
Summary
BI 836826 is a chimeric immunoglobulin G1 antibody targeting CD37, a tetraspanin transmembrane protein predominantly expressed on normal and malignant B cells. This phase I, open-label study used a modified 3 + 3 design to evaluate the safety, maximum tolerated dose (MTD), pharmacokinetics, and preliminary activity of BI 836826 in patients with relapsed/refractory B cell non-Hodgkin lymphoma (NHL; NCT01403948). Eligible patients received up to three courses comprising an intravenous infusion (starting dose: 1 mg) once weekly for 4 weeks followed by an observation period of 27 (Course 1, 2) or 55 days (Course 3). Patients had to demonstrate clinical benefit before commencing treatment beyond course 2. Forty-eight patients were treated. In the dose escalation phase (1–200 mg) involving 37 Caucasian patients, the MTD was 100 mg. Dose-limiting toxicities occurred in four patients during the MTD evaluation period, and included stomatitis, febrile neutropenia, hypocalcemia, hypokalemia, and hypophosphatemia. The most common adverse events were neutropenia (57%), leukopenia (57%), and thrombocytopenia (41%), and were commonly of grade 3 or 4. Overall, 18 (38%) patients experienced infusion-related reactions, which were mostly grade 1 or 2. Preliminary evidence of anti-tumor activity was seen; three patients responded to treatment, including one complete remission in a Korean patient with diffuse large B cell lymphoma. BI 836826 plasma exposure increased more than proportionally with increasing doses. BI 836826 demonstrated preliminary activity; the most frequent adverse events were hematotoxicity and infusion-related reactions which were manageable after amending the infusion schedule. Although BI 856826 will not undergo further clinical development, these results confirm CD37 as a valid therapeutic target in B cell NHL.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Non-Hodgkin lymphomas (NHLs) are a heterogeneous group of malignant disorders with variable clinical and biologic features that together caused over 248,000 deaths worldwide in 2018 [1, 2]. Most NHLs are of B cell origin, and the most common form, diffuse large B cell lymphoma (DLBCL), is an aggressive subtype that is readily curable with immunochemotherapy in the majority of patients but progresses rapidly if left untreated [1, 3]. In contrast, indolent follicular lymphoma (FL) is characterized by favorable prognosis but is still considered to be incurable in the vast majority of cases [4]. Although the addition of the anti-CD20 monoclonal antibody, rituximab, to the NHL therapeutic armamentarium more than 20 years ago greatly improved outcomes, prognosis remains poor for DLBCL patients who are not cured by first-line therapy or FL patients who suffer early progression [3, 4]. New treatments able to overcome resistance to standard immunochemotherapy are therefore urgently needed.
The tetraspanin CD37 is a transmembrane protein that is expressed on B-cells at multiple stages of development, from pre-B to peripheral mature B-cells [5, 6]. CD37 appears to play multiple roles in immune cells, including regulation of apoptosis/survival signaling, B/T cell interaction, and T cell proliferation [5, 7,8,9]. Notably, CD37 is highly expressed on malignant B cells, including most subtypes of NHL [10, 11], making it an attractive therapeutic target. Clinical studies with the anti-CD37 agents otlertuzumab (TRU-016) [12], IMGN529 [13], and AGS67E [14] suggest that targeting CD37 is a viable therapeutic strategy, with evidence of anti-tumor activity seen in patients with DLBCL, FL and other B cell NHLs.
BI 836826 is a chimeric immunoglobulin G1 (IgG1) monoclonal antibody that targets human CD37. It comprises a high-affinity mouse antibody to CD37 with an engineered CH2 domain to improve binding to human Fcγ receptors [6]. Preclinical studies demonstrated strong pharmacodynamic and antitumor effects of BI 836826 [6]. It showed high intrinsic proapoptotic activity accompanied by homotypic aggregation against malignant B cells and promoted antibody-dependent cell-mediated cytotoxicity (ADCC) against lymphoma cells. In blood samples from healthy volunteers, BI 836826 depleted normal B cells and spiked B-lymphoma cells more potently than rituximab. In experiments in vivo, BI836826 dose-dependently reduced peripheral B cells in CD37 transgenic mice and cynomolgous monkeys. It also suppressed tumor growth in a Ramos mouse B cell lymphoma model [6]. In a first-in-human study, BI 836826 showed evidence of anti-tumor activity in patients with relapsed or refractory chronic lymphocytic leukaemia (CLL) [15]. These findings warranted the evaluation of BI 836826 in patients with B cell NHL. Here we report the results of a phase I, dose-escalation study to evaluate the maximum tolerated dose (MTD), safety, and efficacy of BI 836826 in patients with relapsed or refractory B cell NHL (ClinicalTrials.gov identifier: NCT01403948).
Patients and methods
Patients
The study enrolled patients with relapsed or refractory NHL of B cell origin (mature B cell lymphoma according to the World Health Organization [16]) who were not considered candidates for intensive anti-lymphoma therapy. Patients were required to have either aggressive NHL and at least one (at least two for patients enrolled in France) prior anti-CD20-containing immunochemotherapeutic regimens, or indolent NHL with previous anti-CD20 therapy and at least two prior therapies. Other key requirements were age ≥ 18 years, Eastern Cooperative Oncology Group performance status (ECOG PS) of <2, and a life expectancy of ≥3 months. Patients in the expansion cohort were also required to have measurable disease on computed tomography (CT) scan.
Patients were ineligible if they had primary central nervous system (CNS) lymphoma or known CNS involvement, or a prior history of malignancy other than a mature B cell neoplasm, basal or squamous cell carcinoma of the skin, or carcinoma in situ of the uterine cervix or breast, unless free of disease and without treatment for at least 5 years. In addition, patients were excluded if they had inadequate organ function; a significant concurrent medical disease or condition; or chronic or ongoing infectious disease requiring treatment at enrollment or within the previous 2 weeks, including CMV viremia, HIV, or active hepatitis B or C.
The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines, and the study protocol was approved by the Institutional Review Boards or Independent Ethics Committees of all participating institutions. Written informed consent was obtained from all patients.
Study design and treatment
This was an open-label, single-arm, phase I dose-escalation study. The primary objective was to determine the MTD of BI 836826 in patients with NHL of B cell origin. Secondary objectives were to assess the safety and tolerability of BI 836826, to perform pharmacokinetic (PK) and pharmacodynamic (PD) analyses, and to evaluate efficacy.
BI 836826 was administered as an intravenous infusion, initially at a set rate of 83.4 mL/h. Following the occurrence of a Grade 4 infusion-related reaction (IRR), the protocol was amended, and the infusion rate was changed to a slowly increasing dosing schedule beginning at 10 mL/h and increasing every 30 min by 10 mL/h if tolerable, to a maximum of 80 mL/h.
Each patient could receive a maximum of three courses of treatment. The first treatment course was of 7 weeks duration and consisted of one infusion per week for 4 weeks, followed by a 27-day observation period (Supplemental Fig. 1). A second, similar course was administered if BI 836826 was well tolerated and no signs of disease progression were seen at the first response assessment during week 7. Patients received a third course, of up to 12 weeks duration, if they continued to tolerate BI 836826 well, and achieved a response (complete remission [CR], CR unconfirmed [CRu], partial remission [PR]) or stable disease according to CT assessment during the second treatment cycle. Premedication with acetaminophen/paracetamol, antihistamine, and glucocorticoid was mandatory at the start of therapy. Supportive care, such as granulocyte colony-stimulating factors and prophylactic treatment with antibiotics and antivirals was permitted according to local guidelines.
In the first part of the study, BI 836826 was administered to Caucasian patients in escalating dose tiers, starting at 1 mg. Single-patient cohorts were treated until a drug-related adverse event (AE) of Grade ≥ 2 occurred during the MTD evaluation period (the time from the first administration of BI 836826 until 7 days after the second administration). Subsequent cohorts were expanded to three patients following a fixed dose-escalation design with dose de-escalation steps. Dose escalation continued until the MTD was reached, defined as the highest dose of BI 836826 for which the incidence of dose-limiting toxicities (DLTs) was no more than 17% (i.e., one out of six patients) during the MTD evaluation period.
DLTs were defined as any drug-related Grade ≥ 3 non-hematologic AE, except IRRs. Complications resulting from hematologic AEs, such as bleeding due to thrombocytopenia or infection due to neutropenia, were classified as non-hematologic AEs and captured as DLTs. Although not deemed to be DLTs, hematologic AEs (e.g., neutropenia, thrombocytopenia and anemia) were considered for definition of the dose for further development.
Following the determination of the MTD in Caucasian patients, a separate cohort was to be enrolled into an expansion phase at the MTD. Initial enrollment into this cohort included Korean patients. Early results indicated that the tolerability of BI 836826 differed between Korean and Caucasian patients, as three of four Korean patients developed significant drug-related AEs. Consequently, the protocol was amended to explore lower dose levels in a designated escalation cohort of Korean patients, with the aim of defining an MTD in that population.
Study assessments
The primary endpoint of the study was the determination of the MTD and the number of DLTs observed during the period from the first administration of BI 836826 until 7 days after the second administration. Other safety assessments included the incidence and severity of AEs, graded according to the Common Terminology Criteria for Adverse Events, Version 4.0, laboratory parameters, and physical examinations.
Secondary efficacy endpoints were: tumor size reduction; best overall response, according to Standardized or Revised Response Criteria for Malignant Lymphoma [17]; progression-free survival (PFS); and failure-free survival (FFS), defined as the time from first administration of BI 836826 until disease progression, death, or start of next NHL therapy.
The PK parameters of BI 836826 were evaluated using non-compartmental analysis methods and included maximum measured plasma concentration (Cmax); terminal half-life (t1/2); area under the plasma concentration-time curve over the time interval of one treatment course (AUC0-tz); area under the plasma concentration-time curve over the time interval from zero extrapolated to infinity (AUC0-∞); total plasma clearance (CL); and volume of distribution after intravenous infusion at steady state (Vss). Blood samples for PK analysis were taken in cycle 1 before the start of infusion (pre-dose) and at 5, 7, 9, 24, and 72 h, and shortly before the start of the next infusion at 168 h. BI 836826 levels were determined using a validated enzyme-linked immunosorbent assay.
Exploratory PD evaluations were conducted into the potential prognostic biomarker β-2 microglobulin, and to investigate the potential predictive value of polymorphisms in the Fc fragment of the IgG receptor (FCGR) gene. β-2 microglobulin levels were determined using a routine blood sample obtained at screening, while FCGR genotyping was conducted using DNA extracted from a blood sample obtained at the first treatment visit in Cycle 1.
Statistical methods
Statistical analyses were descriptive, and no formal statistical tests were performed for the dose groups. Exploratory analysis of time-to-event endpoints (PFS, FFS) was conducted using Kaplan–Meier methods.
Results
Patients and treatment exposure
A total of 59 patients were enrolled, and 48 treated with BI 836826 in 12 centers across Germany, France, and the Republic of Korea between 25 January 2012 and 28 February 2018.
Thirty-seven Caucasian patients were treated in the dose escalation phase. The median age was 69.0 years (range, 25–83), and 70% of patients were male (Table 1). The patients had been extensively pretreated, with most having received between three and six prior treatments, and more than half had FL. Eleven Korean patients were treated. The median age was 63.0 years (range, 27–79), 36% were male, and more than half had DLBCL (Table 1). All Korean patients had received prior systemic therapy, with most having received two or three prior treatments.
All patients discontinued treatment; reasons for discontinuation among Caucasian patients were progressive disease (59.5%), AEs other than DLTs (13.5%), patient refusal to continue with trial medication (5.4%), completion of all 12 infusions (10.8%), or other reasons (8.1%); one patient (2.7%) was lost to follow-up. Progressive disease was the primary reason for discontinuation among the Korean patients (90.9%); the remaining patient discontinued after receiving the maximum number of infusions. The median number of infusions was 4 (range, 1–12) and 3 (range, 2–12) in Caucasian and Korean patients respectively; the mean cumulative BI 836826 dose was 365.0 mg (standard deviation [SD] = 343.9 mg) and 286.4 mg (SD = 201.2 mg), respectively.
MTD and DLTs
For the Caucasian patients, dose escalation proceeded through 1 mg (n = 1), 3 mg (n = 4), 9 mg (n = 3), 25 mg (n = 4), 50 mg (n = 6), and 100 mg (n = 3) with no DLTs observed. One of three patients initially enrolled in the 200 mg cohort experienced DLTs (Grade 3 oral herpes, stomatitis, and febrile neutropenia). The 200 mg cohort was subsequently expanded to a total of seven patients (one of six patients in the initial cohort withdrew consent and was replaced), and although no further DLTs were reported, five of the seven patients experienced Grade 3/4 leukopenia and/or neutropenia lasting more than 1 week and requiring the next infusion to be delayed (Table 2). As a result, the 200 mg dose was considered to have exceeded the MTD. Dose de-escalation was subsequently undertaken.
Three of six patients treated at a dose of 150 mg experienced a DLT during the MTD evaluation period (grade 4 hypophosphatemia [n = 2] and grade 3 hypokalemia and hypocalcemia [n = 1]). These AEs were resolved within 24 h with supportive care. In addition, four of the six patients experienced grade 3/4 leukopenia and/or neutropenia lasting >7 days after the MTD evaluation period, suggesting that this dose exceeded the MTD. The next lower dose level (100 mg) was subsequently expanded with an additional three patients (to a total of six patients). None of the six patients treated at this dose experienced a DLT during the MTD evaluation period, and the 100 mg dose was therefore defined as the MTD.
Three of four patients enrolled into the 100 mg expansion cohort in Korean patients developed significant drug-related AEs (infections associated with grade 4 neutropenia). Following the protocol amendment, a separate MTD was to be defined for Korean patients based on an escalation scheme starting with 50 mg of BI 836826 administered weekly for 4 weeks, with 75 mg as the next dose step. Of seven patients treated with 50 mg BI 836826 (one patient had violated the exclusion criteria and was replaced), one experienced a DLT of grade 3 herpes zoster virus, and the MTD was not reached. However, prior to the enrollment of any further patients, the clinical development of BI 836826 was terminated, and the study was closed.
Safety
All patients experienced at least one AE. Among Caucasian patients, 7 (18.9%), 21 (56.8%) and 8 (21.6%) experienced a highest AE grade of 3, 4 and 5, respectively. None of the fatal AEs were considered related to BI 836826 (lymphoma progression [n = 6]; pulmonary edema [n = 2]). Of the Korean patients, 1 (9.1%), 8 (72.7%) and 2 (18.2%) had a highest AE grade of 3, 4 and 5, respectively. Neither of the fatal AEs (pneumonia and malignant pleural effusion) were considered related to study drug by the investigator.
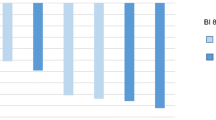
The frequency of treatment-related AEs was 94.6% and 100% among the Caucasian and Korean patients, respectively (Table 3). The most frequent treatment-related grade 3/4 AEs among Caucasian patients were hematotoxicities and IRRs (8.1%; Table 3). Most IRRs were grade 1/2; one patient had a grade 4 IRR, which led to a protocol amendment resulting in a change in the infusion schedule. If necessary, the initial dose could also be split into two infusions. The most common symptoms associated with IRRs were chills and pyrexia. Following the change in infusion schedule, two patients had grade 3 IRRs. The incidence of IRRs was highest in cycle 1, and decreased over time (Fig. 1).

Incidence and severity of infusion-related reactions over time
Similar treatment-related Grade 3/4 AEs were reported in Korean patients, although the incidence of certain AEs differed considerably (Table 3). In particular, treatment-related grade 3/4 febrile neutropenia was much more common in Korean patients than in Caucasian patients (54.5% vs 5.4%).
Four Caucasian patients (10.8%) had serious AEs (SAEs) that were considered treatment-related, including bronchospasm, respiratory failure, IRR, sepsis, atrial fibrillation, oral herpes, febrile neutropenia, and stomatitis (none occurred in more than one patient). Six Korean patients (54.5%) experienced drug-related SAEs. These included five cases of febrile neutropenia (one patient experienced two separate episodes), two of pneumonia, and one case each of neutropenia, herpes zoster, septic shock, and bacteremia.
Eight Caucasian patients (21.6%) had AEs that resulted in discontinuation of study drug. The only AE leading to treatment discontinuation in more than one patient was IRR (two patients [5.4%]; both of these patients were treated prior to the change in the infusion schedule). No AEs occurred in Korean patients that led to treatment discontinuation. Two Caucasian patients and one Korean patient received dose reductions due to AEs.
Based on laboratory data, 20 Caucasian patients (54.1%) experienced grade 4 neutropenia lasting 1 week or longer (a total of 48 neutropenia episodes lasting ≥1 week; median 2.0 neutropenia episodes/patient). Eight patients (21.6%) had grade 4 neutropenia and a concomitant infection, of which four (10.8%) were grade 3/4. Prolonged grade 3/4 neutropenia of 1 week or longer was seen in all dose cohorts except 1 mg. At the MTD (100 mg), one of six patients had one episode of grade 4 neutropenia that lasted 11 days and delayed the administration of the next BI 836826 infusion. This episode did not require a dose reduction or discontinuation of the study drug. The platelet and white blood cell counts for a representative patient are shown in Supplemental Fig. 2. Among the Korean patients, 10 (90.9%) had grade 4 neutropenia episodes lasting more than 1 week (a total of 23 neutropenia episodes lasting ≥1 week, with a median of 2 neutropenia episodes per patient). Four patients (36.4%) had grade 4 neutropenia and a concomitant infection, all of which were grade 3/4.
Eight Caucasian patients, all receiving BI 836826 doses of 100 mg or higher, experienced 11 grade ≥ 3 infection episodes. Of these, two were considered drug-related (one case of sepsis and one episode of oral herpes), and dose reduction was required for the case of oral herpes. Among the Korean patients, six experienced grade ≥ 3 infections; three of seven patients in the 50 mg dose cohort, and three of four patients in the 100 mg dose cohort. All were considered to be DLTs. One patient died as a result of pneumonia.
Efficacy
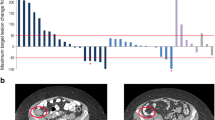
In total, 21 Caucasian and eight Korean patients were evaluated for best change in the sum of the product of diameters (SPD) of their indicator lesions. The median best percentage change from baseline in SPD of indicator lesions was 11.8% (range, −44.7 to 329.8%) for Caucasian patients and 68.7% (range, −100.0 to 146.3%) for Korean patients (Fig. 2).Data from 32 Caucasian patients and 11 Korean patients were evaluable for best overall response (Table 4). According to investigator assessment based on imaging data, one Korean patient (50 mg dose group; DLBCL) achieved CR, while two Caucasian patients (100 mg dose group; FL and mantle cell lymphoma) achieved PR. Nine Caucasian patients (24.3%) and one Korean patient (9.1%) experienced stable disease. Taking into account all assessment methods (physical or imaging assessment by the investigator), a further two Caucasian patients (50 mg dose cohort and 100 mg dose cohort; both FL) had a PR. In the overall population, median PFS and FFS were 47 days (25th and 75th percentiles; 24 days, 169 days), and 46 days (24 days, 116 days), respectively.

Best percentage change from baseline in the sum of product of diameters (SPD) of the indicator lesions, based on imaging data
Pharmacokinetics
Non-compartmental PK parameters, analyzed for the combined population of Caucasian and Korean patients, were determined after the first intravenous infusion of BI 836826 for all patients in the study (Table 5). Maximum plasma concentration increased with dose up to 100 mg, but not in a dose-proportional manner. The highest individual value for Cmax was reached in the 100 mg dose group (Supplemental Fig. 3). The volume of distribution (Vss) slightly decreased from 5.07 L in the 25 mg dose group to 2.82 L in the 100 mg dose cohort, and increased to 4.50 L for 200 mg, displaying overall a rather stable level and indicating that BI 836826 was primarily distributed in plasma at steady state.
T1/2 was variable, ranging from a mean of 8.95 h in the 25 mg dose cohort to 33.8 h in the 200 mg dose group. A preliminary comparison of PK data from patients who received 100 mg BI 836826 suggested that Korean patients achieved approximately 4-fold higher plasma exposure to BI 836826 than Caucasian patients (Table 6).
Pharmacodynamics
Two potential prognostic biomarkers were investigated in the study: β-2 microglobulin and FCGR. At screening, 43.8% of patients had elevated serum β-2 microglobulin levels (≥3.5 mg/L). No correlation was observed between baseline β-2 microglobulin levels and best percentage change in SPD of indicator lesions. Similarly, no association was observed between FCGR genotypes (polymorphisms FCGR2A, FCGR3A) and best overall response.
Discussion
This study defined an MTD and demonstrated preliminary activity of BI 836826 in patients with B cell NHL, thus further validating CD37 as a therapeutic target in this setting. Notwithstanding the fact that few of the 48 treated patients received dose levels where activity may be expected, signals of efficacy were observed in this study. Out of 48 treated patients, one Korean patient had a best overall response of CR, and two Caucasian patients had a best overall response of PR, with SD the best overall response in 10 more patients.
The MTD in Caucasian patients was defined as 100 mg of BI 836826 administered weekly for 4 weeks. The clinical development of BI 836826 was stopped shortly after the 50 mg Korean cohort was completed, which prevented formal establishment of the MTD of BI 836826 in Korean patients. The MTD in this study was lower than observed in a previous phase I trial undertaken in patients with CLL [15]. While a formal MTD of BI 836826 in patients with CLL was not determined, doses of 400 mg administered every 2 weeks were tolerable and recommended for further development. The discrepancy between studies could be related to differences in target expression in different patient populations leading to different pharmacokinetic behavior due to target-mediated drug disposition. In both settings, the half-life of BI 836826 was short but clearance was more rapid in CLL than NHL. As reduction of circulating B cells and other blood cells expressing CD37 (e.g. T cells) underpins the mechanism of action of BI 826826 in both CLL and NHL, differences in clearance rate probably reflect the differences in the expression profile of CD37 in the two diseases, with higher expression in blood, spleen and bone marrow of patients with CLL compared to NHL. The short half-life of BI 826826 was unlikely to be attributable to its chimerism. While anti-drug antibodies were detectable in 19% of patients by the end of treatment, they were mostly detected after several infusions. However, rapid clearance was already observed after the first administration.
All Caucasian patients treated in the dose-escalation phase experienced at least one AE. These AEs were most frequently hematologic, particularly leukopenia, neutropenia, and thrombocytopenia, commonly of Grade 3/4. The occurrence of neutropenia and leukopenia tended to increase with increasing dose and was dose-limiting at the 200 mg and 150 mg doses. Although growth factor support was permitted, prolonged Grade 4 neutropenia lasting at least 1 week was seen in 54% of Caucasian patients and 91% of Korean patients, including eight cases of Grade 4 neutropenia and a Grade 3/4 concomitant infection. The tolerability profile of BI 836826 in this study was similar to that observed in patients with CLL [15].
The rapid kinetics of neutropenia and thrombocytopenia, with steep declines in neutrophil and platelet counts immediately after administration of BI 836826 and recovery prior to the next infusion 7 days later (Supplemental Fig. 2) suggest that cytopenia may be related to direct effects of BI 836826 on mature peripheral leukocytes and platelets, and not due to toxicity on bone marrow precursor cells. CD37 is known to be expressed in megakaryocytes, platelets, and neutrophils [18,19,20]; further, preclinical data suggest that BI 836826 does not impact on myeloid progenitor colony formation (unpublished data).
Fifteen Caucasian patients (40.5%) and three Korean patients (27.3%) experienced IRRs, with one Caucasian patient experiencing a Grade 4 event under the initial protocol-defined infusion rate of 83.4 mL/h over 3 h. These events occurred despite mandatory treatment with acetaminophen/paracetamol, antihistamine, and glucocorticoid prior to the infusion.
While this study suggests that BI 836826 may have a different tolerability profile between Caucasian and Korean patients, it must be noted that only 11 Koreans were treated. Hence the observations may merely reflect the small sample size. Nevertheless it was noteworthy that the PK of BI 836826 in Korean patients differed considerably from the PK seen in Caucasian patients. Exposure and maximum plasma concentration levels reached after intravenous administration of the 100 mg dose showed an almost 4-fold increase compared with the exposure in Caucasian patients. The finding of PK variability between Korean and Caucasian patients may be attributable to possible differences in drug metabolism between East Asians and Caucasians, which is a common phenomenon that may be related to differences in allelic frequencies of drug metabolism genes such as CYP2D6 and the CYP2C subfamily [21].
Although the sponsor has decided not to pursue further clinical development of BI 836826, primarily due to changes in strategy reflecting recent changes in the treatment landscape for CLL, the results of this study support further clinical investigation of CD37 as a therapeutic target in B cell NHL. There was high rates of treatment-related AEs, particularly hematotoxicities, and this needs to be considered for future clinical development programs targeting CD37. However, recent promising results from a phase I/II study of 177Lu-lilotomab satetraxetan, an anti-CD37 antibody-radionuclide conjugate, in which 65% of patients with FL responded, including CR in 24% of patients, also confirm the value of targeting CD37 [22]. As with our study, the incidence of Grade 3/4 neutropenia (53%) was high; grade 3/4 thrombocytopenia was also common (48%). Further clinical investigation of AGS57E is ongoing [14], along with preclinical studies investigating CD37 as a target for chimeric antigen receptor T cell therapy [14, 23].
Data availability
The clinical study report (including appendices, but without line listings) and other clinical documents related to this study may be accessed on request. Prior to providing access, the documents and data will be examined, and, if necessary, redacted and de-identified to protect the personal data of study participants and personnel, and to respect the boundaries of the informed consent of the study participants. See https://trials.boehringer-ingelheim.com/data_sharing/sharing.html#accordion-1-2 for further details. Bona fide, qualified scientific and medical researchers may request access de-identified, analyzable patient-level study data, together with documentation describing the structure and content of the datasets. Researchers should use https://clinicalstudydatarequest.com/ to request access to raw data from this study.
Change history
23 June 2020
The original version of this article unfortunately contained an error. In the Conflict of interest statement, Anne-Marie Quinson is described as declaring no conflict of interest.
31 May 2020
The article Phase I dose escalation study of BI 836826 (CD37 antibody) in patients with relapsed or refractory B-cell non-Hodgkin lymphoma.
References
Howlader N, Morton LM, Feuer EJ, Besson C, Engels EA (2016) Contributions of subtypes of non-Hodgkin lymphoma to mortality trends. Cancer Epidemiol Biomark Prev 25(1):174–179. https://doi.org/10.1158/1055-9965.EPI-15-0921
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68(6):394–424. https://doi.org/10.3322/caac.21492
Sehn LH, Gascoyne RD (2015) Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood 125(1):22–32. https://doi.org/10.1182/blood-2014-05-577189
Kahl BS, Yang DT (2016) Follicular lymphoma: evolving therapeutic strategies. Blood 127(17):2055–2063. https://doi.org/10.1182/blood-2015-11-624288
Lapalombella R, Yeh YY, Wang L, Ramanunni A, Rafiq S, Jha S, Staubli J, Lucas DM, Mani R, Herman SE, Johnson AJ, Lozanski A, Andritsos L, Jones J, Flynn JM, Lannutti B, Thompson P, Algate P, Stromatt S, Jarjoura D, Mo X, Wang D, Chen CS, Lozanski G, Heerema NA, Tridandapani S, Freitas MA, Muthusamy N, Byrd JC (2012) Tetraspanin CD37 directly mediates transduction of survival and apoptotic signals. Cancer Cell 21(5):694–708. https://doi.org/10.1016/j.ccr.2012.03.040
Heider KH, Kiefer K, Zenz T, Volden M, Stilgenbauer S, Ostermann E, Baum A, Lamche H, Küpcü Z, Jacobi A, Müller S, Hirt U, Adolf GR, Borges E (2011) A novel Fc-engineered monoclonal antibody to CD37 with enhanced ADCC and high proapoptotic activity for treatment of B-cell malignancies. Blood 118(15):4159–4168. https://doi.org/10.1182/blood-2011-04-351932
Knobeloch KP, Wright MD, Ochsenbein AF, Liesenfeld O, Löhler J, Zinkernagel RM, Horak I, Orinska Z (2000) Targeted inactivation of the tetraspanin CD37 impairs T-cell-dependent B-cell response under suboptimal costimulatory conditions. Mol Cell Biol 20(15):5363–5369
Bertoni F, Stathis A (2016) Staining the target: CD37 expression in lymphomas. Blood 128(26):3022–3023. https://doi.org/10.1182/blood-2016-11-748137
van Spriel AB, Puls KL, Sofi M et al (2004) A regulatory role for CD37 in T cell proliferation. J Immunol 172(5):2953–2961
Barrena S, Almeida J, Yunta M, López A, Fernández-Mosteirín N, Giralt M, Romero M, Perdiguer L, Delgado M, Orfao A, Lazo PA (2005) Aberrant expression of tetraspanin molecules in B-cell chronic lymphoproliferative disorders and its correlation with normal B-cell maturation. Leukemia 19(8):1376–1383. https://doi.org/10.1038/sj.leu.2403822
Moore K, Cooper SA, Jones DB (1987) Use of the monoclonal antibody WR17, identifying the CD37 gp40-45 Kd antigen complex, in the diagnosis of B-lymphoid malignancy. J Pathol 152(1):13–21. https://doi.org/10.1002/path.1711520103
Pagel JM, Spurgeon SE, Byrd JC, Awan FT, Flinn IW, Lanasa MC, Eisenfeld AJ, Stromatt SC, Gopal AK (2015) Otlertuzumab (TRU-016), an anti-CD37 monospecific ADAPTIR(™) therapeutic protein, for relapsed or refractory NHL patients. Br J Haematol 168(1):38–45. https://doi.org/10.1111/bjh.13099
Stathis A, Flinn IW, Madan S, Maddocks K, Freedman A, Weitman S, Zucca E, Munteanu MC, Lia Palomba M (2018) Safety, tolerability, and preliminary activity of IMGN529, a CD37-targeted antibody-drug conjugate, in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: a dose-escalation, phase I study. Investig New Drugs 36(5):869–876. https://doi.org/10.1007/s10637-018-0570-4
Sawas A, Savage KJ, Perez R et al (2015) A phase 1 study of the anti-CD37 antibody-drug conjugate AGS67E in advanced lymphoid malignancies. Interim results. Blood 126(23):3976–3976
Stilgenbauer S, Aurran Schleinitz T, Eichhorst B, Lang F, Offner F, Rossi JF, Schroyens W, van den Neste E, Ysebaert L, von Wangenheim U, Ursula Kress U, Blum P, Zenz T (2019) Phase 1 first-in-human trial of the anti-CD37 antibody BI 836826 in relapsed/refractory chronic lymphocytic leukemia. Leukemia 33:2531–2535. https://doi.org/10.1038/s41375-019-0475-z
Swerdlow SH, Campo E, Harris NL et al (2008) WHO classification of tumours of haematopoietic and lymphoid tissues, 4th edn. International Agency for Research on Cancer (IARC), Lyon
Cheson BD, Horning SJ, Coiffier B et al (1999) Report of an international workshop to standardize response criteria for non-Hodgkin's lymphomas. NCI Sponsored International Working Group. J Clin Oncol 17(4):1244–1253. https://doi.org/10.1200/JCO.1999.17.4.1244
Burkhart JM, Vaudel M, Gambaryan S, Radau S, Walter U, Martens L, Geiger J, Sickmann A, Zahedi RP (2012) The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 120(15):e73–e82. https://doi.org/10.1182/blood-2012-04-416594
Protty MB, Watkins NA, Colombo D, Thomas SG, Heath VL, Herbert JM, Bicknell R, Senis YA, Ashman LK, Berditchevski F, Ouwehand WH, Watson SP, Tomlinson MG (2009) Identification of Tspan9 as a novel platelet tetraspanin and the collagen receptor GPVI as a component of tetraspanin microdomains. Biochem J 417(1):391–400. https://doi.org/10.1042/bj20081126
Wee JL, Schulze KE, Jones EL, Yeung L, Cheng Q, Pereira CF, Costin A, Ramm G, van Spriel AB, Hickey MJ, Wright MD (2015) Tetraspanin CD37 regulates beta2 integrin-mediated adhesion and migration in neutrophils. J Immunol 195(12):5770–5779. https://doi.org/10.4049/jimmunol.1402414
Kim K, Johnson JA, Derendorf H (2004) Differences in drug pharmacokinetics between East Asians and Caucasians and the role of genetic polymorphisms. J Clin Pharmacol 44(10):1083–1105. https://doi.org/10.1177/0091270004268128
Kolstad A, Madsbu U, Beasley M et al (2018) LYMRIT 37-01: a phase I/II study of 177Lu-lilotomab satetraxetan (Betalutin®) antibody-radionuclide-conjugate (ARC) for the treatment of relapsed non-Hodgkin's lymphoma (NHL) — analysis with 6-month follow-up. Blood 132(Suppl 1):2879–2879. https://doi.org/10.1182/blood-2018-99-110555
Koksal H, Dillard P, Josefsson SE et al (2019) Preclinical development of CD37CAR T-cell therapy for treatment of B-cell lymphoma. Blood Adv 3(8):1230–1243. https://doi.org/10.1182/bloodadvances.2018029678
Acknowledgements
We thank the patients, their families, and all of the investigators who participated in these studies. The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development, and have approved the final version.
Funding
The conduct of this research, study design, data collection, and analysis was financially supported by Boehringer Ingelheim. Medical writing assistance, funded by Boehringer Ingelheim, was provided by Lynn Pritchard, DPhil, of GeoMed, an Ashfield company, part of UDG Healthcare plc, during the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Frank Kroschinsky reports receiving reimbursement for costs of study treatments, documentation, congress fees and travel expenses from Boehringer Ingelheim. Jan Moritz Middeke declares that he has no conflict of interest. Martin Janz declares that he has no conflict of interest. Georg Lenz reports personal fees and/or research support from Amgen, AstraZeneca, BMS, Abbvie, Bayer, Janssen, Novartis, Gilead / Kite, Celgene, Roche, Takeda, Morphosys, ACERTA, AQUINOX, Agios, Verastem, NanoString, and Springer, outside the submitted work. Mathias Witzens-Harig declares that he has no conflict of interest. Reda Bouabdallah declares that he has no conflict of interest. Paul La Rosée reports personal fees from Boehringer Ingelheim (Travel support). Andreas Viardot reports personal fees and non-financial support from Roche, personal fees and non-financial support from Kite/Gilead, non-financial support from Abbvie, personal fees from Amgen, outside the submitted work. Gilles Salles reports personal fees from Amgen, BMS, Abbvie, Janssen, Merck, Novartis, Gilead / Kite, Epizyme, Pfizer, Celgene, Roche, Takeda, Autolus, MOrphosys, ACERTA, and Servier, outside the submitted work. Seok Jin Kim declares that he has no conflict of interest. Tae Min Kim declares that he has no conflict of interest. Oliver Ottmann declares that he has no conflict of interest. Joerg Chromik declares that he has no conflict of interest. Anne-Marie Quinson declares that he has no conflict of interest. Ute von Wangenheim and Ute Burkard are employees of Boehringer Ingelheim. Andreas Berk is an employee of ClinTriCare GmbH & Co. KG, contracted by Boehringer Ingelheim. Norbert Schmitz declares that he has no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised due to a retrospective Open Access order.
Electronic supplementary material
ESM 1
(PDF 236 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kroschinsky, F., Middeke, J.M., Janz, M. et al. Phase I dose escalation study of BI 836826 (CD37 antibody) in patients with relapsed or refractory B-cell non-Hodgkin lymphoma. Invest New Drugs 38, 1472–1482 (2020). https://doi.org/10.1007/s10637-020-00916-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-020-00916-3