
Summary
Introduction This phase 1 study assessed safety, maximum tolerated dose (MTD), pharmacokinetics, cerebrospinal fluid (CSF) distribution, and preliminary clinical activity of the receptor tyrosine kinase inhibitor TAK-285. Methods Patients with advanced, histologically confirmed solid tumors and Eastern Cooperative Oncology Group performance status ≤2 received daily oral TAK-285; daily dose was escalated within defined cohorts until MTD and recommended phase 2 dose (RP2D) were determined. Eleven patients were enrolled into an RP2D cohort. Blood samples were collected from all cohorts; CSF was collected at pharmacokinetic steady-state from RP2D patients. Tumor responses were assessed every 8 weeks per Response Evaluation Criteria in Solid Tumors. Results Fifty-four patients were enrolled (median age 60; range, 35–76 years). The most common diagnoses were cancers of the colon (28 %), breast (17 %), and pancreas (9 %). Escalation cohorts evaluated doses from 50 mg daily to 500 mg twice daily; the MTD/RP2D was 400 mg twice daily. Dose-limiting toxicities included diarrhea, hypokalemia, and fatigue. Drug absorption was fast (median time of maximum concentration was 2–3 h), and mean half-life was 9 h. Steady-state average unbound CSF concentration (geometric mean 1.54 [range, 0.51–4.27] ng/mL; n = 5) at the RP2D was below the 50 % inhibitory concentration (9.3 ng/mL) for inhibition of tyrosine kinase activity in cells expressing recombinant HER2. Best response was stable disease (12 weeks of nonprogression) in 13 patients. Conclusions TAK-285 was generally well tolerated at the RP2D. Distribution in human CSF was confirmed, but the free concentration of the drug was below that associated with biologically relevant target inhibition.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The human epidermal growth factor receptors EGFR (erbB1) and HER2 (erbB2) play critical roles in cell growth and proliferation [1], and have upregulated activity in various cancers [2]. Up to 25 % of patients with breast cancer are HER2-positive (HER2+), and HER2+ patients have a poor prognosis and a higher chance of relapse [3]. Therapy specifically targeting erbB kinases has become an important part of clinical management; the standard of care for patients with HER2+ breast cancer includes trastuzumab, a humanized monoclonal antibody that inhibits HER2 activity [4]. Although therapy with trastuzumab provides clinical benefit, many patients eventually become resistant [5–8]. In addition, more than 25 % of HER2+ patients treated with trastuzumab will develop brain metastases [9]; in that event, monoclonal antibodies are of limited use because they cannot readily cross the blood–brain barrier (BBB) [10]. Lapatinib, a small-molecule HER2 inhibitor, has demonstrated only negligible central nervous system (CNS) penetration across either an intact [11] or a tumor-compromised BBB [12], and has low clinical activity in this setting [13, 14]. Many traditional chemotherapeutic agents likewise have poor CNS distribution [15–17]. Therefore, there is an unmet need for more effective therapies for patients with HER2+ brain metastases.
TAK-285 is an investigational, small-molecule tyrosine kinase inhibitor [18]. It has been shown to be both selective and potent [19]; in vitro 50 % inhibitory concentration (IC50) values for recombinant human EGFR and HER2 are 23 and 17 nmol/L, respectively [20]. Preclinical studies suggest that TAK-285 inhibits the growth of malignant cell lines [21], has antitumor activity in murine xenograft models, and inhibits mutant EGFR kinase activity (L858R and L861Q) [22] (Nakayama et al., unpublished data, 2012); however, TAK-285 did not inhibit the growth of the EGFR mutant NSCLC cell line HCC4006 (data on file). In vitro transport studies suggest that TAK-285 is not a substrate for the BBB efflux transporters P-glycoprotein (P-gp) or breast cancer resistance protein (BCRP), and TAK-285 exhibits high transcellular permeability [23] (Nakayama et al., unpublished data, 2012). In vivo preclinical CNS distribution studies have demonstrated that in rats TAK-285 penetrates the BBB and distributes into brain tissue and interstitial fluid [23, 24].
In a preliminary phase 1 study of TAK-285 in Japanese patients with advanced cancers (N = 26), patients in dose-escalation cohorts received TAK-285 once weekly for 3 weeks followed by 1 week of observation; in a repeated-administration cohort at the maximum tolerated dose (MTD; found to be 300 mg twice daily [BID]), patients received TAK-285 for at least 4 weeks. The treatment was generally well tolerated, and 1 patient with parotid cancer experienced a partial response [25]. The present phase 1 study was undertaken to evaluate the safety (MTD, recommended phase 2 dose [RP2D], dose-limiting toxicities [DLTs]), antitumor activity, and pharmacokinetic properties of TAK-285 in patients with advanced cancer refractory to standard cancer therapy. In addition, this study evaluated CSF distribution of TAK-285 at pharmacokinetic steady-state to determine whether biologically relevant concentrations are achievable in the human CNS at tolerable doses and to assess the utility of TAK-285 as a potential investigational agent for treating and/or preventing brain metastases in patients with HER2+ metastatic breast cancer.
Methods
Patients
Eligible patients were 18 years of age or older with a diagnosis of advanced, histologically confirmed solid tumors refractory to other therapy. Patients were to have Eastern Cooperative Oncology Group performance status ≤ 2, adequate hematologic (absolute neutrophil count ≥ 1,500 cells/mm3, hemoglobin ≥ 9 g/dL, platelet count ≥ 100,000/mm3), hepatic (total bilirubin ≤ 1.5× upper limit of normal [ULN], aspartate aminotransferase [AST] and alanine aminotransferase [ALT] ≤ 2.5× ULN), and renal (serum creatinine ≤ 1.5× ULN) function. Patients enrolled in the RP2D cohort were to have left ventricular ejection fraction ≥ 50 % and be able to tolerate a single lumbar puncture for collection of CSF. Major exclusion criteria included current CNS metastases or primary CNS malignancy, significant electrocardiogram abnormalities including QTc prolongation (> 450 ms for men and >470 ms for women), any other cancer (other than nonmelanoma skin cancer or cervical cancer in situ) unless in complete remission and off all therapy for ≥ 3 years, cardiovascular impairment, pleural or pericardial effusion, active gastrointestinal bleeding or ulceration, systemic treatment with strong or moderate cytochrome P445 (CYP)3A4 inducing/inhibiting drugs within 14 days before study enrollment, and life expectancy less than 12 weeks. The study was conducted in accordance with good clinical practice and the general principles of the Declaration of Helsinki. All patients provided signed informed consent before initiation of any study procedures.
Study design
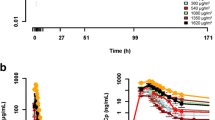
This was a 3-center, multicohort, open-label, nonrandomized, noncomparative, clinical and pharmacokinetic study of TAK-285 in patients with advanced cancer (NCT00535522). The primary objectives of the study were to determine the safety (MTD, RP2D, DLTs) and pharmacokinetic profile (both plasma and CSF concentrations) of the drug. A series of dose-escalation cohorts was used to establish the MTD and RP2D (Fig. 1). The starting dose of TAK-285 was 50 mg daily (QD); the drug was initially administered on days 1 through 21 of a 28-day cycle (50 mg QD, 50 mg BID, and 75 mg BID cohorts). On observation that TAK-285 was generally well tolerated, the dose was escalated to 500 mg, and patients in the remaining cohorts (150 mg BID to 500 mg BID) received the drug daily. Three to 6 patients were recruited at each dose level; once the MTD was established, additional patients were enrolled at that level in an RP2D expansion cohort. The protocol specified that the RP2D expansion cohort would further test the MTD using an initial regimen of dosing on days 1 through 21 during cycle 1 to confirm safety and tolerability, then daily (without days off) during subsequent cycles. The MTD was defined as the dose level immediately below that in which ≥ 2 patients experienced DLTs during the first 28 days of treatment (cycle 1); only DLTs during cycle 1 affected dose escalation decisions for subsequent cohorts. A DLT was defined as any grade ≥ 4 hematologic toxicity, any grade ≥ 3 nonhematologic toxicity (other than nausea, vomiting, and diarrhea that could be controlled with standard supportive care), grade 3 QTc prolongation (> 500 ms assessed by a qualified reader and confirmed on a repeat electrocardiogram), or any TAK-285–related toxicity resulting in a treatment delay of > 21 days.

Dosing chart. BID, twice daily; QD, once daily; RP2D, recommended phase 2 dose
Assessments
The primary endpoints of the study were the safety and pharmacokinetics of TAK-285. Safety of TAK-285 was assessed by physical examination, vital signs, electrocardiogram changes, laboratory evaluations, and occurrence of adverse events (AEs). Adverse events were evaluated at each study visit and were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0. The secondary endpoint was therapeutic efficacy of TAK-285. Disease assessments (computed tomography, magnetic resonance imaging, x-ray and/or bone scans) were performed at baseline, after cycle 2, and every 8 weeks thereafter according to Response Evaluation Criteria in Solid Tumors [26]. Stable disease was defined as no tumor growth for a minimum of 12 weeks.
Pharmacokinetic assessments and data analyses
During cycle 1, blood samples for pharmacokinetic analysis were collected on day 1 from patients in the dose-escalation cohorts starting before treatment, and at 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h after the morning dose. Blood sample collection on day 21 of cycle 1 followed the same schedule, with the additional collection of samples 48 and 72 h after dosing. On days 8 and 15, predose (trough) blood samples were collected in the morning.
For patients in the RP2D cohort, blood samples were collected on days 1, 8, and 15 of cycle 1 as described above. The evening dose on day 1 was not administered in this cohort to permit characterization of the pharmacokinetic profile from 0 to 24 h after day 1 dosing. On day 22, (ie, day 21 of BID administration), samples were collected as on day 1, with additional samples collected at 48, 72, and 120 h after dosing to characterize steady-state terminal distribution half-life (t½). On day 15 of cycle 1, CSF samples were collected by lumbar puncture 3 to 4 h after dosing for CSF TAK-285 concentration and protein binding measurements; concurrent blood samples were also obtained for plasma TAK-285 concentration and protein binding measurements.
TAK-285 concentrations in plasma and CSF were measured by high-performance liquid chromatography/tandem mass spectrometry. Plasma and CSF protein binding were measured using equilibrium dialysis. Pharmacokinetic parameters were calculated from concentration-time data using standard noncompartmental methods (WinNonlin v.5.2) and included the maximum plasma concentration (Cmax), the time of first occurrence of Cmax (Tmax), the area under the plasma concentration-time curve from time zero to the end of the dosing interval (AUC0-τ), steady-state average concentration over the dosing interval (Css,avg), peak-trough ratio (PTR), accumulation ratio (Rac), and t½. In the RP2D cohort, additional pharmacokinetic endpoints included the free fractions of TAK-285 in plasma (fu,p) and CSF (fu,CSF), and the steady-state unbound CSF/unbound plasma concentration ratio (Cu,CSF:Cu,p). The unbound steady-state average concentrations of TAK-285 in CSF (Css,avg,u,CSF) were calculated using Eq. 1 from individual patient values of day 22 plasma Css,avg, total CSF (CCSF) and corresponding total plasma (Cp) TAK-285 concentrations at the time of lumbar puncture on day 15, and the CSF free fraction (fu,CSF).
The above calculation assumes that distributional equilibrium is achieved by day 15 of BID dosing in the CSF compartment and that the CSF-plasma concentration ratio measured at approximately plasma Tmax on day 15 (ie, under pharmacokinetic steady-state conditions in plasma) is representative of the CSF-plasma concentration ratio over the entire dosing interval.
Statistical analysis
Descriptive statistical analyses were conducted for patient demographic and baseline characteristics; summary statistics (mean, median, maximum, minimum, standard deviation, and 95 % confidence interval [CI]) were used to evaluate safety and efficacy. Plasma and CSF pharmacokinetic parameters were summarized using descriptive statistics (arithmetic and geometric means, standard deviation, percentage coefficient of variation [%CV], median, minimum, maximum). Dose-proportionality was assessed using power model analysis. A linear regression was performed on log-transformed Css,avg versus log-transformed daily dose using SigmaPlot for Windows, version 11.0, and the 95 % CI of the estimated slope of this regression was used to assess dose-proportionality.
Results
Patient characteristics
Between August 2007 and June 2011, 54 patients were enrolled and treated with at least 1 dose of TAK-285. Patient characteristics were similar between treatment groups (Table 1) [27]. The most common malignancies were colon (28 %), breast (17 %), and pancreatic cancer (9 %). Most patients had received prior chemotherapy. Five patients (9 %) had received prior treatment with trastuzumab for HER2+ breast cancer.
Dose escalation and safety
Dose levels were escalated in 8 cohorts as follows: 50 mg QD (n = 4), 50 mg BID (n = 7), 75 mg BID (n = 6), 150 mg BID (n = 6), 225 mg BID (n = 4), 325 mg BID (n = 3), 400 mg BID (n = 6), and 500 mg BID (n = 7). Patients in the 50 mg QD, 50 mg BID, and 75 mg BID cohorts received the study drug on days 1 through 21 of a 28-day cycle. Patients in all other dose-escalation cohorts received the drug daily with no days off. Two of the 7 patients at the highest dose level (500 mg BID) experienced DLTs that resulted in dose interruption (Table 2). Therefore, dose escalation was stopped and 400 mg BID was established as the MTD and RP2D. Eleven additional patients were enrolled in the 400 mg BID RP2D expansion cohort. Three patients in the RP2D cohort experienced DLTs during cycle 1 that led to drug interruption or dose reduction (Table 2). Three other patients in the dose-escalation cohorts experienced DLTs leading to drug withdrawal, dose reduction, or dose interruption during cycle 1 (Table 2). One additional patient experienced grade 4 rhabdomyolysis, elevated creatine kinase and AST, and grade 3 elevated ALT after cycle 1, and study drug was withdrawn.
Most patients (98 %) experienced at least 1 AE, and most AEs were grade 1 or 2. The most common treatment-emergent AEs were diarrhea (46 %), fatigue (44 %), and nausea (32 %) (Table 3). Twenty-eight patients (52 %) experienced grade ≥ 3 AEs. The most common grade ≥ 3 AEs were hypokalemia and diarrhea (Table 3). Twenty-five patients (46 %) experienced serious AEs. Serious AEs observed in ≥ 2 patients were disease progression, deep vein thrombosis, ileus, bowel obstruction, abdominal pain, back pain, dyspnea, and hyponatremia. Nine patients experienced AEs that led to discontinuation of the study drug: 3 patients in the 50 mg BID cohort, 2 patients in the 150 mg BID cohort, and 1 patient in each in the 325 mg BID, 400 mg BID, 500 mg BID, and RP2D cohorts. Five patients died during the study (within 30 days of the last dose of study drug): 4 due to disease progression and 1 due to intestinal obstruction. None of these deaths was considered related to study drug.
Pharmacokinetics
After oral administration, absorption of TAK-285 was fast; peak plasma concentrations were achieved 2 to 3 h postdose. Plasma exposures of TAK-285 increased with increasing dose (Fig. 2a and b; Table 4). The extent of accumulation was approximately 3-fold at the MTD of 400 mg BID (Fig. 3; Table 4). In the 400 mg BID dosing group, after cessation of multiple-dose administration, there was an approximately monoexponential decline in plasma concentrations with a mean t½ of 8.9 ± 0.99 h (Fig. 2c). Pharmacokinetic steady-state conditions were achieved by day 8, based on similar trough concentrations on days 8, 15, and 21 (data not shown). Fluctuation over the steady-state dosing interval, measured as PTR, decreased with BID dosing compared with QD dosing (Table 4). On day 21, the PTR was ~2.8 at the MTD of 400 mg BID (Fig. 3).

Mean plasma concentration-time profiles of TAK-285. a and b Overlays of the mean concentration-time profiles from patients in the dose-escalation cohorts (50 mg once daily [QD] to 500 mg twice daily [BID]) measured on (a) day 1 and on (b) day 21. c Semilogarithmic plot of the concentration-time profile at steady-state to display the terminal disposition phase following cessation of multiple dosing. d Relationship between total daily dose of TAK-285 and the steady-state average concentration (Css,avg). The symbols represent individual patients; the solid line is the power model-predicted dose-Css,avg relationship, and the dashed lines represent the 95 % confidence interval of the model-predicted relationship

Mean plasma concentration-time profiles on days 1 and 21 of multiple-dose administration of TAK-285 after twice daily (BID) repeat-dose administration at the maximum tolerated dose (MTD) of 400 mg BID in patients with advanced nonhematologic malignancies. The panel shows an overlay of the day 1 and day 21 steady-state mean concentration-time profiles to display the extent of accumulation
Steady-state exposures of TAK-285 increased in a greater than dose-proportional manner; a 10-fold increase in dose (from 50 mg BID to 500 mg BID) was associated with a 29-fold increase in geometric mean steady-state AUC0-τ (Table 4). The slope of the linear regression of log (Css,avg) versus log (Dose) was estimated to be 1.43 (95 % CI, 1.26–1.59; Fig. 2d).
CSF distribution
To assess the distribution of TAK-285 to the CNS, CSF samples were obtained from 7 patients in the RP2D cohort at 3 to 4 h postdose on day 15. TAK-285 was highly protein bound in plasma, with a mean plasma free fraction of 0.1 % (n = 7) (Table 5). The mean free fraction in CSF was 23.7 % (n = 7). Variability in plasma and CSF protein binding was moderate (%CV, ~35 %). At the time of CSF collection, the mean Cu,CSF:Cu,p ratio was 0.663 (%CV, 23 %; n = 7) indicating that the CSF contained, on average, 66 % of systemically available unbound TAK-285. The range of individual values of the Cu,CSF:Cu,p ratio was approximately 2-fold, indicating relatively low interpatient variability in CNS distribution of TAK-285 when normalized for unbound systemic exposures. Excellent correlation was observed between the measured unbound CSF concentrations and concurrently measured unbound plasma concentrations of TAK-285 (r 2 = 0.95; n = 7; Fig. 4a). The geometric mean Css,avg,u,CSF was 1.54 ng/mL (%CV, 74 %; n = 5); individual values of this parameter varied from 0.51 to 4.27 ng/mL and all were below the HER2 IC50 of 9.3 ng/mL (Fig. 4b).

Cerebrospinal fluid (CSF) distribution of TAK-285. a Relationship between the measured unbound CSF concentration (CCSF) and the concurrently measured unbound plasma concentration of TAK-285 (Cu,p). The symbols represent data from 7 individual patients, the solid line is a linear regression fit to the data, the dashed lines represent the 95 % confidence interval of the fitted linear relationship, and the dotted line is the line of unity for equivalence of unbound CSF and unbound plasma concentrations. b Individual values of the calculated steady-state average unbound concentration of TAK-285 in CSF (Css,avg,u,CSF) in 5 patients, in comparison with the 50 % inhibitory concentration (IC50) for inhibition of human epidermal growth receptor 2 (HER2) by TAK-285. Concentrations achieved in all patients were below the HER2 IC50
Efficacy
No patient experienced a complete or partial response to TAK-285 [27]. The best response was stable disease, which was observed in 9 of 33 patients (27 %) in the dose-escalation cohorts and 4 of 8 patients (50 %) in the RP2D cohort. The clinical benefit rate (proportion of patients with complete response, partial response, or stable disease durable for > 6 cycles) was 9 % (3 patients) in the dose-escalation cohorts and 13 % (1 patient) in the RP2D cohort. Disease stabilization was reported in patients with breast cancer (n = 2; 1 patient each in the 500 mg BID and RP2D cohorts), ovarian cancer (n = 2; 1 patient each in the 50 mg BID and RP2D cohorts), head and neck cancer (n = 1; 75 mg BID), non-small-cell lung cancer (n = 1; 75 mg BID), skin cancer (n = 1; 150 mg BID), angiosarcoma (n = 1; 150 mg BID), clear-cell carcinoma (n = 1; 325 mg BID), gastric cancer (n = 1; 400 mg BID), ampulla of Vater adenocarcinoma (n = 1; 400 mg BID), melanoma (n = 1; RP2D), and squamous cell anal carcinoma (n = 1, RP2D). Both breast cancer patients with stable disease were HER2+; each had been previously treated with trastuzumab and 1 had been previously treated with lapatinib.
Discussion
TAK-285, an orally active multikinase inhibitor, was generally well tolerated at the MTD/RP2D of 400 mg BID. The most frequent AEs encountered at that dose level were diarrhea, fatigue, nausea, anorexia, and rash; therefore, the safety profile of TAK-285 was similar to that of other EGFR/HER2 inhibitors such as lapatinib [28, 29], and no unexpected AEs emerged during the trial. Pharmacokinetic analysis indicated that TAK-285 absorption was fast, with peak drug concentrations achieved 2 to 3 h postdose. Steady-state exposures increased with increasing dose, with evidence for a moderate degree of supra-proportionality in the dose-exposure relationship over the 50 mg BID to 500 mg BID dose range. As the clearance mechanisms of TAK-285 in humans in vivo are not definitively elucidated, the specific reasons for this observation are not currently known. The t½ of TAK-285 was approximately 9 h, supporting BID dosing in this study. Approximately 3-fold accumulation with BID dosing was observed at the MTD of 400 mg (Fig. 2a; Table 4). Steady-state pharmacokinetics was achieved by day 8, consistent with the estimated t½. Pharmacokinetic variability in steady-state systemic exposures of TAK-285 was relatively high (%CV in AUC0-τ of 58 % at the MTD of 400 mg BID; Table 4) despite low variability in the steady-state t½ (%CV of 11 %). These observations suggest that the variability in systemic exposures of TAK-285 is likely explained by interindividual variability in bioavailability (absorption and/or first-pass metabolism by CYP3A4 in the intestine and liver) rather than variability in systemic clearance. The best response to TAK-285 in this study was stable disease in 13 patients. Among the 54 patients enrolled, 9 had breast cancer (7 HER2+), and 2 of the HER2+ patients had stable disease.
A major consideration that led to the present study with TAK-285 is the high incidence of brain metastases in HER2+ breast cancer and the significant unmet need for more effective therapy for these patients. Preclinical studies suggest that TAK-285 crosses the intact BBB in rats and is not a substrate for the BBB efflux transporters MDR1 P-gp or BCRP—features that may distinguish it from the EGFR/HER2 inhibitor lapatinib, which has been evaluated in this setting [11]. Therefore, a unique aspect of this phase 1 study was the characterization of the distribution of TAK-285 into CSF in an expansion cohort dosed at the MTD/RP2D to determine whether bioactive exposures of TAK-285 are achievable in human CNS. The use of CSF distribution as a surrogate of distribution to brain interstitial fluid (ISF) is supported by preclinical data in rats, which suggested quantitatively similar extents of distribution into the CSF and ISF and the lack of meaningful CSF-ISF gradients [23]. TAK-285 displayed good CSF distribution, with the unbound concentrations achieved in CSF averaging 66 % of what would be theoretically achievable in the setting of unrestricted distribution of unbound drug from plasma to CSF. Excellent correlation was observed between the measured unbound CSF concentrations and concurrently measured unbound plasma concentrations of TAK-285 (Fig. 4a). Between-patient variability in unbound CSF TAK-285 concentration is explained largely by between-patient variability in systemic exposures of TAK-285 (Table 4) and is not reflective of variability in CNS distributional processes. The individual ratios of the measured CSF TAK-285 concentrations at 3 to 4 h postdose on day 15 (ie, at pharmacokinetic steady-state) to the corresponding plasma concentrations measured at the same time, together with individual values of TAK-285 free fraction in CSF and plasma Css,avg were used to calculate individual values of steady-state average unbound concentrations achieved in CSF using Eq. 1. An important assumption underlying this calculation is that the measured CSF-plasma concentration ratio at 3 to 4 h postdose on day 15 is a reasonable estimate of the CSF-plasma concentration ratio over the entire steady-state dosing interval. This assumption is supported by the high transmembrane permeability of TAK-285, in vitro data that it is not a substrate for BBB efflux transporters (P-gp, BCRP), preclinical data in rats supporting similar temporal profiles of TAK-285 disposition in brain tissue and systemic circulation [24], and lack of a large fluctuation in plasma TAK-285 concentrations over the steady-state dosing interval (mean PTR of 2.8). Importantly, the results of these calculations indicate that the geometric mean and individual values of steady-state average unbound concentrations achieved in CSF at 400 mg BID were all below the HER2 kinase IC50 (Fig. 4b). These data indicate that biologically relevant levels of target inhibition are not expected to be observed in human CNS after treatment with TAK-285 at the MTD/RP2D.
A key consideration associated with this interpretation is that the CSF distribution of TAK-285 was evaluated in patients without CNS metastases in this study. Higher local levels of distribution of TAK-285 may still be possible within regions of brain metastases, where the BBB may be partially compromised. Even though it is a substrate for efflux transporters, lapatinib has demonstrated limited antitumor activity in patients with brain metastases from HER2+ breast cancer. In a single-arm phase 2 trial (N = 242), the objective response rate to lapatinib monotherapy was 6 % and the clinical benefit rate was 43 %; 8 % of patients experienced a ≥ 50 % reduction in CNS tumor size [14]. Intratumoral levels of lapatinib were not assessed in that study, but it is plausible that access of lapatinib to tumor tissue may be aided by a compromised BBB. In an experimental model of HER2+ brain metastases, intratumoral lapatinib levels were variable and correlated with altered blood-tumor barrier permeability [12]. However, preventing progression of micrometastases will require drug availability within the CNS in regions of preserved BBB integrity, as has been discussed for malignant gliomas [30]. Additionally, the degree of disruption of the BBB because of brain metastasis can be highly variable between patients with metastatic breast cancer. It has been reported that HER2+ brain metastases tend to be associated with preservation of the BBB, whereas BBB disruption frequently occurs in CNS metastases of triple-negative or basal-type breast cancers [31]. Therefore, achievement of bioactive exposures across an intact BBB may still be important for advancement of clinical therapeutics for HER2+ metastatic breast cancer.
The lack of objective responses indicates that TAK-285 offers no advantage over currently available and emerging therapies such as the HER2 dimerization inhibitor pertuzumab. The phase 3 CLEOPATRA trial enrolled 808 patients with metastatic HER2+ breast cancer; patients had not received prior chemotherapy or biologic therapy for their metastatic disease [32]. In that study, the combination of pertuzumab plus trastuzumab and docetaxel significantly extended progression-free survival compared with trastuzumab plus docetaxel alone (18.5 vs 12.4 months; P < .001), and the objective response rate in the pertuzumab arm was 80 % [32]. The pertuzumab/trastuzumab/docetaxel combination received FDA approval in June 2012 [33].
Trials with the investigational HER2-targeted agent trastuzumab emtansine also have shown promising results. In a single-arm phase 2 study that enrolled heavily pretreated patients (N = 110) with refractory HER2+ disease, the overall response rate was 35 % and the clinical benefit rate was 48 % [34]. This agent was compared with the combination of lapatinib and capecitabine in a large phase 3 study in HER2+ patients (N = 991). An interim analysis showed that trastuzumab emtansine was associated with significantly longer progression-free survival (9.6 vs 6.4 months, P < .0001) and fewer serious AEs [35, 36]. If approved, trastuzumab emtansine could be an important therapeutic option for patients with HER2+ disease.
Treatment of advanced metastatic breast cancer remains a challenge; the high incidence of brain metastases among HER2+ patients is of particular concern. The development of new HER2 kinase inhibitors that can cross the BBB and achieve biologically significant levels of CNS exposure is needed and is the subject of ongoing research.
References
Baselga J (2010) Treatment of HER2-overexpressing breast cancer. Ann Oncol 21:vii36–vii40. doi:10.1093/annonc/mdq421
Bianco R, Melisi D, Ciardiello F, Tortora G (2006) Key cancer cell signal transduction pathways as therapeutic targets. Eur J Cancer 42:290–294
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235:177–182. doi:10.1126/science.3798106
Jackisch C (2006) HER-2-positive metastatic breast cancer: optimizing trastuzumab-based therapy. Oncologist 11:34–41. doi:10.1634/theoncologist.11-90001-34
Vogel CL, Cobleigh MA, Tripathy D et al (2002) Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol 20:719–726. doi:10.1200/JCO.20.3.719
Slamon DJ, Leyland-Jones B, Shak S et al (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344:783–792. doi:10.1056/NEJM200103153441101
Cobleigh MA, Vogel CL, Tripathy D et al (1999) Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol 17:2639–2648
Nahta R, Esteva FJ (2006) HER2 therapy: molecular mechanisms of trastuzumab resistance. Breast Cancer Res 8:215. doi:10.1186/bcr1612
Lin NU, Winer EP (2007) Brain metastases: the HER2 paradigm. Clin Cancer Res 13:1648–1655. doi:10.1158/1078-0432.CCR-06-2478
Contessa JN, Hamstra DA (2008) Revoking the privilege: targeting HER2 in the central nervous system. Mol Pharmacol 73:271–273. doi:10.1124/mol.107.042986
Polli JW, Olson KL, Chism JP et al (2009) An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016). Drug Metab Dispos 37:439–442. doi:10.1124/dmd.108.024646
Taskar KS, Rudraraju V, Mittapalli RK et al (2012) Lapatinib distribution in HER2 overexpressing experimental brain metastases of breast cancer. Pharm Res 29:770–781. doi:10.1007/s11095-011-0601-8
Lin NU, Carey LA, Liu MC et al (2008) Phase II trial of lapatinib for brain metastases in patients with human epidermal growth factor receptor 2-positive breast cancer. J Clin Oncol 26:1993–1999. doi:10.1200/JCO.2007.12.3588
Lin NU, Dieras V, Paul D et al (2009) Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin Cancer Res 15:1452–1459. doi:10.1158/1078-0432.CCR-08-1080
Neuwelt EA (2004) Mechanisms of disease: the blood–brain barrier. Neurosurgery 54:131–140, discussion 141-132
Muldoon LL, Soussain C, Jahnke K et al (2007) Chemotherapy delivery issues in central nervous system malignancy: a reality check. J Clin Oncol 25:2295–2305. doi:10.1200/JCO.2006.09.9861
Deeken JF, Loscher W (2007) The blood–brain barrier and cancer: transporters, treatment, and Trojan horses. Clin Cancer Res 13:1663–1674. doi:10.1158/1078-0432.CCR-06-2854
Ishikawa T, Seto M, Banno H et al (2011) Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo[3,2-d]pyrimidine scaffold. J Med Chem 54:8030–8050. doi:10.1021/jm2008634
Takagi S, Hayashi A, Ohta Y (2009) HER2 and HER3 cooperatively regulate tumor cell growth and determine sensitivity to the HER kinase inhibitor TAK-285. Cancer Res 69(2 Suppl 1):Abstract 3155. doi: 10.1158/0008-5472.SABCS-3155
Aertgeerts K, Skene R, Yano J et al (2011) Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J Biol Chem 286:18756–18765. doi:10.1074/jbc.M110.206193
Shell S, Pry K, Trusk P et al (2008) Novel inhibitory mechanisms of TAK-285, a new EGFR/ErbB2 dual inhibitor. Eur J Cancer 6:100, Abstract 313
Iwahara A, Tamura T, Takagi S, Kamiguchi H, Yusa T, Ohta Y (2008) In vivo antitumor efficacy of TAK-285, a novel ErbB1/ErbB2 dual kinase inhibitor. Eur J Cancer 6:99–100. doi:10.1016/S1359-6349(08)72245-6, Abstract 311
Wu J-T, Gordon J, Liao M et al. (2011) Comparison of various nonclinical in vivo/in vitro models to assess CNS penetration of TAK-285, a novel HER2/EGFR inhibitor. Abstract P93. Presented at the 17th North American Regional ISSX Meeting; Atlanta, GA; October 16–20, 2011
Erdo F, Gordon J, Wu JT, Sziraki I (2012) Verification of brain penetration of the unbound fraction of a novel HER2/EGFR dual kinase inhibitor (TAK-285) by microdialysis in rats. Brain Res Bull 87:413–419. doi:10.1016/j.brainresbull.2012.01.002
Doi T, Takiuchi H, Ohtsu A et al (2012) Phase I first-in-human study of TAK-285, a novel investigational dual HER2/EGFR inhibitor, in cancer patients. Br J Cancer 106:666–672. doi:10.1038/bjc.2011.590
Therasse P, Arbuck SG, Eisenhauer EA et al (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216. doi:10.1093/jnci/92.3.205
Chiorean EG, Sausville EA, Heath EI et al. (2011) Phase 1 study of TAK-285, an investigational HER2/EGFR inhibitor, in patients with advanced cancer: updated results and assessment of human CSF distribution. J Clin Oncol 29(suppl). Abstract 2538
Moy B, Goss PE (2007) Lapatinib-associated toxicity and practical management recommendations. Oncologist 12:756–765. doi:10.1634/theoncologist.12-7-756
Roy V, Perez EA (2009) Beyond trastuzumab: small molecule tyrosine kinase inhibitors in HER-2-positive breast cancer. Oncologist 14:1061–1069. doi:10.1634/theoncologist.2009-0142
Agarwal S, Sane R, Oberoi R, Ohlfest JR, Elmquist WF (2011) Delivery of molecularly targeted therapy to malignant glioma, a disease of the whole brain. Expert Rev Mol Med 13:e17. doi:10.1017/S1462399411001888
Yonemori K, Tsuta K, Ono M et al (2010) Disruption of the blood brain barrier by brain metastases of triple-negative and basal-type breast cancer but not HER2/neu-positive breast cancer. Cancer 116:302–308. doi:10.1002/cncr.24735
Baselga J, Cortes J, Kim SB et al (2012) Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med 366:109–119. doi:10.1056/NEJMoa1113216
U.S. Food and Drug Administration (2013) FDA approves Perjeta for type of late-stage breast cancer (2012). http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm307549.htm. Accessed 23 Jan 2013
Krop IE, Lorusso P, Miller KD et al (2012) A phase II study of trastuzumab emtansine in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer who were previously treated with trastuzumab, lapatinib, an anthracycline, a taxane, and capecitabine. J Clin Oncol 30:3234–3241. doi:10.1200/JCO.2011.40.5902
Blackwell K, Miles D, Gianni L et al. (2013) EMILIA: T-DM1 demonstrates greater safety, efficacy for advanced HER2-positive breast cancer (summary of 2012 ASCO oral presentation). http://chicago2012.asco.org/ASCODailyNews/LBA1.aspx. Accessed 23 Jan 2013
Blackwell KL, Miles D, Gianni L et al. (2012) Primary results from EMILIA, a phase III study of trastuzumab emtansine (T-DM1) versus capecitabine (X) and lapatinib (L) in HER2-positive locally advanced or metastatic breast cancer (MBC) previously treated with trastuzumab (T) and a taxane. J Clin Oncol 30(suppl). Abstract LBA1
Acknowledgments
We thank William Sinkins, PhD, ProEd Communications, Inc.®, for his medical editorial assistance with this manuscript.
Support
Financial support for the conduct of this study and for medical editorial assistance was provided by Millennium Pharmaceuticals, Inc.
Conflict of interest
P. LoRusso is a consultant for Millennium Pharmaceuticals, Inc. K. Venkatakrishnan, D. Noe, J.-T. Wu, S. Sankoh, and M. Corvez are employees of Millennium Pharmaceuticals, Inc. E.G. Chiorean and E. Sausville received research funding from Millennium Pharmaceuticals, Inc.
Ethical standards
This study complied with the current laws of the country (United States) in which it was conducted.
Author information
Authors and Affiliations
Corresponding author
Additional information
Trial Registration
NCT00535522 [http://clinicaltrials.gov/ct2/show/NCT00535522]
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
LoRusso, P., Venkatakrishnan, K., Chiorean, E.G. et al. Phase 1 dose-escalation, pharmacokinetic, and cerebrospinal fluid distribution study of TAK-285, an investigational inhibitor of EGFR and HER2. Invest New Drugs 32, 160–170 (2014). https://doi.org/10.1007/s10637-013-9988-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-013-9988-x