
Summary
Purpose: Angiogenesis plays an essential role in tumor development, invasion and metastasis. We evaluated the efficacy and safety of dual angiogenesis blockade with bevacizumab and sorafenib in patients with metastatic breast cancer. Patients and Methods: Patients who had received no more than 2 prior chemotherapy regimens in any setting were treated with sorafenib 200 mg as a single oral dose daily plus bevacizumab intravenously 5 mg/kg every other week. Response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST). The primary endpoint was progression free survival (PFS). Results: Eighteen patients were enrolled. Median age was 56 yo, all had good performance status KPS of 0 or 1, and 17 patients had received 1 or 2 prior chemotherapy regimens. Median PFS was 2.8 months. There were no complete or partial responses; 3 patients had stable disease for >6 months. Toxicity was substantial with 9 (50 %) patients reporting Grade 3 toxicity. Seven (39 %) patients discontinued therapy due to adverse events including hypertension (N = 2), GI toxicity (N = 1), sensory neuropathy (N = 1), rash (N = 1), pain (N = 1) and wound complication (N = 1). Given the lack of clear efficacy and increased toxicity, accrual was terminated. Conclusion: The combination of sorafenib and bevacizumab has substantial toxicity and minimal efficacy in patients with previously treated metastatic breast cancer. Further study of this combination is not recommended.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Metastatic breast cancer is generally incurable with only a few patients achieving long-term survival after standard chemotherapy [1]. Though an expanding arsenal of active agents is available for the treatment of metastatic disease, overall survival has changed little during the last half century.
Extensive laboratory data suggests that angiogenesis plays an essential role in breast cancer development, invasion and metastasis. Of the identified angiogenic factors, vascular endothelial growth factor (VEGF; also known as vascular permeability factor) is the most potent and specific and is a crucial regulator of both normal and pathologic angiogenesis [2]. Bevacizumab, a monoclonal antibody directed against VEGF-A, has modest activity as a single agent but improves response rate and PFS but not overall survival when administered in combination with chemotherapy.
There is also strong evidence supporting a role for platelet-derived growth factor receptor (PDGFR) signaling in breast cancer autocrine and paracrine signaling. PDGFR has been detected in the stromal compartment of many human breast cancers by immunostaining [3]. High levels of immunostaining for PDGF receptor ligands, PDGF-A and PDGF-B, have been commonly detected directly on breast cancer cells in archival human tumors [4].
Sorafenib is a multikinase inhibitor targeting several serine/threonine and receptor tyrosine kinase receptors including VEGF, PDGF and the mitogen activated protein kinases (MAPK) pathway [5]. Sorafenib monotherapy (400 mg twice daily) had little activity in patients with previously treated metastatic breast cancer in two previously reported trials [6]. The addition of sorafenib to chemotherapy drugs is currently being investigated in the TIES (Trials to Investigate the Effects of Sorafenib in Breast Cancer) program. Two of these studies have already been completed and presented—the NU 07B1 and the SOLTI-0701 trials. The SOLTI trial reported by Baselga et al. showed a significant clinical benefit with the combination of capecitabine and sorafenib when compared to capecitabine monotherapy, PFS of median, 6.4 vs. 4.1 mo; hazard ratio 0.58; 1-sided p = 0.0006. This was achieved at the expense of more toxicity. The NU07B1 trial reported by Gradishar et al., compared the safety and efficacy of sorafenib in combination with paclitaxel vs paclitaxel with placebo. Results showed no improvement in PFS which was the primary endpoint [7–9].
Considering that pro-angiogenic peptides are produced in increasing numbers as tumors progress, we hypothesized that combined inhibition of multiple angiogenesis pathways, whether serial or parallel, may increased activity. Results of a phase I trial of a combination of bevacizumab and sorafenib in 39 patients with a variety of tumour types (Azad et al., 2008a, b) has already been reported including ovarian cancer. Their results showed an unexpectedly high partial response rate (PR 1⁄4 46 %) in patients with relapsed EOC compared with 16–21 % response rate reported with bevacizumab alone [10]. Our trial evaluates combined VEGF inhibition with sorafenib and bevacizumab in pre-treated patients with advanced breast cancer.
Patients and methods
Patient eligibility
Eligible patients, ≥18 years old, had histologically confirmed adenocarcinoma of the breast with evidence of metastatic disease, measurable according to RECIST 1.0 criteria. Patients were also required to have an ECOG performance status of 0 or 1 as well as adequate hematologic, renal and hepatic function. Patients with HER2 positive (3+ by immunohistochemistry or gene amplification by fluorescence in situ hybridization) disease must have received prior trastuzumab therapy. Patients could not have received more than 2 prior chemotherapy regimens, considering all adjuvant and neoadjuvant therapy as one regimen. Prior hormonal therapy was allowed. Prior radiation therapy was allowed as long as the irradiated area is not the only source of evaluable disease.
The study was performed in compliance of good clinical practice, the Helsinki Declaration, and federal and institutional guidelines. Ethical review boards at each institution approved the trial protocol prior to patient enrollment. All patients provided written informed consent.
Treatment plan
Sorafenib was administered orally at 200 mg daily; sorafenib dose could be increased to 200 mg twice daily in patients without significant toxicity in the first two cycles. Bevacizumab was given intravenously at 5 mg/kg every other week. One cycle was considered to be 28 days. Treatment continued until disease progression or unacceptable toxicity intervened.
Toxicities were evaluated using the Common Terminology Criteria for Adverse Events (CTCAE) version 3. Patients were evaluated clinically every week for the first cycle than every other week for the subsequent cycle. Hypertension was monitored weekly during the first cycle, then prior to each bevacizumab infusion. Urine protein creatinine ratio or dipstick urinalysis was performed every 8 weeks. Complete blood count and serum chemistries were obtained before each cycle. No Bevacizumab dose reduction was allowed and no dose modification was specified for grades 1/2 toxicities. Bevacizumab was discontinued for any arterial thromboembolic event, all grade 4 non hematologic toxicities, and grades 3 and 4 allergic reactions. Bevacizumab therapy was held for other clinically relevant grade 3 toxicities, then resumed upon resolution or adequate control. Sorafenib was held for all grade 3/4 toxicities and, depending on clinical severity, either permanently discontinued or restarted at a lower dose upon resolution.
Disease status was assessed according to RECIST criteria at the end of every third cycle.
Statistical considerations
The primary endpoint was PFS, defined as the time from first treatment to disease progression or death from any cause. The study was designed to detect an improvement from median PFS of 3 months based on the previous experience with bevacizumab monotherapy to 5 months. Assuming a type I error of 5 % and 90 % power, 39 patients were required. To account for potential dropouts and unevaluable patients, a total sample size of 42 patients was initially planned. Based on the toxicity experience in the first 18 patients, an unplanned analysis of PFS was requested. When this failed to suggest significant benefit for the combined regimen, the study was terminated.
Secondary endpoints included overall response rate (ORR), clinical benefit response defined as the percentage of evaluable patients with confirmed complete response (CR), partial response (PR), or stable disease (SD) for at least 6 months (CR+PR+SD), and safety. The median and range of continuous variables are presented. Frequencies and percents of categorical variables are provided. Exact binomial 95 % confidence intervals were calculated for response. The method of Kaplan-Meier was used to calculate median duration of follow-up, survival, and progression free survival [11].
Results
Patient characteristics
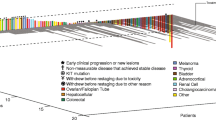
From October 2007 to March 2009, 18 patients were enrolled. Patient characteristics are shown in Table 1. Nearly half of the patients had triple negative (ER-, PR-, HER2-) disease, and most had received 1 or 2 prior chemotherapy regimens.
Efficacy
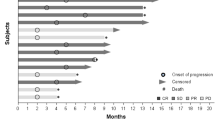
Median PFS was 2.8 months, 95 % CI (1.8–6.1). There were no complete or partial responses. Six patients had stable disease but only three (20 %) were stable for 6 months or more. Three patients died within 30 days of last therapy.
Toxicity
In general, this toxicity was substantial. Although no grade 4 toxicity was noted, 9 (50 % of patients reported grade 3 toxicity (Table 2). Hypertension was common with 14 (78 %) patients experiencing grade 2 or 3 toxicity. GI toxicity, sensory neuropathy, rash, and pain were also relatively common. Seven patients discontinued therapy due to toxicity; reasons for treatment discontinuation were uncontrolled hypertension (n = 2), nausea (n = 1), neuropathy (n = 1), pain (n = 1), rash (n = 1) and wound complications (n = 1). One patient had a grade 3 hypersensitivity reaction; however patient was able to continue with treatment with premedication. No hematologic toxicities were reported, however elevation in the liver transaminases were noted in two patients.
Discussion
This report summarizes the results of the study of the combination of Sorafenib with Bevacizumab in previously treated patients with metastatic breast cancer. Our results show that the combination of the two angiogenic inhibitors in this patient population is poorly tolerated. All patients experienced some kind of toxicity and in half of them the toxicity was reported to be grade 3. The trial was stopped early because of the poor safety profile as well as the lack of significant efficacy of this combination over bevacizumab alone in the short follow up period where it was evaluated.
The significant toxicity of this combination questions the safety of dual VEGF blockade or VEGF/PDGF inhibition. This problem had been encountered in the earlier phase I studies of Sorafenib and Bevacizumab where a recurrent theme was the inability to dose escalate those drugs and where the maximal tolerated dose ended up having to be the initial dose which is usually much lower than the efficacious dose [10, 12]. When it comes to double angiogenesis blockade, our experience also mirrors some of the other combinations like with the combination of bevacizumab with Sunitinib. Several studies with this combination in solid tumors had to be halted secondary to significant side effects including anemia, myelosuppression, fatigue and gastrointestinal complications. In a phase I study by Feldman et al., Sunitinib and bevacizumab was tried in patients with advanced renal cell carcinoma, again significant toxicity was encountered, with similar toxicity profile with the one we encountered with hypertension, fatigue and GI problems being very common [13]. Other studies reflect the same reality. In a recent phase 3 trial the SABRE-B study evaluated the triple combination of paclitaxel, bevacizumab and Sunitinib in metastatic breast cancer, that study accrual was also stopped because poor tolerability and inability to dose escalate sunitinib as planned secondary to the toxicity profile [14]. Many other trials reproduced similar results [15, 16]. The broad spectrum of signaling inhibition of the TKI’s brought increased toxicity with dose reductions and limited drug exposure that ultimately, we believe, hindered activity.
Overall results with VEGF TKIs in MBC have been disappointing. The broader spectrum of activity had provided hope for greater efficacy. However, with that broader profile comes increased toxicity, need for dose reductions and the general inability to combine with chemotherapy.
In conclusion drug development is a challenging task; the combination of biologics is very unpredictable and could be misleading. A deeper understanding of the biology of breast cancer and its underlying molecular identity is warranted prior to embarking into further targeted therapy trials.
References
Greenberg PA et al (1996) Long-term follow-up of patients with complete remission following combination chemotherapy for metastatic breast cancer. J Clin Oncol 14:2197–2205
Ferrara N, Davis-Smyth T (1997) The biology of vascular endothelial growth factor. Endocr Rev 18:4–25
Bhardwaj B et al (1996) Localization of platelet-derived growth factor beta receptor expression in the periepithelial stroma of human breast carcinoma. Clin Cancer Res 2:773–782
Coltrera MD, Wang J, Porter PL, Gown AM (1995) Expression of platelet-derived growth factor B-chain and the platelet-derived growth factor receptor beta subunit in human breast tissue and breast carcinoma. Cancer Res 55:2703–2708
Huang J, Li X, Hilf R, Bambara RA, Muyan M (2005) Molecular basis of therapeutic strategies for breast cancer. Curr Drug Targets Immune Endocr Metabol Disord 5:379–396
Moreno-Aspita A, Hillman D, Wisenfeld TJ, et al (2006) BAY 43–9006 as single oral agent in patients with metastatic breast cancer previously exposed to anthracycline and/or taxane. ASCO abstract 577
Baselga J, Gianni L, Gradishar WJ et al (2009) Phase IIb double-blind, randomized, placebo-controlled trials for the efficacy and safety of sorafenib in patients (pts) with metastatic or locally advanced breast cancer (BC): review of the Trials to Investigate the Effects of Sorafenib in BC (TIES) program. Presented at: 2009 ASCO Annual Meeting. Chicago, IL, USA, 4–8 June 2009
Gradishar WJ, Kaklamani V, Prasad Sahoo T, et al (2009) A double- blind, randomized, placebo-controlled, phase 2b study evaluating the efficacy and safety of sorafenib (SOR) in combination with paclitaxel (PAC) as a first-line therapy in patients (pts) with locally recurrent or metastatic breast cancer (BC). Presented at: 32nd Annual San Antonio Breast Cancer Symposium. December 10–13, 2009. Abstract 44
Baselga J, Grupo Español de Estudio Tratamiento y Otras Estrategias Experimentales en Tumores Sólidos, Roché H et al (2009) SOLTI-0701: A multinational double-blind, randomized Phase IIB study evaluating the efficacy and safety of sorafenib compared to placebo when administered in combination with capecitabine in patients with locally advanced or metastatic breast cancer (BC). Presented at: ECCO 15 and 34th ESMO Multidisciplinary Congress. Berlin, Germany, 20–24 September (2009) (Abstract 3LBA). Updated at: 32nd Annual San Antonio Breast Cancer Symposium. San Antonio, TX, USA, 9–13 December 2009 (Abstract 45). Demonstrates significant activity of sorafenib in combination with capecitabine
Azad NS et al (2008) Combination targeted therapy with sorafenib and bevacizumab results in enhanced toxicity and antitumor activity. J Clin Oncol 26:3709–3714
Kaplan EL, Meier P (1958) Nonparametric estimation from incomplete observations. J Am Stat Assoc 53:457–481
Sosman J, Flaherty K, Atkins M et al (2008) Updated results of phase I trial of sorafenib (S) and bevacizumab (B) in patients with metastatic renal cell cancer (mRCC). J Clin Oncol 26(15S):A5011
Feldman DR et al (2009) Phase I trial of bevacizumab plus escalated doses of sunitinib in patients with metastatic renal cell carcinoma. J Clin Oncol 27:1432–1439
Mayer EL et al (2010) SABRE-B: an evaluation of paclitaxel and bevacizumab with or without sunitinib as first-line treatment of metastatic breast cancer. Ann Oncol 21:2370–2376
Rini BI et al (2009) A phase I study of sunitinib plus bevacizumab in advanced solid tumors. Clin Cancer Res 15:6277–6283
Socinski MA, Scappaticci FA, Samant M, Kolb MM, Kozloff MF (2010) Safety and efficacy of combining sunitinib with bevacizumab+paclitaxel/carboplatin in non-small cell lung cancer. J Thorac Oncol 5:354–360
Conflict of interest
Lida A Mina, MD.: No Conflict of Interest
Cynthia S Johnson: No Conflict of Interest
Cynthia Burkhardt: No Conflict of Interest
Menggang Yu: No Conflict of Interest
Robin Zon, MD: No Conflict of Interest
Kathy D Miller, MD: Honoraria to disclose: Yes Genentech
Research funding to disclose: Genentech and Trial supported by Bayer
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported in part by a Breast Cancer Research Foundation grant to KDM
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Mina, L.A., Yu, M., Johnson, C. et al. A phase II study of combined VEGF inhibitor (bevacizumab+sorafenib) in patients with metastatic breast cancer: Hoosier Oncology Group Study BRE06-109. Invest New Drugs 31, 1307–1310 (2013). https://doi.org/10.1007/s10637-013-9976-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-013-9976-1