. Blood 99:1986–1994. https://doi.org/10.1182/blood.v99.6.1986
Douer D, Yampolsky H, Cohen LJ, Watkins K, Levine AM, Periclou AP et al (2007) Pharmacodynamics and safety of intravenous pegaspargase during remission induction in adults aged 55 years or younger with newly diagnosed acute lymphoblastic leukemia. Blood 109:2744–2750. https://doi.org/10.1182/blood-2006-07-035006
Rizzari C, Zucchetti M, Conter V, Diomede L, Bruno A, Gavazzi L et al (2000) L-asparagine depletion and L-asparaginase activity in children with acute lymphoblastic leukemia receiving i.m. or i.v. Erwinia C. or E. coli L-asparaginase as first exposure. Ann Oncol 11:189–93. https://doi.org/10.1023/a:1008368916800
Borghorst S, Hempel G, Poppenborg S, Franke D, Konig T, Baumgart J (2014) Comparative pharmacokinetic/pharmacodynamic characterisation of a new pegylated recombinant E. coli L-asparaginase preparation (MC0609) in Beagle dog. Cancer Chemother Pharmacol 74:367–78. https://doi.org/10.1007/s00280-014-2506-9
Lee BM, Jecker NS, Marron JM, Rosenberg AR (2021) Striving for equity: Patient prioritization during a critical cancer drug shortage. Pediatr Blood Cancer 2021:e29280. https://doi.org/10.1002/pbc.29280
Veronese FM, Mero A (2008) The impact of PEGylation on biological therapies. BioDrugs 22:315–329. https://doi.org/10.2165/00063030-200822050-00004
Marini BL, Perissinotti AJ, Bixby DL, Brown J, Burke PW (2017) Catalyzing improvements in ALL therapy with asparaginase. Blood Rev 31:328–338. https://doi.org/10.1016/j.blre.2017.06.002
Peters BG, Goeckner BJ, Ponzillo JJ, Velasquez WS, Wilson AL (1995) Pegaspargase versus asparaginase in adult ALL: a pharmacoeconomic assessment. Formulary 30:388–393
Elspar (2012) Lundbeck to stop making Elspar citing cost of securing API supply
Kloos R, van der Sluis IM, Mastrobattista E, Hennink W, Pieters R, Verhoef JJ (2020) Acute lymphoblastic leukaemia patients treated with PEGasparaginase develop antibodies to PEG and the succinate linker. Br J Haematol 189:442–451. https://doi.org/10.1111/bjh.16254
Kloos RQH, Pieters R, Jumelet FMV, de Groot-Kruseman HA, van den Bos C, van der Sluis IM (2020) Individualized Asparaginase Dosing in Childhood Acute Lymphoblastic Leukemia. J Clin Oncol 38:715–724. https://doi.org/10.1200/JCO.19.02292
Brinks V, Jiskoot W, Schellekens H (2011) Immunogenicity of therapeutic proteins: the use of animal models. Pharm Res 28:2379–2385. https://doi.org/10.1007/s11095-011-0523-5
Acknowledgements
We would like to thank Porton Biopharma Limited (PBL) for providing facilities and funding to perform this research. Thanks to Alexandra McEntee for performing plasma activity assays and to Neil Burton (Q3 Analytical Ltd) for amino acid quantitation by LC-MS/MS. Further thanks to Dave Roberts, Christine Hallam, Julie Miller, Nick Foote, Stuart Smith and the PBL Development team members for supporting this work. Special thanks to Professor Vaskar Saha for helpful discussions on the design of the study.
Funding
Porton Biopharma Limited (PBL) provided facilities and funding to perform this research.
Author information
Authors and Affiliations
Contributions
Tapasvi Modi—Methodology, Investigation and Writing; David Gervais—Conceptualization, Writing—review & editing, Visualization, Project administration and Supervision.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The animal experiment protocol was approved by the local ethics committee at Envigo.
Consent of publication
N/A
Informed consent
N/A
Disclosure of potential conflicts of interest
None, TM and DG were employed by PBL (the manufacturer of nErA) at the time of the study.
Research involving Human participants and/or animals—The animal experiment protocol was approved by the local ethics committee at Envigo.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Modi, T., Gervais, D. Improved pharmacokinetic and pharmacodynamic profile of a novel PEGylated native Erwinia chrysanthemi L-Asparaginase. Invest New Drugs 40, 21–29 (2022). https://doi.org/10.1007/s10637-021-01173-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-021-01173-8